

Abstract
Although the transcription factor Sox8 is broadly expressed during embryogenesis in developing ectodermal and mesodermal tissues, mice develop surprisingly normally in the absence of Sox8. Phenotypes in adult Sox8-deficient mice include mild osteopenia, late-onset male infertility, and reduced weight. We show here that progressive degeneration of adipose tissue in adult Sox8-deficient mice significantly contributes to weight reduction. Although serum levels of leptin, IGF-1, and noradrenaline were altered in Sox8-deficient mice, these changes could not explain the observed phenotype. Other serum parameters, including indicators of glucose metabolism, were largely normal. However, expression of the preadipocyte marker Pref-1 was elevated in adipose tissues of Sox8-deficient mice. This increase correlated with an impaired differentiation of Sox8-deficient fibroblasts to adipocytes in culture, a defect that could be rescued by reintroducing Sox8 into the cells. Furthermore, Sox8 levels were higher in mesodermal precursors than in mature adipocytes. We postulate a precursor-intrinsic role of Sox8 during replenishment of the adipocyte pool in adult mice and assume that disturbance of this function significantly contributes to adipose tissue degeneration in Sox8-deficient mice.
Keywords: high-mobility-group, mesoderm, Sry, transgenic mouse model
Transcription factors of the Sox protein family play many important roles during development (1–3). Much less is known about their role in adult homeostasis, tissue maintenance, and regeneration. The strong expression of many Sox proteins during adult life and/or upregulation following tissue damage nevertheless suggests that Sox proteins may continue to be essential regulators after development.
Sox8, Sox9, and Sox10 constitute subgroup E of the Sox protein family in vertebrates. These SoxE proteins furthermore exhibit a strongly overlapping expression during vertebrate development, and there are clear indications that they are functionally similar but not exactly identical (4–8). All three SoxE genes have been deleted in the mouse. These studies have revealed essential roles of Sox9 in chondrocyte, male gonad, neural crest, central nervous system (CNS) and gut development (4, 6, 9–13) and equally important tasks for Sox10 during neural crest and CNS development (14–17). Functions are also reflected in the syndromes that are associated with heterozygous mutations of SOX9 and SOX10 in humans. SOX9 mutations lead to the skeletal malformation syndrome Campomelic Dysplasia and male-to-female sex reversal (18, 19), whereas SOX10 mutations cause Waardenburg-Hirschsprung disease, frequently in combination with peripheral neuropathies and central dysmyelinating leukodystrophies (20, 21).
Compared with these strong loss-of-function phenotypes, Sox8 deletion led to rather mild postnatal phenotypes in the mouse (22). Sox8-deficient mice were viable and able to produce offspring (22), although males developed sterility at older ages (23). Dramatic phenotypic manifestations are also not apparent in humans with SOX8 deletions. This is evident from patients with α-thalassemia/mental retardation syndrome ATR-16 in which large deletions are detected in chromosomal region 16pter-p13.3 that also affect the SOX8 gene. None of the main symptoms in ATR-16 patients can, however, be linked to the loss of SOX8 (24).
On a mixed genetic background, adult Sox8-deficient mice were slightly smaller, but severely lighter than their wild-type littermates, whereas they were equally smaller and lighter on an inbred C57Bl/6J background (22, 25). On the C57Bl/6J background, weight reduction correlated with a reduced bone mass that was caused by precocious osteoblast differentiation thus identifying Sox8 as a negative regulator of this process (25). While certainly a contributing factor, the lower bone mass was not sufficient to explain the full extent of the observed weight reduction. Especially on a mixed genetic background, the osteopenic phenotype is too mild to account for the complete difference in weight. Here we report that Sox8-deficient mice additionally have a lipodystrophy-like phenotype and that postnatal loss of adipose tissue mass is a major cause for weight reduction.
MATERIALS AND METHODS
Animal husbandry, genotyping, and tissue preparation
Mice with a Sox8lacZ allele were kept as heterozygotes or homozygotes on a mixed C57Bl/6J:C3HeB/FeJ background under standard housing conditions (22). Genotyping was performed by PCR as described (22). Embryos were obtained at 11.5 and 16.5 days post coitum (dpc) from staged pregnancies. Two- and four-month-old animals were sacrificed, and various tissues were collected. Length of animals was measured from nose to anus.
Actimot, metabolic cages, and leptin sensitivity test
TSE ActiMot / MoTil TSE system (Bad Homburg, Germany) was used for activity analysis. Two-month-old male Sox8-deficient and wild-type littermates were monitored for 24 h. The total observation time was divided into resting periods, active periods (with movements between 5 cm/s and 20 cm/s), and hyperactive periods (with movements exceeding 20 cm/s). Age-matched Sox8-deficient and wild-type mice were kept in metabolic cages (Tecniplast, Neumarkt-Sankt Veit) for five days to analyze food and water intake and to collect urinary samples. To determine leptin sensitivity, mice were intraperitoneally injected after two days in metabolic cages with 0.4 μg recombinant mouse leptin (Sigma, Taufkirchen) in PBS per g body weight or PBS alone twice a day (9 AM and 6 PM). Food intake and body weight was measured daily at 9 AM.
Glucose tolerance test
Glucose tolerance tests were performed after overnight fasting for 15 h. The animals were injected intraperitoneally with 10% glucose in aqua ad iniectabilia (sterile filtered) with 1.5 mg glucose per g body weight. Tail-vein blood samples were obtained before and 15, 30, 60, 90, and 120 min after injection and analyzed with a glucometer (Precision Xtra Plus, MediSense) or using the Ultra Sensitive Rat Insulin ELISA kit (Crystal Chem Inc.) as described below.
Serology
Blood samples were obtained by decapitation of etherized 2 and four-month-old Sox8-deficient and wild-type animals after 4 h fasting at 3:00 PM. After blood clotting and centrifugation, serum was stored at –20°C.
Serum levels were determined by ELISA for insulin using the Ultra Sensitive Rat Insulin ELISA kit with mouse insulin standard (Crystal Chem Inc.), for leptin using the Mouse Leptin ELISA Kit (Crystal Chem Inc.), and for Igf-1 using the Mouse/Rat Igf1 ELISA (DSL-10-29200, Diagnostics Systems Laboratories Inc.). Serum triglycerides and free fatty acids were measured using the Serum Triglyceride Determination kit (Sigma) and the NEFA C kit (Wako Diagnostics), respectively.
Noradrenaline measurements
Urine was collected from two-month-old Sox8-deficient and wild-type males on the second day of metabolic cage housing. HCl was added for stabilization of catecholamines, and probes were stored at –20°C before analysis. Noradrenaline was determined after alumina extraction by high-performance liquid chromatography combined with electrochemical detection and normalized to creatinine content of urine. Creatinine levels were similar between wild-type and Sox8-deficient mice.
Magnetic resonance imaging
T1-weighted MRI scanning was performed on a 4.7 T BRUKER Biospec scanner and used to study the distribution of fat in wild-type and Sox8-deficient mice of two and four months of age.
β-galactosidase staining
For β-galactosidase staining, transverse sections from five-day-old mice or whole 11.5 dpc-old embryos were incubated in 1% X-gal solution for several h at 37°C. After staining, samples underwent fixation in 4% paraformaldehyde. Whole mount embryos were further processed on a vibratome and cut into 100 μm sections.
Histological staining procedures
Collected tissues of adult animals underwent overnight fixation in 4% paraformaldehyde, dehydration, and paraffin-embedding. 5 μm sections were stained with Meyer's hemalum solution and eosin and examined by light microscopy.
Tissue culture, transfection, and adipocyte differentiation
To prepare primary mouse embryonic fibroblasts (MEF) Sox8-deficient and wild-type embryos were collected at 16.5 dpc and decapitated. After removal of the liver, embryos were washed three times in PBS and finely minced with a razor blade in trypsin solution. The resulting mixture was stirred in the presence of glass beads for 20 min before trypsin was inactivated by DMEM containing 20% fetal calf serum (FCS), 2 mM glutamine, 0.1 mM nonessential amino acid, 0.1 mM β-mercaptoethanol, and 10 U penicillin and streptomycin, each. After centrifugation at 900 rpm, cells were resuspended at a density of 5 × 106 cells per 15 cm dish in the same DMEM-based medium with FCS reduced to 10%. Subconfluent cells were passaged once and cryopreserved in liquid nitrogen, as soon as they again reached 60% confluence.
