

Abstract
We established a novel technique for differential activity-based gel electrophoresis (DABGE) of lipolytic enzymes from two different biological samples. For this purpose, a set of three fluorescent suicide inhibitors was developed. These probes possess the same substrate analogous structures but carry different cyanine dyes (Cy2b, Cy3, and Cy5) as reporter fluorophores. For comparison of enzyme profiles, two samples are individually labeled with a different probe followed by mixing, gel electrophoresis, fluorescence imaging, and identification of the tagged proteins by MS/MS. Protocols for quantitative determination of active enzymes were developed on the basis of lipolytic proteomes that had been admixed with defined amounts of known lipases and esterases. A detailed analysis of the fluorescence intensities showed that the found enzyme ratios very closely reflected the relative amounts of the labeled enzymes that were used for spiking. The DABGE method was used to compare the lipolytic proteomes of brown and white adipose tissue showing specific enzyme patterns of both samples. This study represents the first application of this technology for comparative analysis of lipases and esterases. Further applications of this technique can be expected to provide entirely new information on lipid enzymology in health and disease with high precision.
Keywords: lipid metabolism, fluorescent lipid, lipid-associated disorders, functional proteomics, fluorescent phosphonate
In the postgenomic era, researchers are now challenging the proteome with methods like two-dimensional gel electrophoresis or multidimensional chromatography followed by mass spectrometry and various other methods for abundance-based proteome profiling. However, the amount of proteins present at a certain state of the cell might not correlate with the enzyme activities responsible for the metabolic fluxes, cell management, and signal transduction. Therefore, elucidation of changes in protein activity is the ultimate goal of functional proteomics.
Methods have been developed for specific detection of enzymes on the basis of their catalytic activities. For this purpose, activity recognition probes (ARPs) (1) or activity-based probes are used. Basically, an ARP is a molecule consisting of i) a recognition site targeting a certain enzyme species, ii) a properly positioned reactive site that forms a covalent bond with the target, and iii) a tag for visualization and/or isolation of the covalently bound target (2–4). Many reactive groups have been developed so far for identifying different types of enzyme activity, i.e., fluorophosphonates for serine hydrolases (5), p-nitrophenyl organophosphonates for lipases and esterases (3, 6), epoxides for cysteine proteases (7), and sulfonate esters for various enzyme classes (8, 9).
Two-dimensional (2D) gel electrophoresis is a well-established technique for simultaneous separation and display of hundreds to thousands of proteins (10). The proteins are separated in two dimensions according to their isoelectric point (isoelectronic focusing) and molecular size (SDS-PAGE). But despite of the substantial advances in this technology, some of the more significant systemic shortcomings have remained unsolved. The most troublesome of these is the inherent lack of reproducibility between gels (11, 12). To overcome these issues, Ünlü, Morgan, and Minden (13) developed an approach involving multiplexing of samples, called 2D difference gel electrophoresis (DIGE™), which has since been commercialized. For comparison, the protein extracts are prior to electrophoresis covalently labeled with different fluorescent cyanine dyes, which are N-hydroxy succinimidyl ester derivates of Cy2, Cy3, and Cy5. The dyes react with the ɛ-amino group of lysine residues on proteins forming an amide bond. The labeled samples are then mixed before separation by 2D gel electrophoresis. Variation in spot intensities due to gel-specific experimental factors, for example, protein migration from the Immobiline™ Dry Strip Gel strip into the SDS gel, will be the same for each sample within a single DIGE system. Consequently, the relative amount of a protein in a gel in one sample compared with another will be unaffected (14). This procedure greatly facilitates screening for proteins that are up- or downregulated in a given sample compared with a reference sample (e.g., mutants vs. wild type, “diseased” vs. “healthy,” etc.).
Here, we report on a novel approach using fluorescent ARPs for differential activity-based gel electrophoresis (DABGE) of two functional lipolytic and esterolytic proteomes. Lipolytic enzymes hydrolyze acyl esters inside and outside cells, thus fulfilling specific functions in lipid metabolism and signaling. This protein family includes tri-, di-, and monoacylglycerol lipases, cholesterol ester hydrolases, retinyl esterases, and (lyso-) phospholipases. The activities of these biocatalysts are more or less dependent on the chemical structure and the supramolecular presentation of the substrate. Affinity tags specifically designed for lipases should resemble hydrophobic molecules. Fluorescently labeled p-nitrophenyl- and fluoroalkylphosphonates meet the above criteria. Depending on their polarities and recognition sites, they are excellent baits to profile different serine hydrolase activities in complex proteomes. These compounds have to be properly solubilized in order to be “accepted” by their target enzymes. The experimental approach is based on selective labeling of two samples A and B with different ARPs followed by protein separation and dual color analysis of protein-lipid complex fluorescence in one gel. For this purpose, we developed ARPs with the same chemical properties but different emission wavelengths for comparison of two different lipolytic proteomes in one electrophoresis gel. The respective probes are fluorescent inhibitors that possess the same substrate analogous structures but carry different cyanine dyes (Cy2b, Cy3, and Cy5) as reporter fluorophores. In addition to the composition of the protein patterns, the relative activities of the individual enzymes can be determined by the differential experimental design. This method was validated using artificial “proteomes” containing defined amounts of known enzymes. Furthermore, we employed the method for quantitative comparison of the lipolytic proteomes of brown adipose tissue (BAT) and white adipose tissue (WAT), demonstrating that DABGE is also an efficient method for the differential analysis of complex biological samples.
EXPERIMENTAL PROCEDURES
Fluorescent dye and inhibitor synthesis
Organic solvents were obtained from Carl Roth (Karlsruhe, Germany). Chemicals for organic synthesis were purchased from Sigma-Aldrich (Taufkirchen, Germany). Some of these chemicals were specially purified immediately before use: triethylamine absolute (distillation over CaH2/ninhydrin to remove water and primary or secondary amines) and 2-methylbenzoxazole (distillation). Flash chromatography was carried out on Silica gel 60 (0.040–0.063 mm); TLC was performed on TLC aluminum sheets coated with silica gel 60 F254, both from Merck (Darmstadt, Germany). Chemicals for gel electrophoresis and the dye reagent for the Bradford protein assay were purchased from Bio-Rad Laboratories (Hercules, CA).
NMR spectra were recorded using a Varian INOVA-500 spectrometer (Palo Alto, CA); multiplicities are abbreviated as follows: s, singlet; d, duplet; dd, double duplet; t, triplet; dt, double triplet; q, quadruplet; p, pentet; m, multiplet. All spectra were recorded at room temperature.
Mass spectra were obtained using a MALDI micro MX™ (Waters, Milford, MA) equipped with a nitrogen UV laser (337 nm wavelength) and a time-lag focusing unit. Analysis was carried out in reflectron mode at 15 kV source voltage and 1950 V pulse voltage. Calibration was performed with a suitable polyethylene glycol mixture as standard. Samples were prepared by mixing solutions of matrix [10 mg/ml α-cyano-4-hydroxy-cinnamic acid in ethanol/acetonitrile/aqueous 0.1% trifluoroacetic acid (TFA) 495/495/10 (v/v/v)] and analyte [0.01–1 mg/ml in CHCl3/MeOH 2/1 (v/v)] in the ratio 10:1 (v/v). One microliter of this mixture was spotted on a target plate (stainless steel) and allowed to air dry prior to analysis.
Synthesis of the cyanine dyes
The Cy3 and Cy5 dyes are structurally identical, whereas the Cy2b dye shows some substantial structural differences. The synthesis of Cy2b was performed as described by Hung et al. (15). The methods for the synthesis of Cy3 and Cy5 were derived from procedures published by Jung and Kim (16), Mader et al. (17), and Mujumdar et al. (18). All reactions were carried out under nitrogen.
For synthesis of the Cy2b dye, 3-(5-carboxypentyl)-2-methylbenzoxazolium bromide (generated from 2-methylbenzoxazole and 6-bromohexanoic acid in dry 1,2-dichlorobenzen) was reacted with 2-(2-phenylacetamideo-E-1-ethenyl)-3-ethylbenzoxazolium iodide (obtained by reaction of 3-ethyl-2-methylbenzoxazolium iodide and N,N'-diphenylformamide in dry acetic anhydride under nitrogen). The reaction was performed in triethylamine and absolute ethanol and yielded the Cy2b dye, which was purified by flash column chromatography [n-hexane/methanol 45:55 (v/v)].
To synthesize the Cy3 dye, 2,3,3-trimethylindolenin was added to 6-bromohexanoic acid to give 1-(5-carboxypentyl)-2,3,3-trimethyl-3H-indolium bromide. This compound was condensated with N,N'-diphenylformamide in acetic anhydride/acetylchloride to yield 2-(2-phenylacetamido-E-1-ethenyl)-3,3-dimethyl-1-(5-carboxypentyl)indolium bromide. The resulting product was reacted with 1-propyl-2,3,3-trimethyl-3H-indolium iodide (synthesized from 2,3,3-trimethylindolenine and 1-iodopropane). The reaction was performed in acetic anhydride in the presence of sodium acetate. The obtained Cy3 dye was purified by flash column chromatography [n-hexane/dichloromethane/methanol 40:50:10 (v/v/v)].
For the synthesis of the Cy5 dye, 2-(4-phenylacetamino-1E,3E-butadien-1-yl)-1,3,3-trimethylindolium iodide (prepared by condensation of 1,2,3,3,-tetramethyl-3H-indolium iodide with malondialdehyde dianil hydrochloride in acetic anhydride/acetic acid) was reacted with 1-(5-carboxypentyl)-2,3,3-trimethyl-3H-indolium bromide in acetic anhydride/pyridine. The resulting Cy5 dye was purified by flash column chromatography [dichloromethane/methanol 60:10 (v/v)].
General procedure for the activation of Cy-carboxylic acids with N,N,N',N'-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate
The purified dyes were dissolved in absolute acetonitrile. Three equivalents of N,N,N',N'-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate together with six equivalents of pyridine (distilled over ninhydrin and CaH2) were added and stirred at room temperature overnight. Organic solvents were removed in vacuo, and the crude products were purified by flash chromatography [CH2Cl2/methanol 95:5 (v/v)]. Results from MS analysis of Cy5-, Cy3-, and Cy2b-N-hydroxysuccinimide (NHS) were as follows: Cy5-NHS [1 μM in CHCl3/methanol 2:1 (v/v)]: ESI-Q1 MS (positive mode): m/z =580.78, calculated 580.737 for [C36H42N3O4]+, Cy3-NHS [1 μM in CHCl3/MeOH 2:1 (v/v)]: ESI-Q1 MS (positive mode): m/z =582.92, calculated 582.752 for [C36H44N3O4]+, and Cy2-NHS [1 μM in CHCl3/MeOH 2:1 (v/v)]: ESI-Q1 MS (positive mode): m/z =516.28, calculated 516.565 for [C29H30N3O6]+.
Synthesis of p-nitrophenyl phosphonates
Synthesis of 2,5-dioxopyrrolidin-1-yl-11-(ethoxy(4-nitrophenoxy)phosphoryl) undecyl carbonate (Compound 1, Fig. 1)
Fig. 1.

Chemical synthesis of Cy-tagged phosphonic acid esters.
The p-nitrophenyl phosphonate (1) was synthesized by a multistep reaction as described by Reetz et al. (19). Briefly, 11-bromoundecanol was added to a solution of 3,4-dihydro-2H-pyran and toluene-4-sulfonic acid in CH2Cl2 to give 2-[(11-bromoundecyl)oxy]tetrahydro-2H-pyrane. This was added to a mixture of diethyl phosphite and sodium hydride to yield diethyl-11-[(2-tetrahydro-2H-pyranyl)oxy]undecylphosphonate. A solution of this phosphonate was treated with Amberlite IR-120 to yield diethyl(11-hydroxyundecyl)phosphonate. After removal of the solvent, the product was dissolved in acetonitrile. Triethylamine and di(N-succinimidyl)carbonate were consecutively added to give the activated carbonate. The phosphonate was activated with oxalyl dichloride in N,N-dimethylformamide and finally reacted with p-nitrophenol. After purification by flash chromatography, the p-nitrophenyl phosphonate NHS-ester was obtained.
Synthesis of 2-(tert-butoxycarbonylamino)ethylcarbamyl-11-(ethoxy(4-nitrophenoxy) phosphoryl) undecyl ether (Compound 3, Fig. 1)
Three hundred milligrams (553 μmol) of Compound 1 were dissolved in 1 ml acetonitrile. A solution of a slight molar excess of N-tert-butyloxycarbonyl-ethylenediamine (100 μL, 101 mg, 632 μmol) and 160 μL triethylamine in 2 ml acetonitrile was added dropwise during 1 h at room temperature. The progress of the reaction was monitored by TLC. After stirring overnight, the solvent was removed at 40°C in vacuo, and the product was purified by flash chromatography with dichloromethane/ethyl acetate 1:1 (v/v) as mobile phase. After removal of the solvent at 40°C in vacuo, the oily product (Compound 2) (C27 H46 N3 O9 P: 587.643) was stored at 4°C. The yield was 243 mg (75%).
Five hundred microliters of TFA were added to the crude Compound 2 to remove the tert-butyloxycarbonyl group, and the reaction mixture was left for 40 min on ice. The reaction was monitored by TLC. TFA was removed in vacuo, and the obtained product (Compound 3) was extracted twice with diethylether. After complete removal of the solvent, Compound 3 (C24H39F3N3O9P: 601.550) was obtained as a nearly colorless oil and stored at 4°C.
1H-NMR (500 MHz, CD3OD): δ (ppm) = 8.25 d(2H), 7.43 d(2H), 4.26 m(2H), 4.05 t(2H), 3.38 t(2H), 3.29 s(1H), 3.05 t(2H), 2.05 m(2H), 1.68 p(2H), 1.62 m(2H), 1.45 m(2H) 1.30 m(15H).
General procedure for the preparation of ARP 4a-c
The phosphonate (Compound 3) was dissolved in absolute N,N-dimethylformamide (1 ml). Ten equivalents of anhydrous triethylamine and 0.8 equivalents of the respective NHS-activated dye were added. After stirring at room temperature for 2 h, all volatile compounds were removed in vacuo and the residue was purified by flash chromatography using CH2Cl2/MeOH 95:5 (v/v) as solvent.
ARP 4a [1 μM in CHCl3/MeOH 2:1 (v/v)]: ESI-Q1 MS (positive mode): m/z = 888.38, calculated 889.005 for [C47H63N5O10P]+, MALDI: m/z = 888.46
1H-NMR (500 MHz, CDCl3): δ (ppm) = 8.42–8.37 (t, 1H, J = 13Hz), 8.17–8.15 (d, 2H, J = 8Hz), 7.42–7.39 (m, 2H), 7.33–7.17 (m, 4H), 6.36–6.34 (d, 1H, J = 13Hz), 6.23–6.21 (d, 1H, J = 13Hz), 4.20–3.79 (m, 10H), 3.32–3.20 (m, 4H), 2.29–2.26 (t, 2H, J = 6Hz), 1.86–1.81 (m, 2H), 1.71–1.43 (m, 10H), 1.34–1.30 (m, 2H), 1.26–1.23 (t, 3H, J = 7Hz), 1.20–1.16 (m, 10H), 0.82–0.79 (t, 3H, J = 7Hz).