For differentiation into adipocytes, these MEF were rapidly thawed, passaged once more and grown on 10 cm dishes in DMEM, 10% FCS, 2 mM glutamine, 0.1 mM nonessential amino acid, 0.1 mM β-mercaptoethanol, and 10 U penicillin and streptomycin. Alternatively, mouse 3T3 L1 fibroblasts were used. Upon confluence, MEF or 3T3 L1 cells were placed for the first two days in medium additionally containing 1 μM insulin, 0.4 mg/ml dexamethasone and 0.5 mM isobutylmethylxanthine, and from the third day onwards in medium supplemented with 1 μM insulin.
For transfection experiments, Sox8-deficient MEF or 3T3 L1 cells were seeded in 24 well plates. Cells were transfected with 1 μg expression plasmid per well using lipofectamine 2000 (Invitrogen, Carlsbad) at 60% confluence, and once confluent, differentiated for five days. In addition to pCMV5-EGFP and pCMV5-Sox8-IRES2-EGFP, plasmids were used that expressed a Sox8-specific shRNA (positions 274 to 394 of mouse Sox8, acc no. NM_011447) in wild-type or scrambled version in the context of the pSUPER.neo + gfp vector (Oligoengine, Seattle).
Immunoprecipitation and western blot
Extracts were prepared from cells immediately after they reached confluence or after seven days of differentiation. After overnight incubation with a Sox8-specific antibody (26), Sox8 was immunoprecipitated from extracts using protein A-Sepharose beads (Amersham Biosciences). After centrifugation and repeated washing, precipitates were subjected to SDS polyacrylamide gel electrophoresis. Separated proteins were blotted onto nitrocellulose, and membranes were incubated with antisera against Sox8 (1:2,000 dilution) or acetylated α-tubulin (1:2,000; Sigma, Taufkirchen, Germany) before detection with horseradish-peroxidase–coupled secondary antibodies (1:3,000 dilution) and the ECL kit (Amersham Biosciences). Western blots were also performed to detect Sox8 in 293 cells transfected with pCMV5-Sox8 alone or in combination with Sox8-specific shRNA expression plasmids.
Oil Red O staining
Tissues collected from Sox8-deficient and wild-type mice underwent overnight fixation in 1% paraformaldehyde before they were embedded in OCT compound, frozen at –80°C, and transversely cut into 10 μm sections on a cryotome (Leica, Bensheim). Oil Red O stock solution (0.5% in isopropanol) was diluted in water (3:2) and filtered through a 0.45 μm filter. After washing in PBS, tissue sections were stained with 1 ml Oil Red O solution in a humid chamber for 1 h at room temperature. After washing with water, sections were treated for 1 h with 4% paraformaldehyde and mounted in mowiol.
Differentiated fibroblasts were stained with Oil Red O nine days after induction when untransfected and five days after induction when transfected. After fixation in 4% paraformaldehyde for 10 min at room temperature, dishes were stained with Oil Red O for 30 min. To specifically determine the fraction of transfected fibroblast that had undergone differentiation, counter-staining was performed with anti-GFP primary (1:10,000 dilution; Molecular Probes) and Alexa-coupled secondary (1:1,000 dilution; Molecular Probes) antibodies. In some experiments, the Oil Red O retained on each dish was also photometrically quantified by absorbance at 520 nm after excess staining solution was removed by washing briefly first with 70% ethanol and then with water and extracting the dye with 1 ml of 4% Nonident P-40 in isopropanol for 5 min.
RNA preparation and RT-PCR analyses
RNA was prepared from MEF and 3T3 L1 fibroblasts before and during the differentiation regimen and from several tissues of two- and four-month-old wild-type and Sox8-deficient mice using Trizol reagent (Invitrogen). Two micrograms of total RNA from each sample were reverse-transcribed in a total volume of 25 μl into cDNA using oligo-dT primers and Moloney murine leukemia virus reverse transcriptase (New England Biolabs). Polymerase chain reactions were performed in a quantitative manner with 0.2 μl of each cDNA on a Roche Lightcycler according to the manufacturer's instructions using the ABsolute™ QPCR SYBR® Green Mix (Thermo Fisher Scientific Inc.) with an annealing temperature of 60°C. Amounts of all PCR products were normalized to Rpl8 (5). The following gene-specific primer pairs were used: 5′- GTGTGGACCGAGGGGCTTTTACTTC-3′ and 5′- GCTTCAGTGGGGCACAGTACATCTC-3′ for Igf1, 5′- TGGAGACCCCTGTGTCGGTT-3′ and 5′- AGCATTCAGGGCTAACATCCAACT-3′ for leptin, 5′- GACGTTACTACAACTGAAGAGC-3′ and 5′- CATTCTTTTCCTGATACTGGTC-3′ for adiponectin, 5′- GGATGAAGAACCTTTCATTTCC-3′ and 5′- AAGACTGCTGTGCCTTCTGG-3′ for resistin, 5′-GCCTGGCAGATATCATCACC-3′ and 5′ TGAGTCGTAGAGGCCAATCC-3′ for Ucp-1, 5′-CAGACCTTAAGGGACAACACG-3′ and 5′-GTGCATCGGATGTCGTAGG-3′ for CideA, 5′-CTGAACCCTGGAGCCTCTC-3′ and 5′-CGTCTCTGAACTTGAGTATTTGC-3′ for Cpt1b, 5′-TGTCAATGGAGTCTGCAAGG-3′ and 5′-GCAGTCCTTTCCAGAGAACC-3′ for Pref-1, 5′-GAACAGCAACGAGTACCGGGTA-3′ and 5′-CCATGGCCTTGACCAAGGAG-3′ for C/EBPα, 5′-CGAGGTTTCCACAAATAAAACC-3′ and 5′-GACTTCTTCAGAGACTTGTCATGG-3′ for LPL, 5′-TCTGCTCAAGTATGG TGTCC-3′and 5′-AGTCCTTGTAGATCTCCTGG-3′ for PPARγ2, and 5′-TGGAGTCTGGTGCCTATGCCTGT-3′ and 5′-GCCGAGCACTGCATCAGCTT TGT-3′ for Sox8.
Software and Statistics
Adipocyte cell diameter was measured with Scion Image (Scion Corporation). At least 100 adipocytes from four individuals were counted per genotype to determine mean value and standard deviation. Statistical significance between values obtained for wild-type and Sox8-deficient mice (*, P ≤ 0.05; **, P ≤ 0.005; ***, P ≤ 0.0005) was determined by the Student's t-test.
RESULTS
Sox8-deficient mice have reduced fat depots at older ages
Adult Sox8-deficient mice vary in their phenotype depending on the genetic background. On a C57Bl/6J background, Sox8-deficient mice are severely smaller and lighter, whereas weight reductions are more prominent than length reductions on a mixed C57Bl/6J:C3HeB/FeJ background. In fact, length reductions in Sox8-deficient mice amount to a mere 7% on the mixed background (Fig. 1A). On a C57Bl/6J background, the osteopenic phenotype of Sox8-deficient mice is much more pronounced (25) and Sox8-deficient males have been reported to develop progressive infertility with increasing age (23). To avoid confounding effects from the osteopenic phenotype and potential breeding problems, we performed our study with mice on a mixed background.
Fig. 1.

Appearance of Sox8-deficient mice at four months of age. (A, B) Comparison of length (A) and weight (B) of Sox8-deficient (black bars, n = 8) and wild-type (white bars, n = 10) males. Length was determined from the tip of the nose to the rostrum. The weight and length difference between wild-type and Sox8-deficient mice were statistically significant (***, P ≤ 0.0005). (C) MRI of wild-type (left) and Sox8-deficient (right) mice. A 3D-reconstruction from T-1 weighted images shows the dramatic reduction in adipose tissues.
By four months of age, Sox8-deficient mice were much lighter than age-matched wild-types (Fig. 1B). Their weight corresponded only to 70% of the wild-type. These weight differences were not detectable at birth and developed postnatally (22). To identify the potential cause of this weight reduction, we performed MRI. Visualization of lipid-rich tissues on a T1-weighted image instantaneously revealed that Sox8-deficient mice at four months of age exhibited an overall dramatic decrease of adipose tissues (Fig. 1C). This difference was not apparent at two months of age (data not shown).