ARP 4b [1 μM in CHCl3/MeOH 2:1 (v/v)]: ESI-Q1 MS (positive mode): m/z = 954.55,
calculated 955.192 for [C54H77N5O8P]+, MALDI: m/z = 954.59
1H-NMR (500 MHz, CDCl3): δ (ppm) = 8.44–8.38 (t, 1H, J = 13Hz), 8.25–8.22 (d, 2H, J = 8Hz), 7.42–7.34 (m, 8H), 7.13–7.09 (m, 2H), 6.93–6.89 (d, 1H, J = 13Hz), 6.78–6.74 (d, 1H, J = 13Hz), 4.26–4.03 (m, 8H), 3.96–3.93 (m, 2H), 3.40–3.30 (m, 4H), 2.38–2.35 (m, 3H), 1.98–1.75 (m, 8H), 1.73 (s, 6H), 1.72 (s, 6H), 1.59 (bs, 8H), 1.33–1.30 (m, 3H), 1.25 (bs, 8H), 1.12–1.09 (t, 3H, J = 7Hz).
ARP 4c [1 μM in CHCl3/MeOH 2:1 (v/v)]: ESI-Q1 MS (positive mode): m/z = 952.45,
calculated 953.176 for [C54H75N5O8P]+, MALDI: m/z = 952.58
1H-NMR (500 MHz, CDCl3): δ (ppm) = 8.42–8.22 (d, 2H, J = 9Hz), 7.82–7.78 (m, 2H), 7.39–7.34 (m, 6H), 7.24–7.20 (m, 2H), 7.11–7.06 (m, 2H9, 6.93-6.86 (t, 1H, J = 12Hz), 6.42–6.38 (d, 2H, J = 12Hz), 6.34–6.29 (d, 2H, J = 12Hz), 4.25–4.12 (m, 2H9, 4.06–3.94 (m, 4H), 3.59 (s, 3H), 3.38–3.32 (m, 4H9, 2.38–2.35 (t, 2H, J = 6Hz), 1.96–1.89 (m, 2H), 1.85-1.81 (m, 2H), 1.79–1.75 (m, 2H9, 1.69 (s, 12H), 1.58–1.51 (m, 4H), 1.41–1.36 (m, 2H), 1.33–1.30 (t, 3H, J = 7 Hz), 1.29–1.21 (m, 12H).
Enzymes
The following commercially available lipases and esterases (Fluka/Sigma-Aldrich, Germany) were used as reference enzymes: Candida antarctica lipase B (CAL-B), Chromobacterium viscosum lipase (CVL), Geotrichum candidum lipase (GCL), and Mucor mihei esterase (MME). To prepare stock solutions, these proteins were dissolved in 10 mM TRIS/HCl buffer containing 0.25 M sucrose, pH 7.4.
Preparation of mouse tissue homogenates
Mouse adipose and liver tissues were kindly provided by R. Zechner (Institute of Molecular Biosciences, University of Graz, Austria). Animals were maintained on a regular light-dark cycle (14 h light, 10 h dark) and kept on a standard laboratory chow diet containing 4.5% fat and 21% protein (SSNIFF, Germany) with free access to water. Fat pads and liver were collected from fed (free access to food over night) male animals aged between 3 and 6 months between 9.00 and 10.00 AM. All procedures in this study were in conformity with the Public Health Service Policy on the use of Laboratory Animals and were approved by local ethical committees.
BAT, WAT, and liver were surgically removed and washed in PBS. Homogenization was performed on ice in lysis buffer (10 mM Tris/HCl buffer, pH 7.4, containing 0.25 M sucrose, 1 mM EDTA, 1 mM DTT, 20 μg/ml leupeptin, 2 μg/ml antipain, and 1 μg/ml pepstatin) using a motor-driven teflon-glass homogenizer (8 strokes, at 1,500 rpm; Schuett Labortechnik, Germany). Cell debris and lipid fraction were removed by centrifugation at 1,000 g for 15 min to obtain cytoplasmatic extracts. Protein concentration was determined using the Bio-Rad protein assay based on the method of Bradford (20).
Spiking of tissue homogenates with reference enzymes
A standard sample was prepared by mixing Cy2b-, Cy3- and Cy5-labeled homogenates (see below) of brown adipose tissue or liver (15 and 45 μg total protein for 1D and 2D gel electrophoresis, respectively) with 150 ng reference enzyme. Reference enzymes were CAL-B, CVL, and MME. Samples containing higher amounts of reference enzyme were prepared by adding 300, 450, and 750 ng for 1D and 2D PAGE to the homogenate (15 and 45 μg total protein, respectively).
Activity tagging of lipolytic enzymes in tissue homogenates
Incubations of proteomes with activity tags were conducted as follows: For a sample containing 50 μg of protein, the following reagent was prepared: 5 μl of a 10 mM solution of Triton X-100 in CHCl3 (final sample concentration 1 mM) and 5 μl of ARP dissolved in CHCl3 (1 nmol/10 μl, final sample concentration 10 μM) were mixed and the organic solvent was removed under a stream of argon. Fifty microliters of homogenate (1.0 mg/ml protein) were added, and the resulting mixture was incubated at 37°C under light protection for 2 h. Proteins were precipitated with 10% trichloroacetic acid on ice for 1 h and collected by centrifugation at 4°C and 14,000 g for 15 min. The pellet was washed once with ice-cold acetone and resuspended in sample buffer for 1D SDS-PAGE (20 mM KH2PO4, 6 mM EDTA, 60 mg/ml SDS, 100 mg/ml glycerol, 0.5 mg/ml bromophenol blue, and 20 μl/ml mercaptoethanol, pH 6.8) or sample buffer for 2D PAGE (7 M urea, 2 M thiourea, 4% CHAPS, and 2% Pharmalyte 3-10). Prior to loading onto the gel, the samples for 1D SDS-PAGE were heated to 95°C for 5 min. BAT and WAT from three different mice were analyzed in three independent experiments (including an original and a dye-swap experiment) each.
SDS-PAGE and 2D gel electrophoresis
SDS-PAGE was performed essentially according to the method of Fling and Gregerson (21) in a Tris/glycine buffer system. Proteins (15 μg protein/lane) were applied onto a 5% stacking gel and separated in a 10% resolving gel at 20 mA constant current (Bio-Rad Mini PROTEAN 3), respectively. The 2D gel electrophoresis was performed as described by Gorg et al. (22–24). In the first dimension, 45 μg protein were separated by isoelectric focusing in 7 cm immobilized nonlinear pH 3–10 gradients (Immobiline™ Dry Strip Gel strips; GE Healthcare, Germany) using Multiphor II (GE Healthcare). A discontinuous voltage gradient was used starting at 0 V and increased to 200 V within the first minute. The voltage was then further increased to 3,500 V during the following 1.5 h and held at this level for another 1.5 h.
In the second dimension, proteins were separated by 10% SDS-PAGE on 7-cm gels at 20 mA constant current for 1.5 h.
Visualization
Gels were fixed in 7.5% acetic acid and 10% ethanol and scanned at a resolution of 100 μm (Bio-Rad Molecular Imager™ FX Pro Plus). Fluorescence detection was carried out using the following wavelengths: Cy2b excitation at 488 nm and emission at 530 nm, Cy3 excitation at 532 nm and emission at 605 nm, and Cy5 excitation at 635 nm and emission at 695 nm.
The photomultiplier voltage of the molecular imager was individually set for each Cy-tagged inhibitor using the same sample to reach comparable fluorescence signal intensities.
For visualization of the whole protein, pattern gels were afterwards stained with SYPRO Ruby following the manufacturer's instructions (Molecular Probes) and scanned at 605 nm and an excitation wavelength of 488 nm. The signals obtained with SYPRO Ruby varied depending on the preceding incubation with the Cy-tagged inhibitors. Proteins giving fluorescent lanes/spots with the ARPs showed less intense lanes/spots with SYPRO Ruby.
Quantification of the fluorescence signals was performed using Quantity One 1D analysis software (Bio-Rad, Vienna, Austria) and Progenesis PG 220 version 2006 2D analysis software (Nonlinear Dynamics, Newcastle upon Tyne, UK).
LC-MS/MS analysis
Fluorescent protein spots were excised from gels and tryptically digested according to the method by Shevchenko et al. (25). Peptide extracts were dissolved in 0.1% formic acid and separated by nano-HPLC-system (FAMOS™ autosampler, SWITCHOS™ loading system, and ULTIMATE™ dual gradient system; LC-Packings, Amsterdam, The Netherlands) as described (26), but using the following gradient: solvent A: water, 0.3% formic acid; solvent B: acetonitrile/water 80/20 (v/v), 0.3% formic acid; 0 to 5 min: 4% B, after 40 min 55% B, then for 5 min 90% B, and 47 min reequilibration at 4% B. The sample was ionized in a Finnigan nano-ESI source equipped with NanoSpray tips (PicoTip™ Emitter; New Objective, Woburn, MA) and analyzed in a Thermo-Finnigan LTQ linear ion trap mass spectrometer (Thermo, San Jose, CA). The MS/MS data were analyzed by searching the National Center for Biotechnology Information nonredundant public database with SpectrumMill Rev. 03.03.078 (Agilent, Darmstadt, Germany) software. Acceptance parameters were two or more identified distinct peptides according to Carr et al. (27). Identified protein sequences were subjected to BLAST and motif search for identification of potential serine hydrolases.
RESULTS
We report on novel ARPs for comparative analysis of lipolytic enzymes of two different samples in one electrophoresis gel (DABGE). For this purpose, the synthesis of the ARPs, the design of the DABGE procedure, and the application of this method to the comparative analysis of lipases and esterases in BAT and WAT has been developed.
Synthesis of activity recognition probes
The ARPs were synthesized as outlined in Fig. 1. The NHS-activated ethyl-p-nitrophenyl phosphonate (Compound 1) served as central synthon (19). Reaction of this compound with equimolar amounts of tert-butyloxycarbonyl-protected ethylene diamine followed by removal of the protective group from the intermediate (Compound 2) led to the phosphonate (Compound 3) containing a free amino group.
The final ARPs (4a-c) were obtained by coupling of the respective fluorophore-NHS esters to this compound.
Properties of the cyanine-tagged inhibitors 4a-c
The cyanine-tagged inhibitors differ solely with respect to the fluorescent dye. The fluorophores Cy3 and Cy5 are highly similar, whereas chromophore structure and polarity of Cy2b are slightly different.
The fluorescence properties of these markers allow good spectral separation of their signals and storage in a multichannel readout format. The Cy2b-, Cy3-, and Cy5-tagged inhibitors absorb and emit fluorescence at different wavelengths (Cy2b: λex, 491 nm, λem, 509 nm; Cy3: λex, 553 nm, λem, 569 nm; Cy5, λex, 645 nm, λem, 664 nm). The fluorescent ARPs 4a-c mimic natural carboxylic acid ethyl esters but contain an alkylphosphonyl instead of an acyl residue. The reactive group is a p-nitrophenyl phosphonate ester, which reacts with the nucleophilic serine of lipid and other hydrolases, thereby forming a covalent, stable and fluorescent probe-protein complex.
After separation by PAGE or chromatography, the labeled proteins are detected through their fluorescence and identified by mass spectrometry.
Optimization of ARP labeling: effects of incubation time and probe and protein concentration
To ensure quantitative labeling of all lipases in an unknown sample by the ARPs, various reaction parameters were optimized. Homogenates of murine BAT were spiked with CAL-B, CVL, or MME as reference enzymes, respectively, followed by incubation with the inhibitors for different times (between 5 and 150 min) (Fig. 2A–C). Here, the experiments using CVL as reference enzyme are shown. Similar results were obtained when CAL-B or MME were used (data not shown).
Fig. 2.

Optimization of time-dependent enzyme labeling by Cy-tagged ARPs. A–D: Homogenized brown adipose tissue (15 μg protein /lane) was spiked with CVL (150 ng/15 μg; highlighted by frames) as reference enzyme as described (see Experimental Procedures) followed by incubation with fluorescent ARP (A: 4a; B: 4b; C: 4c, final label concentration was 10 μM; D: whole protein stain using SYPRO Ruby). Incubation times were 5 min (1), 10 min (2), 30 min (3), 60 min (4), 90 min (5), 120 min (6), and 150 min (7). The reaction was stopped by precipitation of the protein with trichloroacetic acid followed by SDS gel electrophoresis. E–H: Effect of probe concentration on enzyme labeling by Cy-tagged ARPs. Mouse liver homogenate (15 μg/lane) was spiked with CVL as reference enzyme (150 ng/15 μg) followed by labeling with fluorescent ARP (E, 4a; F, 4b; G, 4c; H, SYPRO Ruby stain). Final probe concentrations were: 1, 0.5 μM; 2, 1.0 μM; 3, 2.5 μM; 4, 5.0 μM; 5, 7.5 μM; 6, 10.0 μM; 7, 15.0 μM; 8, 20.0 μM; 9, 25.0 μM. I–L: Effect of protein concentration on enzyme labeling by Cy-tagged ARPs. Mouse liver homogenate was spiked with CVL as reference enzyme (150 ng/15 μg) and labeled with fluorescent ARPs [I, 4a; J, 4b; K, 4c (10 μM final concentration); L, SYPRO Ruby stain]. The total protein contents of the samples were: 1, 0.5 μg; 2, 1.0 μg; 3, 2.0 μg; 4, 4.0 μg; 5, 6.0 μg; 6, 8.0 μg; 7, 10.0 μg; 8, 12.0 μg; 9, 15.0 μg.
Some proteins were immediately labeled after addition of the fluorescent ARP to the sample. Other enzymes showed much slower reaction rates with the probes. After 90 min, protein labeling appeared to be complete since the number and intensity of the fluorescent lanes remained constant. Thus, an incubation time of 120 min was used for all further studies. Notably, the labeling patterns obtained with the Cy3- and Cy5-tagged inhibitors were similar but significantly different to the Cy2b-tagged samples. This may be due to the fact that the structure of the Cy2b chromophore differs from the Cy3 and Cy5 chromophores.
In addition, probe concentrations were optimized for the DABGE experiments. For this purpose, tissue homogenates were labeled with different concentrations of the individual ARPs between 0.5 and 25 μM final concentration (protein content of the sample was 1 mg/ml). The extent of protein labeling strongly depended on probe concentration as shown in Fig. 2E–G. While some proteins were labeled at very low concentrations, others needed higher concentrations. A final probe concentration of 10 μM was chosen for all further experiments because protein patterns did not change above this value.