To better quantify the effect, we determined the weight of several adipose tissues that can be dissected and compared the values for Sox8-deficient and wild-type mice after correcting for the differences in body weight between the two genotypes. At two months of age, neither the epididymal adipose tissue, the kidney adipose tissue, nor the interscapular adipose tissue exhibited significant weight differences between the two genotypes (Fig. 2A). Although similar results were obtained for males and females, only the results for males are shown. The weight of several organs, including kidney and testis, was equally comparable (Fig. 2B). In contrast, at four months of age, the weight of the epididymal adipose tissue was reduced to 38% of the wild-type, and the weight of the kidney adipose tissue to 22% of the wild-type in Sox8-deficient males (Fig. 2C). The interscapular adipose tissue also showed a reduction to 57% of the wild-type in Sox8-deficient males. At the same time, weight of kidney and testis remained comparable between the two genotypes (Fig. 2D), arguing that the Sox8-deficient mice indeed suffer from a selective loss of adipose tissue. Taking into account that intraperitoneal and kidney adipose tissue primarily consist of white adipose tissue (WAT), whereas the interscapular adipose tissue is predominantly brown adipose tissue (BAT), it appears that WAT is more strongly affected than BAT.
Fig. 2.

Weight of select fat depots and organs in Sox8-deficient mice. At two (A, B) and four (C, D) months of age, epididymal WAT (EWAT), kidney WAT (KWAT) and interscapular BAT (A, C) as well as testis and kidney (B, D) were weighed in Sox8-deficient (black bars, n = 10) and wild-type (white bars, n = 11) males as indicated below the bars. Determined weights were normalized to body weight and are expressed as mg per g body weight. Data are presented as mean ± SEM. Differences to the wild-type were statistically significant only for the fat depots in four-month-old mice (*, P ≤ 0.05; **, P ≤ 0.005).
Absolute food intake of Sox8-deficient and wild-type mice within a 24 h period was very similar both at two and at four months of age (Fig. 3A and data not shown). Motility of Sox8-deficient mice was only slightly altered when compared with the wild-type, as the total time spent resting was slightly reduced in Sox8-deficient mice at the expense of small but statistically significant increases in phases of activity and hyperactivity (Fig. 3B). Additional differences in the temporal distribution of phases of activity over the observation period or alterations in the spatial motility patterns were not observed between the two genotypes (data not shown). These results argue that the observed reduction in adipose fat tissue is not simply a trivial secondary consequence of alterations in feeding or activity behavior of Sox8-deficient mice.
Fig. 3.

Feeding and activity behavior of Sox8-deficient mice. (A) Food intake of Sox8-deficient mice (black bars, n = 6) and age-matched wild-type mice (white bars, n = 6) at four months was determined over a period of 24 h in metabolic cages and is presented as gram per day. (B) The activity of wild-type (white bars, n = 13) and Sox8-deficient mice (black bars, n = 17) was determined during 24 h in an ActiMot monitoring cage and divided into periods of resting, activity, and hyperactivity. The fraction of each period to the total observation time is presented as mean ± SEM. Differences to the wild-type were statistically significant only for the motility data (*, P ≤ 0.05; **, P ≤ 0.005).
Adipose tissues are progressively reduced in adult Sox8-deficient mice
Fat depots were not only smaller in Sox8-deficient mice than in wild-type mice at four months of age. In contrast to the wild-type, they also did not increase in weight from two to four months (Fig. 4A). Whereas weight of the epididymal WAT increased by 127% in the wild-type mice, there was a 33% reduction in Sox8-deficient mice during that period. For the kidney WAT, differences were similar, with a 520% weight increase in wild-type mice and a 25% reduction in Sox8-deficient mice. Although weight differences between both ages were less pronounced for the interscapular BAT, the tendency was the same with a 59% weight increase in wild-type mice between two and four months of age and a 25% weight reduction during the same period in Sox8-deficient mice. These results imply that the fat depots degenerate in older Sox8-deficient mice. Weight of the kidney as a representative organ changed only slightly during the same period in both wild-type and Sox8-deficient mutant (Fig. 4A). The selective degeneration of fat depots in Sox8-deficient mice is reminiscent of a lipodystrophic phenotype.
Fig. 4.

Reduced adipose tissues in Sox8-deficient mice. (A) Weight gains and losses of kidney, epididymal WAT (EWAT), kidney WAT (KWAT), and interscapular BAT (as indicated below the bars) were determined in wild-type (white bars, n = 11) and Sox8-deficient (black bars, n = 10) males during the period between two and four months of age. Data are presented as mean ± SEM. Weight changes in adipose tissues were statistically significant as determined by the Student's t-test (*, P ≤ 0.05; **, P ≤ 0.005). (B) Hemalum-eosin staining of epididymal adipose tissue from wild-type (+/+) and Sox8-deficient (lacZ/lacZ) males at four months of age. (C,D) Determination of adipocyte diameters in hemalum-eosin stained adipose tissue sections from Sox8-deficient and wild-type males at four months of age. The average adipocyte diameter in both genotypes (C) and the overall distribution of cell diameters (μm) (D) are shown. Data were collected from four males per genotype and are presented as mean ± SEM. Differences in the mean adipocyte diameter between the two genotypes were statistically significant as determined by the Student's t-test (*, P ≤ 0.05).
Hemalum-eosin staining revealed a similar WAT histology in Sox8-deficient and wild-type mice at four months of age (Fig. 4B). The most conspicuous difference was in the size of adipocytes. This is evident from the mean diameter of adipocytes, which in Sox8-deficient mice is only 67% of wild-type mice (Fig. 4C). Binning of adipocytes according to their cell diameters revealed this difference as well (Fig. 4D). Less than 8% of all adipocytes in Sox8-deficient mice had a diameter larger than 40 μm, whereas approximately 35% of all adipocytes fulfilled this criterion in the wild-type mice. On the other end of the scale, only 8% of all wild-type adipocytes were smaller than 20 μm in diameter, whereas approximately 39% of all Sox8-deficient adipocytes fell into this group. In accord with this finding, Oil Red O staining revealed smaller lipid droplets in Sox8-deficient adipocytes than in wild-type adipocytes (data not shown). At the same time, there was no indication from histology (Fig. 5A, B, E, F) or Oil Red O staining (Fig. 5C, D, G, H) that a compensatory fat deposition had occurred in the liver or muscle tissue of Sox8-deficient mice. In the liver, lipid contents was even strongly reduced (Fig. 5C, D).
Fig. 5.
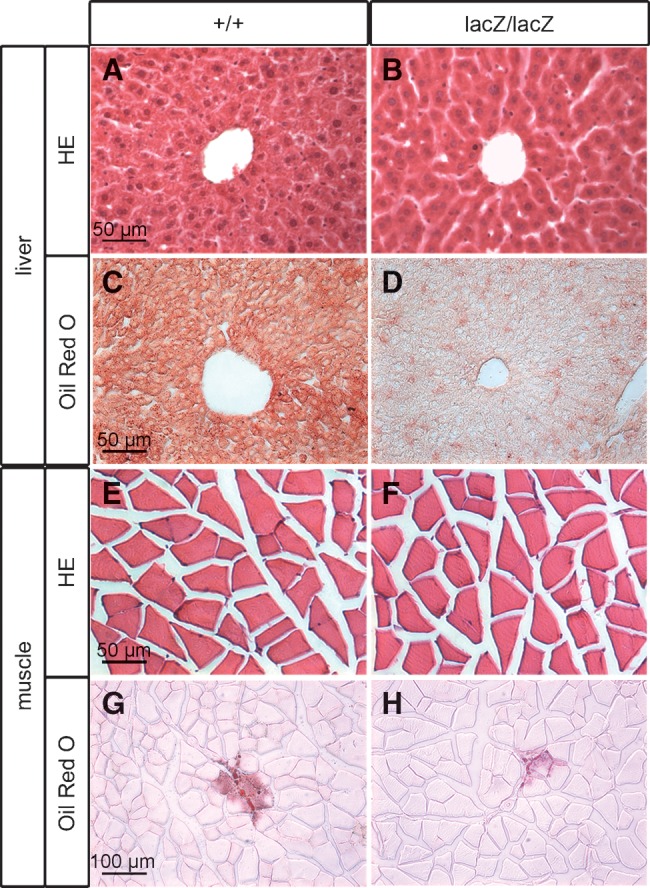
Lipid content of various tissues in Sox8-deficient mice. Hemalum-eosin staining (A, B, E, F) and Oil Red O staining (C, D, G, H) of liver (A–D) and muscle (E–H) from Sox8-deficient and wild-type males at four months of age. Although the lipid content was significantly reduced in Sox8-deficient adipose tissues, there was no compensatory lipid accumulation in liver or muscle.