Finally, the protein concentration of the sample was optimized for the DABGE assay. All samples in this series were incubated with the Cy-tagged ARPs (10 μM final concentration) for 120 min (sample protein concentrations between 0.02 and 1.00 mg/ml). The results are shown in Fig. 2I–K. The samples containing low protein concentrations showed weak fluorescence signals, whereas higher amounts of protein gave intensely fluorescent lanes. Under our experimental conditions, irrespective of the amount of protein loaded, the fluorescent bands were always well resolved after 1D-PAGE. For all further experiments, protein concentrations of 1 mg/ml were used since under these conditions labeling appeared to be best.
The optimization studies outlined above led to the following standard protocol, which is recommended for labeling of biological samples with cyanine-tagged ARPs: protein concentration 1 mg/ml, probe concentration 10 μM, and reaction time 2 h.
Proof of principle: DABGE analysis of artificial proteomes using cyanine-tagged ARPs
Artificial proteomes were prepared containing defined amounts of three different commercial enzymes (CVL, MME, and GCL), which were labeled using the Cy-tagged ARPs. It was the aim of this study to compare the relative amounts of these enzymes in two samples A and B characteristic for different enzyme ratios. The amount of one enzyme, namely, MME, was always kept constant in both samples A and B to mimic a “housekeeping” protein fraction, which shows a constant “expression level” in both samples. The amounts of the other two enzymes (CVL and GCL) were varied to mimic different enzyme “expression” levels. The ratios of lipases in sample A versus B were as follows: CVL 1:3, GCL 2:1, and MME 1:1. The individual samples A and B were labeled with the Cy3 and Cy5 probes, respectively. Independently, a mixture of samples A and B containing the same total amount of protein was labeled with Cy2b inhibitor. Finally, equal amounts of the Cy3-, Cy5-, and Cy2b-labeled aliquots were mixed, and the resultant mixture was subjected to 2D-PAGE. In a dye-swap experiment, the same procedure was performed except that sample A and sample B were labeled with the Cy5- and Cy3-ARP, respectively (experimental setup; Fig. 3).
Fig. 3.

Experimental setup of a DABGE experiment. To prepare the original sample (dye-swap), sample A is incubated with Cy3-tagged (Cy5-tagged) and sample B with Cy5-tagged (Cy3-tagged) inhibitor. A 1:1 mixture of samples A and B is reacted with Cy2b-tagged inhibitor. The samples are mixed, followed by protein precipitation, gel electrophoretic separation, and detection of the fluorescence signal.
Figure 4 shows the results of a DABGE analysis of lipases in the artificial proteome are displayed. MME shows yellow spots in both gels (Fig. 4A, B) because its concentrations in samples A and B are the same (A/B ratio = 1/1). CVL is red in the left gel (green in the dye-swap, right panel) because its A/B ratio is 1/3. Conversely, GCL appears green in the left gel (red in the dye-swap) because its A/B ratio is 2/1.
Fig. 4.

DABGE of lipases in artificial proteomes. Three commercially available enzymes (CVL, GCL, and MME) were used to mimic a lipolytic proteome. Two separate enzyme mixtures were prepared differing in the CVL/GCL ratio. The amount of MME was the same in both samples. The ratios of lipases in sample A versus B were as follows: CVL 1:3, GCL 2:1, and MME 1:1. The individual samples A and B were labeled with the Cy3 and Cy5 probes, respectively. Independently, a mixture of samples A and B containing the same amount of total protein was labeled with Cy2b inhibitor. Finally, equal amounts of the Cy3-, Cy5-, and Cy2b-labeled aliquots were mixed and the resultant mixture was subjected to 2D-PAGE. In a dye-swap experiment, the same procedure was performed except that sample A and sample B were labeled with the Cy5- and the Cy3-ARP, respectively. A: Original experiment. B: Dye-swap. C: Quantitative analysis of the fluorescent intensities of the labeled lipases showed that the obtained enzyme ratios very closely reflected the amounts of the respective labeled enzymes.
The enzyme ratios determined from the fluorescence intensities were very close (<10% mean error) to the theoretical values (Fig. 4C).
DABGE of complex proteomes: spiking of active biological samples with reference enzymes
For calibration, mouse liver homogenates were admixed with the following reference enzymes: CAL-B, CVL, and MME. GCL was not included because of extensive overlaps with the enzymes of the biological sample. In three independent experiments, one of the three enzymes was added to mouse liver homogenate followed by labeling with Cy-ARPs as described above. To establish a quantitative relationship between enzyme concentration and apparent fluorescence intensity, we analyzed samples containing various amounts of reference lipases. Aliquots of the spiked homogenates were taken such that sample A contained 1-fold, 2-fold, 3-fold, and 5-fold amounts of reference protein, relative to the respective protein in sample B, which was always kept constant (1-fold). Protein samples A and B were then incubated with Cy3-tagged and Cy5-tagged ARP, respectively. A mixture of A:B = 1:1 was labeled with Cy2b-tagged inhibitor. The labeled samples were mixed, and the protein was precipitated and then separated by 1D or 2D gel electrophoresis. For a dye-swap experiment, samples A and B were labeled with Cy5-tagged and Cy3-tagged ARPs, respectively. Again, a mixture of both protein samples was labeled with Cy2b-tagged inhibitor as a reference (Fig. 3).
Figure 5 shows the results of the spiking experiment using MME as reference enzyme. CAL-B or CVL spiking gave similar results (data not shown).
Fig. 5.

Quantitative analysis of a reference protein in DABGE of a native proteome. A: Mouse liver homogenate was admixed with MME as a reference enzyme. Samples A containing various relative amounts of MME (1×, 2×, 3×, and 5×) and sample B containing the same relative amount of MME (1×) were prepared. The individual samples A and B were incubated with Cy3- and Cy5-tagged ARPs, respectively. Independently, a mixture of sample A and B with the same total protein amounts was labeled with Cy2b-tagged inhibitor. Finally, equal amounts of the Cy3-, Cy5-, and Cy2b-labeled aliquots were mixed and the proteins were separated either by 1D SDS-PAGE (A) (15 μg protein/lane) or 2D SDS-PAGE (C, D) (45 μg protein /gel). A: Relative amounts of MME in sample A versus B were: lane 1, 1:1; lane 2, 2:1; lane 3, 3:1; lane 4, 5:1 (dye-swap: lane 5, 2:1; lane 6, 3:1; lane 7, 5:1). MME in lane 1 appeared yellow, which is caused by the overlay of green and red; lanes 2–4 appeared green [sample labeled with Cy3-tagged ARP (false color green)]; and the lanes 5–7 appeared red [sample labeled with Cy5-tagged ARP (false color red)]. B: The relative amounts of MME in each lane were determined from the respective fluorescence intensities that were plotted against the “theoretical” ratios used for sample preparation. C, D: The relative amount of MME in sample A versus B was 3:1; thus, MME appeared green in C (sample A was labeled with Cy3-tagged ARP) and red in D (sample A labeled with Cy5-tagged inhibitor). E: Quantitative analysis of fluorescence intensities of the labeled lipases showed that the obtained enzyme ratios very closely reflected the relative amounts of the respective labeled enzymes used for spiking.
Figure 5A shows an overlay of the fluorescent images obtained after protein separation by 1D gel electrophoresis. Lanes containing identical amounts of MME in A and B appeared yellow. The lanes of the samples A, which were admixed with 2-fold, 3-fold, and 5-fold MME, appear green (Cy3-tagged ARP). In the dye-swap experiment, the corresponding lanes appear red (Cy5-tagged ARP). The Cy2b-tagged enzymes served as an internal intensity reference for the variable Cy3 and Cy5 fluorescence signals. The enzyme ratios experimentally determined from quantitative analysis of the fluorescent intensities of the Cy3- and Cy5-labeled proteins very closely reflected the relative amounts of the respective enzymes in the protein mixtures, the mean error being <10% (Fig. 5B).
Typical fluorescence images of the same lipolytic and esterolytic proteomes that were separated by 2D gel electrophoresis are shown in Fig. 5C (original experiment) and 5D (dye-swap experiment). For this experiment, liver homogenate was spiked with 3-fold MME before labeling with the Cy-tagged ARPs (sample A). A homogenate containing 1-fold MME served as sample B. In Fig. 5C, the MME spot (3-fold) appears green because sample A was labeled with the Cy3-tagged inhibitor. All other enzymes (natural components of the biological matrix), which were the same in samples A and B, appear yellow (mixture of red and green). The MME spot (3-fold) in Fig. 5D is red because sample A was labeled with the Cy5 inhibitor (dye-swap).
Fluorescence intensities of cyanine-labeled proteins were again analyzed in terms of enzyme ratios that very closely reflected the amounts that were used for spiking the biological samples. The mean error was <10% in all experiments (Fig. 5E).
Application of the DABGE technique to comparative enzyme analysis of adipose tissues
The DABGE technique was used to compare lipolytic enzymes in BAT and WAT. BAT and WAT were labeled with Cy3- and Cy5-tagged ARPs, respectively, and vice versa in a dye-swap experiment. The 1:1 (protein amount) mixtures of BAT and WAT homogenates were labeled with Cy2b-tagged inhibitor as an internal standard. The labeled samples were mixed, and the proteins were precipitated followed by 1D or 2D gel electrophoresis.
Figure 6A and C show the results obtained after protein separation by 1D gel electrophoresis. Enzymes more abundant in BAT are shown as green and red bands in the original and in the dye-swap experiment, respectively. Red (original) or green (dye-swap) color was observed if the labeled enzymes were less abundant in BAT than in WAT. If enzyme concentrations were the same in BAT and WAT, yellow lanes were observed. Fig. 6B (original experiment) and D (dye-swap experiment) show the results after 2D protein separation.
Fig. 6.

DABGE analysis of lipolytic enzymes in BAT and WAT. Fluorescence patterns of tagged proteins after 1D and 2D electrophoresis. A shows the results of the original experiment comparing BAT and WAT, whereas C shows the corresponding gel obtained after dye-swap. In a comparative experiment, proteins were separated by 2D gel electrophoresis leading to images B and D. The fluorescence images show that one fluorescent band of the 1D gel contains several different enzymes corresponding to several spots in the 2D image (e.g., lane n of the 1D PAGE contains the enzymes corresponding to the spots n1 and n2 in 2D PAGE). Higher amounts of active proteins in BAT as compared with WAT appear green in A and B (and red in C and D) and vice versa. Similar amounts in both tissues appear yellow. Relative enzyme ratios of BAT versus WAT and the protein identification are listed in Table1.
To identify the enzymes, the lanes or spots were cut out, tryptically digested, and analyzed by nano-HPLC-MS/MS. The relative ratios of the labeled enzymes in BAT versus WAT were determined from the fluorescence intensities of Cy3 and Cy5 in BAT and WAT using the following formula:
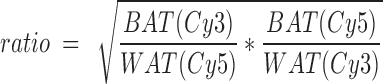 |
.
In case of calculated ratios smaller than 1, the negative inverse ratios were generated. Thus, positive values mean that the respective active enzymes are more abundant in BAT and vice versa. The combined results are shown in Table 1. The lipolytic and esterolytic proteomes of BAT and WAT differ to a great extent. The enzyme compositions of BAT and WAT vary greatly. For instance, much higher abundances of active esterase 31-like, esterase 1, and monoglyceride lipase were detected in WAT than in BAT. Several thiolases (e.g., mitochondrial acyl-CoA thioesterase 1, 3-ketoacyl-CoA thiolase, and mitochondrial acetoacetyl-CoA thiolase) and the lysophospholipases 1 and 2 are more abundant in BAT than in WAT. On the other hand, other enzymes are equally active in BAT and WAT (e.g., hormone-sensitive lipase, epoxide hydrolase, and adipose tissue triglyceride lipase). Albumin was also detected by the fluorescent phosphonates. This observation is in li-ne with reports indicating that albumin exerts esterolytic activity (28).
TABLE 1.
The differential lipolytic proteome of mouse adipose tissue
| No. | Name | NCBI No. | MW (kDa) | pI | Identified Peptides | Sequence Coverage (%) | Relative Ratios | |
|---|---|---|---|---|---|---|---|---|
| a | Fatty acid synthase | 28461372 | 242.4 | 6.13 | 80 | 49 | −1.6 | |
| b | Oxoglutarate dehydrogenase | |||||||
| 1 | Oxoglutarate dehydrogenase | 15489120 | 116.5 | 6.36 | 48 | 55 | 3.7 | |
| c | Hormone sensitive lipase | |||||||
| 1 | Hormone sensitive lipase | 26325924 | 88.0 | 8.15 | 9 | 17 | 1.5 | |
| d | Esterase 1, Albumin 1 | |||||||
| 1 | Esterase1 | 6679689 | 61.1 | 5.02 | 7 | 17 | −2.1 | |
| 2 | Albumin1 | 74137565 | 68.7 | 5.79 | 27 | 58 | −1.6 | |
| e | Albumin1, Leukotriene-A4 hydrolase, Carnitine palmitoyltransferase 2 | |||||||
| 1 | Albumin1 | 74137565 | 68.7 | 5.79 | 40 | 71 | −1.9 | |
| 2 | Leukotriene-A4 hydrolase | 74148023 | 69.1 | 5.98 | 7 | 14 | 2.1 | |
| 3 | Carnitine palmitoyltransferase 2 | 74139371 | 74.0 | 8.59 | 14 | 20 | 1.4 | |
| f | Esterase 31-like, Carnitine acetyltransferase | |||||||
| 1 | Esterase 31-like | 21362301 | 63.0 | 5.65 | 4 | 7 | −7.3 | |
| 2 | Carnitine acetyltransferase | 59802828 | 70.9 | 8.53 | 6 | 12 | −1.6 | |
| g | Epoxide Hydrolase, Adipose triglyceride lipase, Carboxylesterase ML1, Triacylglycerol lipase, 3-Oxoacid CoA transferase 1 | |||||||
| 1 | Epoxide Hydrolase | 563510 | 62.5 | 5.86 | 3 | 8 | −1.3 | |
| 2 | Adipose triglyceride lipase | 26327465 | 53.7 | 6.06 | 6 | 17 | −1.3 | |
| 3 | Carboxylesterase ML1 | 15488664 | 61.6 | 6.03 | 15 | 35 | −1.4 | |
| 4 | Triacylglycerol lipase | 16716505 | 61.9 | 6.30 | 15 | 36 | −1.4 | |
| 5 | 3-Oxoacid CoA transferase 1 | 74222276 | 56.0 | 8.73 | 11 | 29 | 1.0 | |
| h | Enolase 1 | |||||||
| 1 | Enolase1 | 53734652 | 50.2 | 8.01 | 19 | 57 | 1.6 | |
| i | Apolipoprotein A4, S-adenosylhomocysteine hydrolase, CGI-58 like (Abhd5), Fumaryl acetoacetase, Acyl-CoA thioesterase 2 | |||||||
| 1 | Apolipoprotein A4 | 38198665 | 45.0 | 5.33 | 12 | 29 | −1.3 | |
| 2 | S-adenosylhomocysteine hydrolase | 74185262 | 47.8 | 6.03 | 8 | 22 | −1.5 | |
| 3 | CGI-58 like (Abhd 5) | 22477988 | 39.2 | 6.33 | 3 | 10 | −1.7 | |
| 4 | Fumaryl acetoacetase | 14789698 | 46.1 | 6.92 | 3 | 9 | −1.8 | |
| 5 | Acyl-CoA Thioesterase 2 | 31980998 | 50.6 | 8.74 | 5 | 12 | −1.4 | |
| j | Mitochondrial Acyl-CoA thioesterase 1, 3-Ketoacyl-CoA Thiolase, Acetyl-CoA acyltransferase 1, Acetyl-CoA acyltransferase 2 | |||||||
| 1 | Mitochondrial Acyl-CoA thioesterase 1 | 74196709 | 49.7 | 6.88 | 15 | 34 | 4.5 | |
| 2 | 3-Ketoacyl-CoA thiolase | 29126205 | 41.8 | 8.33 | 14 | 55 | 3.2 | |
| 3 | Mitochondrial acetoacetyl-CoA thiolase | 19354555 | 44.8 | 8.71 | 7 | 23 | 5.8 | |