Furthermore, WAT depots in four-month-old Sox8-deficient mice exhibited a significant upregulation of Pref-1 as a marker of preadipocytes (Fig. 6A). In contrast, expression of markers for differentiated adipocytes, such as C/EBPα and PPARγ, and of adipokines, such as LPL, resistin, and adiponectin, was slightly decreased as determined by quantitative RT-PCR on epididymal WAT (Fig. 6A). Taking the relatively low expression of Pref-1 and the relatively high expression of adipocyte markers into account, it is not surprising that only changes in the first but not the latter reached statistical significance. They may point to the fact that WAT of four-month-old Sox8-deficient mice contains increased numbers of preadipocytes. Alternatively, the adipocytes themselves may express aberrantly high Pref-1 levels, which could then be indicative of a mild differentiation defect. Expression of BAT markers such as CideA, Ucp-1 and Cpt1b was not increased in WAT depots of Sox8-deficient four-month-old males (Fig. 6B). This finding rules out that WAT has been replaced to a significant extent by BAT in Sox8-deficient mice.
Fig. 6.

Expression of adipocyte markers in Sox8-deficient mice. Expression of the preadipocyte marker Pref-1, the mature white adipocyte markers PPARγ2, C/EBPα, LPL, resistin and adiponectin (A) and the brown adipocyte markers CideA, Ucp-1 and Cpt1b (B) was determined by quantitative RT-PCR in the epididymal adipose tissue of four-month-old wild-type (n = 5) and Sox8-deficient (n = 5) male mice. The amounts of PCR products were normalized to Rpl8 and the mean value obtained for the wild-type was arbitrarily set to 1. Statistical significance between the two genotypes was only detected for Pref-1 as determined by the Student's t-test (*, P ≤ 0.05).
Basic metabolic parameters are not significantly changed in Sox8-deficient mice
Despite the dramatically reduced storage of lipids in the various fat depots, triglyceride concentrations in the serum of four-month-old mice were only slightly reduced (Fig. 7A) and free fatty acid concentrations were even comparable to the wild-type mice when determined after 4 h fasting (Fig. 7B). Blood glucose levels were slightly reduced under the same conditions, although the difference did not reach statistical significance (Fig. 7C).
Fig. 7.

Serology of Sox8-deficient mice. Serum levels of triglycerides (A), free fatty acids (B), glucose (C, D) and insulin (E) were determined in wild-type (+/+, white bars or white squares) and Sox8-deficient (lacZ/lacZ, black bars or black rhomboids) males at four months of age either after 4 h fasting (A,B,C) or after overnight fasting and intraperitoneal injection of 1.5 mg glucose per gram body weight at the indicated time points (D, E). Eight males were used per genotype in A–C; five males in D, E. Statistical significance between the two genotypes was only detected for serum triglycerides and in the glucose tolerance test as determined by the Student's t-test (*, P ≤ 0.05; **, P ≤ 0.005).
To study glucose metabolism in Sox8-deficient mice in more detail, we performed a glucose tolerance test in four-month-old males. Throughout this test, blood glucose levels remained significantly lower in Sox8-deficient mice than in the age-matched wild-type mice (Fig. 7D). In the wild-type mice, glucose levels increased from 6 mM to a maximum of 14 mM, whereas levels changed from 4 mM to a maximum of 10 mM in Sox8-deficient mice. Despite this difference in absolute levels, the relative increase and the kinetics with which glucose was cleared from the blood were comparable between the two genotypes (Fig. 7D). Similar to the blood glucose levels, insulin levels were also slightly lower in four-month-old Sox8-deficient mice than in age-matched wild-type mice, both after overnight fasting and 30 min into the glucose tolerance test (Fig. 7E). However, the relative increase of serum insulin in Sox8-deficient mice after glucose challenge was again comparable to the wild-type mice. We thus failed to obtain any evidence for dramatic alterations in the basic glucose metabolism of Sox8-deficient mice.
Sox8-deficient mice exhibit several hormonal changes
To analyze whether changes in hormonal regulation could be responsible for the observed loss of adipose tissue in Sox8-deficient mice, serum levels were analyzed for a number of hormones. Among the tested hormones, three exhibited significant changes.
At four months of age, IGF-1 levels were markedly reduced in Sox8-deficient mice when compared with wild-type mice (Fig. 8B). Consistent with these lower serum levels, Igf-1 expression was also significantly reduced in muscle and liver of Sox8-deficient mice when compared with the corresponding organs from wild-type mice by quantitative RT-PCR (Fig. 8C). A reduction in Igf-1 expression was also detected in the epididymal WAT of Sox8-deficient mice, although the difference did not reach statistical significance (Fig. 8C). Notably, however, IGF-1 serum levels were already reduced in two-month-old Sox8-deficient mice (Fig. 8A), thus making it unlikely that the changes in IGF-1 are the main cause for the loss of adipose tissues.
Fig. 8.

IGF-1 and noradrenaline status of Sox8-deficient mice. (A, B, D) Serum levels of IGF-1 (n = 6-8) (A, B) and noradrenaline (n = 21) (D) were determined in wild-type (+/+, white bars) and Sox8-deficient (lacZ/lacZ, black bars) males at 2 (A) and 4 (B, D) months of age. (C) Igf-1 expression was determined by quantitative RT-PCR in muscle, liver and epididymal WAT (EWAT) of four-month-old wild-type (n = 4) and Sox8-deficient (n = 4) mice as indicated below the bars. The amounts of PCR products were normalized to Rpl8 and the mean value obtained for the wild-type was arbitrarily set to 1. With exception of EWAT, all alterations in Sox8-deficient mice were statistically significant as determined by the Student's t-test (*, P ≤ 0.05).
In contrast to IGF-1 levels, noradrenaline levels were increased in the Sox8-deficient mice (Fig. 8D). Taking into account that we detected only mild changes in the overall activity of Sox8-deficient mice, there is no indication that the altered noradrenaline levels are the main cause for the observed loss of adipose tissue.
The third hormone with significant changes in Sox8-deficient mice was leptin. Serum levels of leptin were already slightly reduced at two months of age in Sox8-deficient mice (Fig. 9A) and on average 9.3-fold lower than in the wild-type mice at four months of age (Fig. 9C). These reductions are reflected by even more dramatic decreases in leptin expression levels in epididymal WAT as a representative fat depot (Fig. 9B, D). Again the difference between both genotypes was much more pronounced at four months than at two months of age. The most straightforward explanation is that the reduced leptin levels are a consequence of the observed alterations in the adipose tissue itself. In this context, it deserves to be mentioned that obvious histological changes were not detected in the leptin-responsive hypothalamic nuclei of Sox8-deficient mice by hemalum-eosin staining (Fig. 9E) and that hypothalamic Sox8 expression is restricted to cells of the oligodendrocyte lineage in the fiber tracts. This is evident by staining for the lacZ marker in Sox8+/lacZ mice (Fig. 9F), the distribution of which closely correlates with endogenous Sox8 occurrence (7, 22).
Fig. 9.