| k | Acetyl-CoA acetyltransferase 3, Acetyl-CoA acetyltransferase 2 | |||||||
| 1 | Acetyl-CoA acetyltransferase 3 | 29612600 | 41.5 | 8.09 | 2 | 12 | 4.2 | |
| 2 | Acetyl-CoA acetyltransferase 2 | 30348956 | 41.3 | 6.60 | 4 | 13 | 1.1 | |
| l | Esterase D, Monoglyceride lipase, Williams-Beuren syndrome critical region | |||||||
| 1 | Esterase D | 12846304 | 34.9 | 8.51 | 7 | 37 | −1.6 | |
| 2 | Monoglyceride lipase | 74201613 | 37.5 | 8.91 | 7 | 24 | −3.1 | |
| m | Esterase 1 homolog, Glutathione S-transferase μ2, Glutathione S-transferase θ2, Glutathione S-transferase ζ1 | |||||||
| 1 | Esterase 1 homolog | 20070420 | 28.1 | 9.00 | 9 | 43 | 1.3 | |
| 2 | Glutathione S-transferase μ2 | 22477996 | 25.7 | 6.91 | 14 | 64 | 1.4 | |
| 3 | Glutathione-S-transferase θ2 | 2495110 | 27.7 | 7.03 | 5 | 27 | −1.4 | |
| 4 | Glutathione-S-transferase ζ1 | 21594192 | 24.3 | 7.68 | 3 | 18 | 1.8 | |
| n | Lysophospholipase 1, Lysophospholipase 2 | |||||||
| 1 | Lysophospholipase 1 | 15488808 | 24.7 | 6.14 | 4 | 20 | 3.3 | |
| 2 | Lysophospholipase 2 | 45768815 | 24.8 | 6.74 | 3 | 10 | 2.2 | |
Letters and numbers correspond to the fluorescent protein lanes (1D PAGE) and spots (2D PAGE) in Fig. 5. All proteins in this list were identified by nano-HPLC-MS/MS analysis. The relative ratios were calculated using the formula indicated in Results. Active enzymes are considered more abundant if relative ratios are lower than −1.9 (WAT) and higher than −1.9 (BAT).
In summary, our results show that the new DABGE method is a very useful tool for differential activity profiling of closely related proteomes since activity patterns of lipolytic and/or esterolytic enzymes can be compared in one gel with high sensitivity, precision, and reproducibility.
DISCUSSION
Matched sets of activity-directed suicide enzyme inhibitors were developed for comparative analysis of closely related lipolytic proteomes in the same electrophoresis gel. The characteristic feature of this so-called DABGE technology is the combination of the main advantages of two powerful technologies that are currently used in proteomics research: 1) The new method can be used for direct comparison of complex proteomes within the same gel, using a set of spectrally different markers that are matched in structure and charge [DIGE™ principle (13, 29, 30)]; and 2) selection of a small number of active enzymes of interest from thousands of proteins within the entire proteome (activity-based proteomics). For this purpose, only the functional target proteins are labeled with specific enzyme probes. Therefore, this procedure highly improves conventional DIGE™ in which all proteins of a sample are unselectively labeled by fluorescent dyes. As a consequence, the differential proteomes in DABGE are less complex than the differential DIGE™ patterns, thus providing more specific information on up- and downregulation of protein activities.
A set of suitable activity-based probes, represented by the Cy-Ethyl suicide inhibitor series, was developed for DABGE. The enzyme-specific probes inactivate lipases, carboxyl esterases, thioesterases, and amide hydrolases by reacting covalently with the nucleophilic serine in the active center in these proteins. The ARPs 4a, b, and c contain fluorophores commonly used for differential 2D gel electrophoresis (DIGE™). The cyanine-based fluorophores Cy2b, Cy3, and Cy5 show a maximum of structural similarity. Their extinction coefficients are far higher than those of many other fluorophores. Their molecular weights are very similar (ARP 4a, 889 Da; ARP 4b, 955 Da; ARP 4c, 953 Da) and thus hardly affect relative migration of activity-tagged enzymes in polyacrylamide gels. This is especially important for small enzymes.
Conditions of activity-based fluorescence labeling were optimized to ensure maximal tagging of enzymes in a sample by the ARPs. This implies that the resultant labeling patterns reflect the abundance of the labeled enzymes in the sample and not the kinetics of protein labeling. Since enzyme labeling by the ARPs is stoichiometric, the differential functional proteomes can be quantified on the basis of their fluorescence intensities.
A typical analysis was performed using a model containing defined amounts of known enzymes (CVL, GCL, and MME). The experimentally determined values are in accordance with the theoretical ratios that were used for preparing the artificial proteomes.
To determine possible matrix effects of the DABGE technique in complex biological samples, we admixed mouse liver homogenate as a natural proteome with defined amounts of reference enzymes. Again, the experimentally determined enzyme ratios matched the ratios of the reference enzymes contained in the homogenates. Finally, the DABGE method was used to identify the quantitative differences between the lipolytic and esterolytic proteomes of two complex biological samples, namely, BAT and WAT. We found that both tissues showed specific, yet distinctive, enzyme patterns. WAT and BAT perform essentially opposite functions in vivo. Whereas WAT is accumulating excess energy as triacylglycerol, BAT is dissipating energy through adaptive thermogenesis (31). Current investigations in our laboratory aim at elucidating the biological function of the lipolytic enzymes that are expressed differently in BAT and WAT. In particular, the relationships between apparent enzyme activities of the individual protein components, moles of active enzyme, enzyme mass, and individual gene expression on the RNA level will be studied. They may specifically depend not only on the genetic background of the functional proteomes but also on the (patho)physiological conditions of interest.
We conclude that the DABGE technique is very useful for differential activity profiling of closely related proteomes since changes in activity of different lipolytic and/or esterolytic enzymes can be observed simultaneously with high accuracy. Thus, it can be employed for comparative analysis of lipolytic activities in samples differing with respect to genetic background, the environment, or subcellular localization. It can be expected that this technique will find many applications in various fields of biomedicine, biotechnology, and routine laboratory practice.
Abbreviations
1D, one-dimensional
2D, two-dimensional
ARP, activity recognition probe
BAT, brown adipose tissue
CAL-B, Candida antarctica lipase B
CVL, Chromobacterium viscosum lipase
Cy, cyanine dye
DABGE, differential activity-based gel electrophoresis
DIGE, differential gel electrophoresis
GCL, Geotrichum candidum lipase
MME, Mucor mihei esterase
NHS, N-hydroxysuccinimide
TFA, trifluoroacetic acid
WAT, white adipose tissue
The authors gratefully acknowledge financial support by the Austrian Federal Ministry of Science and Research/FFG in the framework of the GEN-AU program (GOLD project).