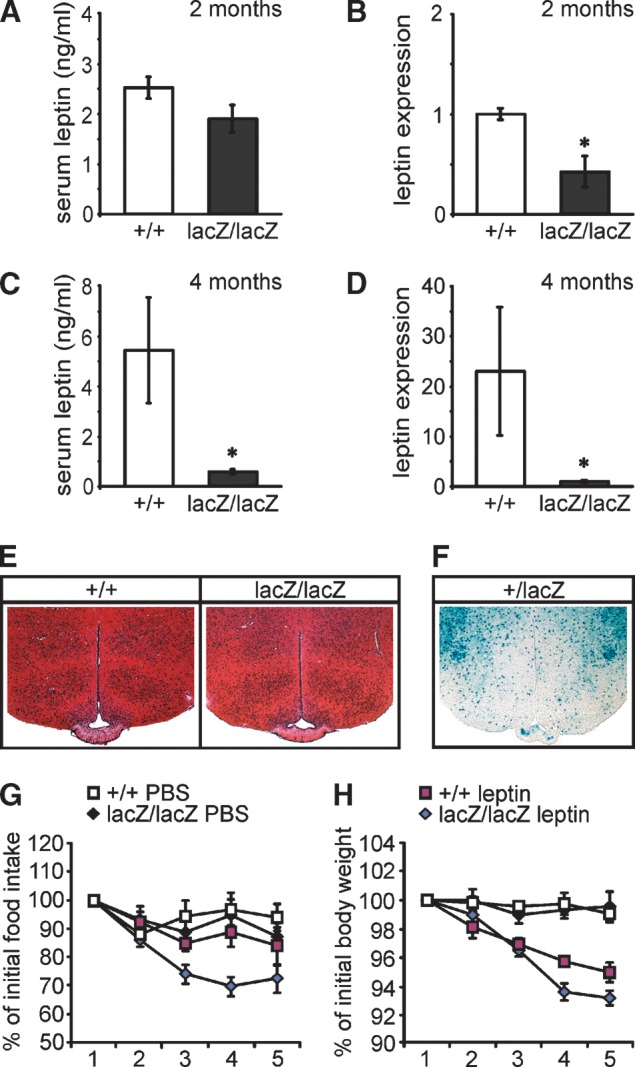
Leptin status of Sox8-deficient mice. (A, C) Serum levels of leptin were determined in wild-type (+/+, white bars, n = 7) and Sox8-deficient (lacZ/lacZ, black bars, n = 8) males at two months (A) and at four months (C) of age. (B, D) Leptin expression was determined by quantitative RT-PCR in epididymal WAT of two-month-old (B) and four-month-old (D) wild-type (n = 5) and Sox8-deficient (n = 7) mice. The amounts of PCR products were normalized to Rpl8, and the mean value obtained for Sox8-deficient mice was arbitrarily set to 1. Alterations of expression levels in Sox8-deficient mice were statistically significant at two and four months as determined by the Student's t-test (*, P ≤ 0.05), alterations in serum levels only at four months. (E) Hemalum-eosin staining of hypothalamic brain sections from adult wild-type and Sox8-deficient males at four months of age. (F) X-gal staining of hypothalamic brain sections from adult Sox8+/lacZ males showing Sox8 expression in oligodendrocytes in the fiber tracts, but not in neurons of the leptin-responsive nuclei. The bluish precipitate formed during X-gal staining is indicative of Sox8 expression as the lacZ marker introduced into the Sox8 locus faithfully reproduces Sox8 expression. (G, H) Daily food intake (G) and change in body weight (H) was determined over a five-day period with intraperitoneal injections of 0.4 μg leptin per gram body weight twice a day. Six males were used per genotype. Food intake and body weight over the five-day period are shown as percent of the respective values at the onset of the experiment.
The dramatically reduced leptin levels in Sox8-deficient mice at four months were compensated by increased leptin sensitivity. When injected with leptin twice a day over a 5 day period, Sox8-deficient mice reduced their food intake faster than wild-type mice and to lower levels (Fig. 9G). This translated into proportionately stronger weight losses (Fig. 9H). In summary, several hormonal changes were observed in Sox8-deficient mice. None, however, offers an obvious explanation for the reduction of adipose tissue in Sox8-deficient mice.
Differentiation of embryonic fibroblasts to adipocytes is influenced by the presence of Sox8
To investigate a cell-autonomous cause for the observed loss of adipose tissue, we prepared MEF from wild-type and Sox8-deficient embryos and subjected them to the standard regimen for adipogenic differentiation (Fig. 10A). After nine days of differentiation, lipid droplets were visible in only half as many Sox8-deficient cells as in wild-type cells (Fig. 10B). Lipid content was also reduced so that only half as much Oil Red O was retained on plates with Sox8-deficient cells than on plates with wild-type cells (Fig. 10C). As a consequence of these reductions, Oil Red O staining of plates with Sox8-deficient cells was clearly less intense than staining of plates with wild-type cells (Fig. 10A). This difference was still observed when the differentiation was continued for longer times in tissue culture (data not shown).
Fig. 10.

Adipogenic differentiation of fibroblasts in the presence and absence of Sox8. (A) Oil Red O (ORO) staining of differentiated MEF cultures after nine days of adipogenic differentiation. (B) Quantification of adipocytes as lipid-droplet containing cells and (C) photometrical quantification of ORO content after nine days of differentiation of MEF from wild-type (+/+, white bars) and Sox8-deficient (lacZ/lacZ, black bars) embryos. The experiment was repeated two times with two different preparations per genotype. Data are presented as mean ± SEM. Differences to the wild-type were statistically significant as determined by the Student's t-test (*, P ≤ 0.05). (D–G) Expression of Pref-1 (D), C/EBPα (E), PPARγ2 (F), and LPL (G) was determined in undifferentiated subconfluent (SC) and confluent MEFs (day 0) from wild-type (white bars) and Sox8-deficient (black bars) embryos after 1, 2, 3, and 7 days of adipogenic differentiation by quantitative RT-PCR as indicated below the bars. The amounts of PCR products were normalized to Rpl8. Data represent the mean ± SEM from two different experiments. (H) MEF from Sox8-deficient mice were transfected with expression plasmids for EGFP or for Sox8 in combination with EGFP, and the percentage of transfected cells that had developed into adipocytes after five days of differentiation was determined. The percentage of adipocytes obtained from EGFP-transfected cells was arbitrarily set to 1. (I) 3T3 L1 fibroblasts were transfected with an expression plasmid for EGFP (EGFP), an expression plasmid for Sox8 and EGFP (Sox8), or a pSUPER.neo + gfp vector that contained an shRNA for Sox8 in wild-type (sh) or scrambled (sc) version. The percentage of transfected cells that had developed into adipocytes after five days of differentiation was determined. The number of adipocytes obtained from cells transfected only with EGFP was arbitrarily set to 1. Transfection experiments were repeated three times and data are presented as mean ± SEM. (J) Western blot on extracts from 293 cells transfected with a Sox8 expression plasmid, either alone or in combination with a pSUPER.neo + gfp vector that contained an shRNA for Sox8 in wild-type (sh) or scrambled (sc) version.
Furthermore, quantitative RT-PCR studies on these differentiating MEF showed that expression of Pref-1 as a marker for preadipocytes was higher in Sox8-deficient MEF than in wild-type MEF during the early phase of the differentiation process, before becoming lower in the late phase (Fig. 10D). In contrast, expression of C/EBPα, PPARγ2, and LPL as markers for terminally differentiating adipocytes was significantly reduced in Sox8-deficient MEFs, especially during the late phase of the adipogenic differentiation process (Fig. 10E, F, G). PPARγ2 levels were, for instance, 5.5-fold lower and C/EBPα levels 3.8-fold lower in Sox8-deficient MEF than in wild-type MEF after seven days of differentiation (Fig. 10E, F).
To further study a cell-autonomous role of Sox8, we performed additional gain- and loss-of-function experiments. When Sox8 was reintroduced by transfection into MEF from Sox8-deficient mice, differentiation of these MEF into adipocytes was rescued as determined by Oil Red O staining (Fig. 10H). A similar increase in adipocyte differentiation was also observed in transfected 3T3 L1 fibroblasts (Fig. 10I). On the other hand, shRNA-dependent reduction of Sox8 in 3T3 L1 fibroblasts decreased the capacity of 3T3 L1 fibroblasts to differentiate into adipocytes (Fig. 10I). No such effect was observed with a scrambled shRNA which, in contrast to the Sox8-specific shRNA, failed to reduce Sox8 amounts (Fig. 10J).
Sox8 is not so much expressed in mature adipocytes as in cells and at sites from which adipocytes arise
By RT-PCR, Sox8 transcripts were detectable in wild-type MEF and in 3T3 L1 fibroblasts (Fig. 11A, C). Compared with wild-type testis as a positive control, transcript levels were lower. Expression was downregulated during the differentiation process, as Sox8 transcript levels were higher in subconfluent and confluent MEF and 3T3 L1 fibroblasts before the induction of differentiation than during the differentiation process (Fig. 11A, C). A similar downregulation during adipocyte differentiation was also observed on the protein level (Fig. 11B, D). In adult adipose tissue such as epididymal WAT, Sox8 transcript levels were even lower (Fig. 11A). There was furthermore no significant X-gal staining in adipose tissue of Sox8+/lacZ mice (Fig. 11E) indicating that the majority of cells in this tissue do not express Sox8 at significant levels. This was also corroborated in immunostainings with a Sox8-specific antibody (data not shown). Sox8 was, however, broadly expressed during mouse development in mesodermal progenitors such as dermamyotomal cells at 11.5 dpc (Fig. 11F). We therefore conclude that Sox8 is mainly expressed in mesodermal precursor cells and only at low levels in adipocytes.
Fig. 11.
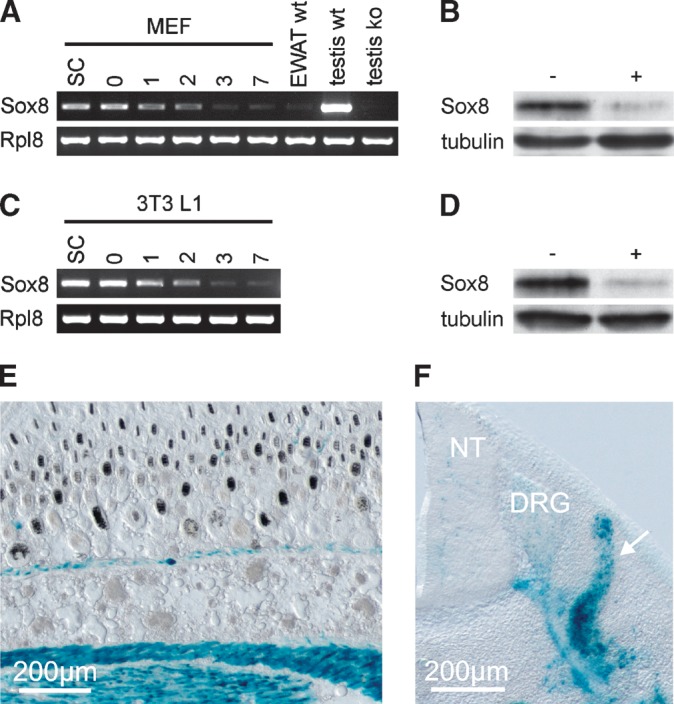
Sox8 expression in mesodermal cells. (A,C) Sox8 transcript levels were determined by RT-PCR in subconfluent (SC), confluent (0), and differentiating (1–3, 7) wild-type MEF, adult epididymal WAT (A), and 3T3 L1 fibroblasts (C). cDNA from testis of wild-type (wt) mice served as positive control, whereas cDNA from testis of Sox8-deficient (ko) mice represented the negative control. (B, D) Sox8 protein levels were determined after immunoprecipitation by Western Blot in undifferentiated (–) and differentiated (+) wild-type MEF (B) and 3T3 L1 fibroblasts (D). Acetylated α-tubulin served as loading control. (E, F) X-gal staining of subcutaneous adipose tissue on transverse skin sections of Sox8+/lacZ animals at postnatal day 5 (E) and of dermamyotomal progenitors (white arrow) on transverse sections of Sox8+/lacZ embryos at 11.5 dpc (F). The bluish precipitate formed during X-gal staining is indicative of Sox8 expression because the lacZ marker introduced into the Sox8 locus faithfully reproduces Sox8 expression. DRG, dorsal root ganglion; NT, neural tube.
DISCUSSION
Sox8-deficient mice exhibit a substantial weight reduction (22), but the cause has not yet been fully determined. Although previous studies have shown that osteopenia contributes to weight loss on a C57Bl/6J background (25), it is too subtle on mixed genetic backgrounds to account for the weight reduction (22). The present study now shows that Sox8-deficient mice also have strongly reduced adipose tissues at older ages, which may in fact account for a good part of the overall weight reduction in these animals.
On a mixed C57Bl/6J:C3HeB/FeJ background, the adipose tissue phenotype was observed in the absence of a bone phenotype. Although both tissues can influence each other in the adult (27), it thus appears unlikely that the reduction in adipose tissues is secondary to the bone phenotype (25). Nor is there any reason to believe that the reduction in adipose tissues is a consequence of the progressive infertility observed in Sox8-deficient males at older ages (23), because alterations in the adipose tissue precede those in the testis and are also observed in females which do not exhibit any fertility problems. We thus have to conclude that the reduction of adipose tissues represents an independent phenotype in Sox8-deficient mice.
Available data furthermore show that adipose tissues in Sox8-deficient mice and wild-type littermates are very similar at birth and during the first two months. Differences were detected by four months of age with fat depots of Sox8-deficient mice being essentially smaller and lighter than at two months of age. Sox8-deficient mice thus suffer from a condition that is reminiscent of lipodystrophy. Although WAT weight reductions are stronger than BAT weight reductions, both types of adipose tissues are affected. This argues that the observed phenotype is due to a general effect of Sox8 on adipose tissues. In line with such an assumption, we did not find any evidence for a transformation of white adipocytes into brown adipocytes or vice versa.
As Sox8 is constitutively deleted in the mouse, the observed reduction in adipose tissues could be cell-autonomous or noncell-autonomous. If noncell-autonomous, effects are likely to be mediated by hormones. Indeed, we were able to detect several hormonal changes in the Sox8-deficient mouse such as a decrease in IGF-1 and leptin levels as well as an increase in noradrenaline levels. Insulin levels were only slightly lower than in the wild-type and there was no sign of insulin resistance or otherwise dramatically altered glucose metabolism in Sox8-deficient mice. Although increased noradrenaline levels correlated with a slight increase in activity, the effect was too small to account for the observed decrease in adipose tissue. Although IGF-1 has been linked to adipocyte differentiation (28), it is similarly difficult to explain the adipose tissue phenotype as a consequence of the altered IGF-1 levels, as reduction of IGF-1 levels failed to temporally correlate with the loss of adipose tissue.
Reduction of serum leptin levels are likely secondary to the adipose tissue phenotype. Interestingly, there was even a decrease of leptin expression per adipocyte, whereas expression of other adipostatic hormones such as resistin and adiponectin was unchanged on a per cell basis. Reduced leptin levels in the serum are therefore not only caused by the overall reduction of adipose tissue, but also by a decreased leptin expression in adipocytes. Considering the low amounts of Sox8 in adipocytes, it is unlikely that Sox8 directly regulates leptin expression. Serum insulin and noradrenaline have, however, been shown to influence both leptin expression (29, 30). Reduced insulin and increased noradrenaline serum levels may thus be at least partly responsible for leptin changes in Sox8-deficient mice.
There were no obvious anatomical alterations in the ventral medial hypothalamus where the leptin-responsive neurons are localized (31, 32). Taken together with the fact that Sox8 expression in the CNS is largely restricted to the oligodendrocyte lineage both during development and in the adult (7, 22), leptin-responsive regions of the CNS are morphologically not affected by the Sox8 deletion.
The reduced levels of circulating leptin in Sox8-deficient mice would be expected to translate into hyperphagia (33). Food intake of Sox8-deficient mice at four months was however normal, a fact that can be explained by the increased leptin sensitivity of these mice. In summary, the hormonal changes observed in Sox8-deficient mice may contribute to the reduction in adipose tissues. No single hormonal change, however, offers a satisfying explanation for the phenotype. The cause for the phenotype is thus likely to be complex.
Several results of our study also point to a cell-autonomous component of the adipose tissue phenotype. For one, expression of the only widely accepted preadipocyte marker Pref-1 (34) was significantly increased in WAT of Sox8-deficient mice and in vitro at most time points during adipogenic differentiation of Sox8-deficient MEF. At the same time, differentiating Sox8-deficient MEF expressed significantly decreased levels of many markers of mature adipocytes such as C/EBPα, PPARγ2 and LPL (28, 35). Although decreased expression of mature adipocyte markers did not reach statistical significance in WAT, data are compatible with an intrinsic adipocyte differentiation defect in Sox8-deficient mice. Such a defect would also lead to an overall reduction in adipocyte size which we indeed observed in our studies. Additional evidence for a cell-autonomous function of Sox8 in the adipocyte lineage comes from the observation that reintroduction of Sox8 in MEF from Sox8-deficient mice rescues their adipocyte differentiation defect in culture and that a comparable adipocyte differentiation defect was observed in 3T3 L1 fibroblasts after Sox8 levels were reduced by shRNA.