Published, JLR Papers in Press, March 11, 2009.
References
- 1.Cravatt B. F., and E. J. Sorensen. 2000. Chemical strategies for the global analysis of protein function. Curr. Opin. Chem. Biol. 4 663–668. [DOI] [PubMed] [Google Scholar]
- 2.Campbell D. A., and A. K. Szardenings. 2003. Functional profiling of the proteome with affinity labels. Curr. Opin. Chem. Biol. 7 296–303. [DOI] [PubMed] [Google Scholar]
- 3.Scholze H., H. Stutz, F. Paltauf, and A. Hermetter. 1999. Fluorescent inhibitors for the qualitative and quantitative analysis of lipolytic enzymes. Anal. Biochem. 276 72–80. [DOI] [PubMed] [Google Scholar]
- 4.Speers A. E., and B. F. Cravatt. 2004. Chemical strategies for activity-based proteomics. ChemBioChem. 5 41–47. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y., M. P. Patricelli, and B. F. Cravatt. 1999. Activity-based protein profiling: the serine hydrolases. Proc. Natl. Acad. Sci. USA. 96 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oskolkova O. V., R. Saf, E. Zenzmaier, and A. Hermetter. 2003. Fluorescent organophosphonates as inhibitors of microbial lipases. Chem. Phys. Lipids. 125 103–114. [DOI] [PubMed] [Google Scholar]
- 7.Greenbaum D., K. F. Medzihradszky, A. Burlingame, and M. Bogyo. 2000. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem. Biol. 7 569–581. [DOI] [PubMed] [Google Scholar]
- 8.Adam G. C., B. F. Cravatt, and E. J. Sorensen. 2001. Profiling the specific reactivity of the proteome with non-directed activity-based probes. Chem. Biol. 8 81–95. [DOI] [PubMed] [Google Scholar]
- 9.Adam G. C., E. J. Sorensen, and B. F. Cravatt. 2002. Proteomic profiling of mechanistically distinct enzyme classes using a common chemotype. Nat. Biotechnol. 20 805–809. [DOI] [PubMed] [Google Scholar]
- 10.O'Farrell P. H. 1975. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 250 4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 11.Knowles M. R., S. Cervino, H. A. Skynner, S. P. Hunt, C. de Felipe, K. Salim, G. Meneses-Lorente, G. McAllister, and P. C. Guest. 2003. Multiplex proteomic analysis by two-dimensional differential in-gel electrophoresis. Proteomics. 3 1162–1171. [DOI] [PubMed] [Google Scholar]
- 12.Viswanathan S., M. Ünlü, and J. S. Minden. 2006. Two-dimensional difference gel electrophoresis. Nat. Protoc. 1 1351–1358. [DOI] [PubMed] [Google Scholar]
- 13.Ünlü M., M. Morgan, and J. S. Minden. 1997. Difference gel electrophoresis. A single gel method for detecting changes in protein extracts. Electrophoresis. 18 2071–2077. [DOI] [PubMed] [Google Scholar]
- 14.Karp N. A., D. P. Kreil, and K. S. Lilley. 2004. Determining a significant change in protein expression with DeCyder™ during pair-wise comparison using two-dimensional difference gel electrophoresis. Proteomics. 4 1421–1432. [DOI] [PubMed] [Google Scholar]
- 15.Hung S. C., J. Ju, R. A. Mathies, and A. N. Glazer. 1996. Cyanine dyes with high absorption cross section as donor chromophores in energy transfer primers1. Anal. Biochem. 243 15–27. [DOI] [PubMed] [Google Scholar]
- 16.Jung M. E., and W-J. Kim. 2006. Practical syntheses of dyes for difference gel electrophoresis. Bioorg. Med. Chem. 14 92–97. [DOI] [PubMed] [Google Scholar]
- 17.Mader O., K. Reiner, H-J. Egelhaaf, R. Fischer, and R. Brock. 2004. Structure property analysis of pentamethine indocyanine dyes: identification of a new dye for life science applications. Bioconjug. Chem. 15 70–78. [DOI] [PubMed] [Google Scholar]
- 18.Mujumdar R. B., L. A. Ernst, S. R. Mujumdar, C. J. Lewis, and E. Wagner. 1993. Cyanine dye labeling reagents: sulfoindocyanine succinimidyl esters. Bioconjug. Chem. 4 105–111. [DOI] [PubMed] [Google Scholar]
- 19.Reetz M. T., C. J. Ruggeberg, M. J. Droge, and W. J. Quax. 2002. Immobilization of chiral enzyme inhibitors on solid supports by amide-forming coupling and olefin metathesis. Tetrahedron. 58 8465–8473. [Google Scholar]
- 20.Bradford M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- 21.Fling S. P., and D. S. Gregerson. 1986. Peptide and protein molecular weight determination by electrophoresis using a high-molarity tris buffer system without urea. Anal. Biochem. 155 83–88. [DOI] [PubMed] [Google Scholar]
- 22.Gorg A., W. Postel, S. Gunther, and J. Whitaker. 1985. Improved horizontal two-dimensional electrophoresis with hybrid isoelectric focusing in immobilized pH gradients in the first dimension and laying-on transfer to the second dimension. Electrophoresis. 6 599–604. [Google Scholar]
- 23.Gorg A., W. Weiss, and M. J. Dunn. 2004. Current two-dimensional electrophoresis technology for proteomics. Proteomics. 4 3665–3685. [DOI] [PubMed] [Google Scholar]
- 24.Gorg A., W. Postel, and S. Gunther. 2007. Two-dimensional electrophoresis. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis. 9 531–546. [DOI] [PubMed] [Google Scholar]
- 25.Shevchenko A., M. Wilm, O. Vorm, and M. Mann. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68 850–858. [DOI] [PubMed] [Google Scholar]
- 26.Birner-Gruenberger R., H. Susani-Etzerodt, M. Waldhuber, G. Riesenhuber, H. Schmidinger, G. Rechberger, M. Kollroser, J. G. Strauss, A. Lass, R. Zimmermann, et al. 2005. The lipolytic proteome of mouse adipose tissue. Mol. Cell. Proteomics. 4 1710–1717. [DOI] [PubMed] [Google Scholar]
- 27.Carr S., R. Aebersold, M. Baldwin, A. Burlingame, K. Clauser, and A. Nesvizhskii. 2004. The need for guidelines in publication of peptide and protein identification data. Mol. Cell. Proteomics. 3 531–533. [DOI] [PubMed] [Google Scholar]
- 28.Córdova J., J. C. Ryan, B. B. Boonyaratanakornkit, and D. S. Clark. 2008. Esterase activity of bovine serum albumin up to 160°C: a new benchmark for biocatalysis. Enzyme Microb. Technol. 42 278–283. [Google Scholar]
- 29.Minden, J., and A. Waggoner. 2000. Difference gel electrophoresis using matched multiple dyes. 08/425480 United States patent 6127134.
- 30.Tonge R., J. Shaw, B. Middleton, R. Rowlinson, S. Rayner, J. Young, F. Pognan, E. Hawkins, I. Currie, and M. Davison. 2001. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 1 377–396. [DOI] [PubMed] [Google Scholar]
- 31.Hansen J. B., and K. Kristiansen. 2006. Regulatory circuits controlling white versus brown adipocyte differentiation. Biochem. J. 398 153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]