Sox8 is therefore a novel transcription factor with functions in adipocyte development. Our expression studies furthermore indicate that Sox8 likely exerts its cell-autonomous function in the adipocyte lineage during early stages. This assumption is based on the fact that Sox8 is expressed during early embryonic development in parts of the paraxial mesoderm from which adipocytes may arise and in undifferentiated MEF and 3T3 L1 fibroblasts, whereas it is downregulated in differentiating fibroblasts and in mature adipocytes of WAT and BAT. A more definitive proof of the early expression of Sox8 in the adipocyte lineage is hampered by the fact that thorough lineage tracing studies do not exist that tracked the developmental origins of adipocytes (36). An early expression of Sox8 in the adipocyte lineage is, however, also supported by its occurrence in all mesodermal precursor cells that have been analyzed to date, including osteo-chrondroprogenitors, chondroblasts, osteoblasts, myoblasts, and Sertoli cell precursors (6, 37–39). If some adipocytes are generated from the neural crest, as has been suggested (40), Sox8 may also be a relevant factor as it is expressed in the early neural crest (8, 22).
Other transcription factors with roles in adipocytes include the C/EBP family of bZIP proteins, the PPAR family of nuclear receptors, their RXR heterodimerization partners, FoxC2, SREBP, and STAT proteins (28, 35, 36). Some of these transcription factors, such as C/EBPβ and C/EBPδ, are transiently expressed and functionally active during the early phases of adipocyte differentiation (41), whereas most, including C/EBPα and PPARγ, function in later phases where they shut off proliferation and allow lipid accumulation (42–44). Taking the expression pattern of Sox8 into account, functional interaction can thus only exist between Sox8 and those transcription factors that are functionally active at early phases in the lineage. However, Sox8 may already be active before C/EBPβ and C/EBPδ proteins. With regard to its active phase, Sox8 may most closely resemble the nonhistone chromatin protein HMG-IC which is only expressed in the adipocyte lineage during embryonic and fetal stages and whose disruption confers resistance to obesity (45).
In line with the different expression of Sox8 and most other known transcriptional regulators of adipocyte development, the phenotype of Sox8-deficient mice is significantly different from the phenotypes of mouse models for these other transcription factors (35). Especially noteworthy is the fact that, compared with the other mouse mutants, the adipose tissue defect in Sox8-deficient mice is less severe. Adipocyte development is not completely blocked in the absence of Sox8. Sox8 thus rather serves as a modulator than as a master regulator of adipocyte development. This agrees with the general observation that Sox proteins usually serve as cofactors and rarely function on their own (2, 46, 47).
The proposed function of Sox8 in early stages of adipocyte differentiation is also somewhat at odds with the fairly late onset of the adipose tissue phenotype in Sox8-deficient mice. However, it is known that adipose tissue has a high turnover throughout adult life and that adipocytes are constantly replaced by cells newly generated from a precursor pool (48). We imagine that Sox8 may be expressed in a precursor pool and that dysregulation of a precursor pool in the absence of Sox8 could explain the phenotype. In support of such an assumption, Sox8 is expressed in satellite cells which represent a comparable mesodermal precursor pool in the muscle (39). A pool of proliferating and renewing adipogenic precursors has recently been identified from the stromal vascular fraction of the adult adipose tissue (49, 50). It will be interesting to see in future studies whether Sox8 is expressed in this precursor population and whether selective ablation of Sox8 in these cells recapitulates the phenotype. Nevertheless it needs to be emphasized that the situation may be even more complex as evidence points to precursor cell heterogeneity and the existence of several precursor pools (36) of which only some may rely on Sox8.
Acknowledgments
The authors thank Jutta Prade for expert technical assistance.
Abbreviations
BAT, brown adipose tissue
dpc, days post coitum
FCS, fetal calf serum
MEF, mouse embryonic fibroblasts
WAT, white adipose tissue
This work was supported by Grant SFB473 (M.W.) from the Deutsche Forschungsgemeinschaft.
Published, JLR Papers in Press, March 12, 2009.
References
- 1.Bowles J., G. Schepers, and P. Koopman. 2000. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 227 239–255. [DOI] [PubMed] [Google Scholar]
- 2.Wegner M. 1999. From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res. 27 1409–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guth S. I. E., and M. Wegner. 2008. Having it both ways: Sox protein function between conservation and innovation. Cell. Mol. Life Sci. 65 3000–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheung M., and J. Briscoe. 2003. Neural crest development is regulated by the transcription factor Sox9. Development. 130 5681–5693. [DOI] [PubMed] [Google Scholar]
- 5.Kellerer S., S. Schreiner, C. C. Stolt, M. R. Bösl, and M. Wegner. 2006. Functional equivalency of transcription factors Sox8 and Sox10 is tissue-specific. Development. 133 2875–2886. [DOI] [PubMed] [Google Scholar]
- 6.Chaboissier M-C., A. Kobayashi, V. I. P. Vidal, S. Lützkendorf, H. J. G. van de Kant, M. Wegner, D. G. de Rooij, R. R. Behringer, and A. Schedl. 2004. Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development. 131 1891–1901. [DOI] [PubMed] [Google Scholar]
- 7.Stolt C. C., P. Lommes, R. P. Friedrich, and M. Wegner. 2004. Transcription factors Sox8 and Sox10 perform non-equivalent roles during oligodendrocyte development despite functional redundancy. Development. 131 2349–2358. [DOI] [PubMed] [Google Scholar]
- 8.O'Donnell M., C. S. Hong, X. Huang, R. J. Delnicki, and J. P. Saint-Jeannet. 2006. Functional analysis of Sox8 during neural crest development in Xenopus. Development. 133 3817–3826. [DOI] [PubMed] [Google Scholar]
- 9.Akiyama H., M-C. Chaboissier, R. Behringer, D. H. Rowitch, A. Schedl, J. A. Epstein, and B. de Crombrugghe. 2004. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA. 101 6502–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akiyama H., M-C. Chaboissier, J. F. Martin, A. Schedl, and B. de Crombrugghe. 2002. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16 2813–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bi W., J. M. Deng, Z. Zhang, R. R. Behringer, and B. de Crombrugghe. 1999. Sox9 is required for cartilage formation. Nat. Genet. 22 85–89. [DOI] [PubMed] [Google Scholar]
- 12.Stolt C. C., P. Lommes, E. Sock, M-C. Chaboissier, A. Schedl, and M. Wegner. 2003. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev. 17 1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mori-Akiyama Y., M. van den Born, J. H. van Es, S. R. Hamilton, H. P. Adams, J. Zhang, H. Clevers, and B. de Crombrugghe. 2007. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology. 133 539–546. [DOI] [PubMed] [Google Scholar]
- 14.Britsch S., D. E. Goerich, D. Riethmacher, R. I. Peirano, M. Rossner, K. A. Nave, C. Birchmeier, and M. Wegner. 2001. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 15 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbarth B., V. Pingault, N. Bondurand, K. Kuhlbrodt, I. Hermans-Borgmeyer, A. Puliti, N. Lemort, M. Goossens, and M. Wegner. 1998. Mutation of the Sry-related Sox10 gene in Dominant megacolon: a mouse model for human Hirschsprung disease. Proc. Natl. Acad. Sci. USA. 95 5161–5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Southard-Smith E. M., L. Kos, and W. J. Pavan. 1998. Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat. Genet. 18 60–64. [DOI] [PubMed] [Google Scholar]
- 17.Stolt C. C., S. Rehberg, M. Ader, P. Lommes, D. Riethmacher, M. Schachner, U. Bartsch, and M. Wegner. 2002. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 16 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner T., J. Wirth, J. Meyer, B. Zabel, M. Held, J. Zimmer, J. Pasantes, F. D. Bricarelli, J. Keutel, E. Hustert, et al. 1994. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene Sox9. Cell. 79 1111–1120. [DOI] [PubMed] [Google Scholar]
- 19.Foster J. W., M. A. Dominguez-Steglich, S. Guioli, C. Kwok, P. A. Weller, M. Stevanovic, J. Weissenbach, S. Mansour, I. D. Young, P. N. Goodfellow, et al. 1994. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 372 525–530. [DOI] [PubMed] [Google Scholar]
- 20.Inoue K., M. Khajavi, T. Ohyama, S-i. Hirabayashi, J. Wilson, J. D. Reggin, P. Mancias, I. J. Butler, M. F. Wilkinson, M. Wegner, et al. 2004. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 36 361–369. [DOI] [PubMed] [Google Scholar]
- 21.Pingault V., N. Bondurand, K. Kuhlbrodt, D. E. Goerich, M-O. Prehu, A. Puliti, B. Herbarth, I. Hermans-Borgmeyer, E. Legius, G. Matthijs, et al. 1998. Sox10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 18 171–173. [DOI] [PubMed] [Google Scholar]
- 22.Sock E., K. Schmidt, I. Hermanns-Borgmeyer, M. R. Bösl, and M. Wegner. 2001. Idiopathic weight reduction in mice deficient in the high-mobility-group transcription factor Sox8. Mol. Cell. Biol. 21 6951–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Bryan M. K., S. Takada, C. L. Kennedy, G. Scott, S. Harada, M. K. Ray, Q. Dai, D. Wilhelm, D. M. de Kretser, E. M. Eddy, et al. 2008. Sox8 is a critical regulator of adult Sertoli cell function and male fertility. Dev. Biol. 316 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pfeifer D., F. Poulat, E. Holinski-Feder, F. Kooy, and G. Scherer. 2000. The SOX8 gene is located within 700 kb of the tip of chromosome 16p and is deleted in a patient with ATR-16 syndrome. Genomics. 63 108–116. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt K., T. Schinke, M. Haberland, M. Priemel, A. F. Schilling, C. Mueldner, J. M. Rueger, E. Sock, M. Wegner, and M. Amling. 2005. The high-mobility-group transcription factor Sox8 is a negative regulator of osteoblast differentiation. J. Cell Biol. 168 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maka M., C. C. Stolt, and M. Wegner. 2005. Identification of Sox8 as a modifier gene in a mouse model of Hirschsprung disease reveals underlying molecular defect. Dev. Biol. 277 155–169. [DOI] [PubMed] [Google Scholar]
- 27.Lee N. K., H. Sowa, E. Hinoi, M. Ferron, J. D. Ahn, C. Confavreux, R. Dacquin, P. J. Mee, M. D. McKee, D. Y. Jung, et al. 2007. Endocrine regulation of energy metabolism by the skeleton. Cell. 130 456–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosen E. D., C. J. Walkey, P. Puigserver, and B. M. Spiegelman. 2000. Transcriptional regulation of adipogenesis. Genes Dev. 14 1293–1307. [PubMed] [Google Scholar]
- 29.Cong L., K. Chen, J. Li, P. Gao, Q. Li, S. Mi, X. Wu, and A. Z. Zhao. 2007. Regulation of adiponectin and leptin secretion and expression by insulin through a PI3K–PDE3B dependent mechanism in rat primary adipocytes. Biochem. J. 403 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trayhurn P., J. S. Duncan, D. V. Rayner, and L. J. Hardie. 1996. Rapid inhibition of ob gene expression and circulating leptin levels in lean mice by the beta 3-adrenoceptor agonists BRL 35135A and ZD2079. Biochem. Biophys. Res. Commun. 228 605–610. [DOI] [PubMed] [Google Scholar]
- 31.Baskin D., J. Breininger, and M. Schwartz. 1999. Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes. 48 828–833. [DOI] [PubMed] [Google Scholar]
- 32.Cheung C., D. Clifton, and R. Steiner. 1997. Proopimelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. 138 4489–4492. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz M. W., S. C. Woods, D. Porte, R. Seeley, and D. G. Baskin. 2000. Central nervous system control of food intake. Nature. 404 661–671. [DOI] [PubMed] [Google Scholar]
- 34.Villena J. A., K. H. Kim, and H. S. Sul. 2002. Pref-1 and ADSF/resistin: two secreted factors inhibiting adipose tissue development. Horm. Metab. Res. 34 664–670. [DOI] [PubMed] [Google Scholar]
- 35.Valet P., G. Tavernier, I. Castan-Laurell, J. S. Saulnier-Blache, and D. Langin. 2002. Understanding tissue development from transgenic animal models. J. Lipid Res. 43 835–860. [PubMed] [Google Scholar]
- 36.Gesta S., Y-H. Tseng, and C. R. Kahn. 2007. Developmental origin of fat: tracking obesity to its source. Cell. 131 242–256. [DOI] [PubMed] [Google Scholar]
- 37.Akiyama H., J. E. Kim, K. Nakashima, G. Balmes, N. Iwai, J. M. Deng, Z. Zhang, J. F. Martin, R. R. Behringer, T. Nakamura, et al. 2005. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. USA. 102 14665–14670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmid R. S., S. Shelton, A. Stanco, Y. Yokota, J. A. Kreidberg, and E. S. Anton. 2004. Alpha3 beta1 integrin modulates neuronal migration and placement during early stages of cerebral cortical development. Development. 131 6023–6031. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt K., G. Glaser, A. Wernig, M. Wegner, and O. Rosorius. 2003. Sox8 is a specific marker for muscle satellite cells and inhibits myogenesis. J. Biol. Chem. 278 29769–29775. [DOI] [PubMed] [Google Scholar]
- 40.Billon N., P. Iannarelli, M. C. Monteiro, C. Glavieux-Pardanaud, W. D. Richardson, N. Kessaris, C. Dani, and E. Dupin. 2007. The generation of adipocytes by the neural crest. Development. 134 2283–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka T., N. Yoshida, T. Kishimoto, and S. Akira. 1997. Defective adipocyte differentiation in mice lacking the C/EBPβ and C/EBPδ gene. EMBO J. 16 7432–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang N-D., M. J. Finegold, A. Bradley, C. N. Ou, S. V. Abdelsayed, M. D. Wilde, L. R. Taylor, D. R. Wilson, and G. J. Darlington. 1995. Impaired energy homeostasis in C/EBPα knockout mice. Science. 269 1108–1112. [DOI] [PubMed] [Google Scholar]
- 43.Kubota N., Y. Terauchi, H. Miki, H. Tamemoto, T. Yamauchi, K. Komeda, S. Satoh, R. Nakano, C. Ishii, T. Sugiyama, et al. 1999. PPARγ mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell. 4 597–609. [DOI] [PubMed] [Google Scholar]
- 44.Rosen E. D., P. Sarraf, A. E. Troy, G. Bradwin, K. Moore, D. S. Milstone, B. M. Spiegelman, and R. M. Mortensen. 1999. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell. 4 611–617. [DOI] [PubMed] [Google Scholar]
- 45.Anand A., and K. Chada. 2000. In vivo modulation of Hmgic reduces obesity. Nat. Genet. 24 377–380. [DOI] [PubMed] [Google Scholar]
- 46.Kamachi Y., M. Uchikawa, and H. Kondoh. 2000. Pairing SOX off with partners in the regulation of embryonic development. Trends Genet. 16 182–187. [DOI] [PubMed] [Google Scholar]
- 47.Wegner M. 2005. Secrets to a healthy Sox life: Lessons for melanocytes. Pigment Cell Res. 18 74–85. [DOI] [PubMed] [Google Scholar]
- 48.Spalding K. L., E. Arner, P. O. Westermark, S. Bernard, B. A. Buchholz, O. Bergmann, L. Blomqvist, J. Hoffstedt, E. Naslund, T. Britton, et al. 2008. Dynamics of fat cell turnover in humans. Nature. 453 783–787. [DOI] [PubMed] [Google Scholar]
- 49.Rodeheffer M. S., K. Birsoy, and J. M. Friedman. 2008. Identification of white adipocyte progenitor cells in vivo. Cell. 135 240–249. [DOI] [PubMed] [Google Scholar]
- 50.Tang W., D. Zeve, J. M. Suh, D. Bosnakovski, M. Kyba, R. E. Hammer, M. D. Tallquist, and J. M. Graff. 2008. White fat progenitor cells reside in the adipose vasculature. Science. 322 583–586. [DOI] [PMC free article] [PubMed] [Google Scholar]