

Abstract
Given the increased prevalence of cardiovascular disease in the world, the search for genetic variations controlling the levels of risk factors associated with the development of the disease continues. Multiple genetic association studies suggest the involvement of procollagen C-proteinase enhancer-2 (PCPE2) in modulating HDL-C levels. Therefore biochemical and mechanistic studies were undertaken to determine whether there might be a basis for a role of PCPE2 in HDL biogenesis. Our studies indicate that PCPE2 accelerates the proteolytic processing of pro-apolipoprotein (apo) AI by enhancing the cleavage of the hexapeptide extension present at the N terminus of apoAI. Surface Plasmon Resonance and immunoprecipitation studies indicate that PCPE2 interacts with BMP-1 and pro-apoAI to form a ternary pro-apoAI/BMP-1/PCPE2 complex. The most favorable interaction among these proteins begins with the association of BMP-1 to pro-apoAI followed by the binding of PCPE2 which further stabilizes the complex. PCPE2 resides, along with apoAI, on the HDL fraction of lipoproteins in human plasma supporting a relationship between HDL and PCPE2. Taken together, the findings from our studies identify a new player in the regulation of apoAI post-translational processing and open a new avenue to the study of mechanisms involved in the regulation of apoAI synthesis, HDL levels, and potentially, cardiovascular disease.
Keywords: apoAI synthesis, BMP-1, cardiovascular disease, genetics, HDL, PCPE2
Dyslipidemia is a major risk factor for premature development of cardiovascular disease. A high ratio of atherogenic lipoproteins (VLDL, LDL, and IDL) to HDL leads to endothelial dysfunction, focal inflammation, and oxidative stress resulting in the initiation and progression of atherosclerosis. Plasma levels of HDL are inversely correlated with the onset of coronary artery disease as attested to by the outcome of several large prospective epidemiological studies (1, 2). The protective effect of HDL is due, at least in part, to the role of this class of lipoproteins in modulating inflammation and mediating cholesterol transport from peripheral cells to sites of reutilization and degradation (3). Apolipoprotein AI (apoAI); the main protein moiety of HDL; drives the formation and speciation of HDL resulting in a very heterogeneous class of particles, particularly with respect to particle size, density, protein content, and surface charge, which determines their functional role and metabolic fate (4). Understanding the processes involved in the synthesis and maturation of HDL species will ultimately provide the knowledge required to understand and appreciate the functional significance of each of the HDL subpopulations and their impact in the development of atherosclerosis.
The importance of HDL-C in disease makes it amenable to candidate-gene and whole-genome association studies (5–9). Because of the accuracy with which HDL-C can be determined and its high heritability, studies with HDL-C often yield positive results with cholesteryl ester transfer protein (CETP) generally found to be the most highly associated gene. Beyond CETP, many other genes, some with well-known functions and some with less-characterized functions, have been identified as associated with HDL-C. Known HDL-C effectors such as ABCA1, LPL, LCAT, LIPC, LIPG, and the apoE/apoC cluster are frequently found along with less characterized genes like GALNT2 and MVK/MMAB (10, 11).
The genetic component of HDL-C levels is not fully explained by known genes, indicating that there are likely many more genes with weaker effects or genes only important in some populations. It is likely that many such genes are hidden among the weaker signals in candidate- and whole-genome studies. With some of these, the genetic effects may never be strong enough to be absolutely convincing, either due to the smaller direct impact on HDL-C or the lack of common, high-effect variants. Thus, further proof that such genes are important in the modulation of HDL-C and disease will require direct functional studies driven by the suggestive genetic data.
One gene examined previously as a candidate gene (5) is procollagen C-proteinase enhancer-2 (originally named PCOLCE2 and now known as PCPE2) that was found to be weakly associated with plasma HDL-C levels. PCPE2, a 52 kDa protein, was initially identified by expressed sequence tag (EST) sequencing (12). The cDNA encodes a 415-amino acid protein that has 43% identity to the type I procollagen C-proteinase enhancer protein (PCPE1) (13). PCPE2, like PCPE1, contains two CUB (complement C1r/C1s, Uegf, Bmp1) domains, which are thought to be important in protein-protein interaction (14) and binding to the COOH-terminal propeptide of type I procollagen leading to the C-terminal processing of fibrillar procollagens by tolloid proteinases such as bone morphogenetic protein-1 (BMP-1) (13). In addition, the recent finding identifying BMP-1 as the key enzyme responsible for the cleavage of pro-apoAI into its mature form (15) raises the intriguing possibility that PCPE2 alone or in combination with BMP-1 participates in the processing of pro-apoAI and therefore influence the biogenesis of HDL. Therefore, we genotyped PCPE2 single-nucleotide polymorphisms (SNPs) in human populations to determine the extent of the association between PCPE2 and levels of HDL-C and undertook a series of experiments to elucidate the molecular mechanism whereby PCPE2 participates in the regulation of apoAI synthesis and, potentially, HDL-C levels. The distribution of PCPE2 among plasma lipoproteins was assessed using human plasma.
MATERIALS AND METHODS
Genetic analysis
Three separate populations were used for genetic analysis. The first population was from the ACCESS trial (16), which included 3916 individuals with cardiovascular disease who participated in a statin efficacy trial. Individuals within the highest and lowest 15% of HDL-C values at the initiation of the trial were chosen for analysis. Clinical and demographic data for genotyped individuals is provided elsewhere (5). The second population was from the TNT trial (17), which included 10,001 individuals with coronary heart disease (CHD) who were followed for five years to determine the effect of atorvastatin on new cardiovascular events. A whole-genome scan was performed on a subset of those individuals (9), and this included several SNPs in and near the PCPE2 gene. Genotyping for three SNPs was also carried out within the complete population from whom DNA was available. Clinical and demographic data for those genotyped are provided elsewhere (9). The third population was selected from the University of California San Francisco (UCSF) Genomic Resource in Arteriosclerosis (GRA). Subjects were identified from the GRA as individuals with HDL-C less than the 10th percentile and above the 90th percentile. Because only the extremes were included in this population, HDL-C was treated as a dichotomous trait in the analysis. In all populations, whole blood from participating subjects was obtained with appropriate institutional review and appropriate informed consent documentation that provided an assessment of the risks and benefits associated with study participation. Except for the whole-genome scan, TaqMan assays were used for genotyping and obtained from ABI (Foster City, CA).
Cloning and expression of PCPE2 cDNA
A full-length human PCPE2 cDNA was cloned by reverse transcription PCR using human adipose tissue poly A+ mRNA (Clontech, Mountain View, CA) and specific 5′ and 3′ terminal primers to the published PCPE2 cDNA sequence (Gene Bank accession number AF098269). The PCR product was digested using NheI and KpnI restriction endonucleases and cloned into a pcDNA5/FRT/V5-His TOPO vector (Invitrogen, Carlsbad, CA) following the conditions described by the manufacturer. The orientation of the cDNA insert was determined by restriction digestion mapping. Both strands were sequenced to ensure an error-free clone. Plasmids containing the full-length human PCPE2 cDNA were transfected into CHO cells using Lipofectamine according to the manufacturer's conditions. CHO cells stably expressing PCPE2 (CHO-PCPE2) were selected using DMEM/F12 medium supplemented with 10% FBS and 400 μg/ml Hygromycin B. Desired clones were isolated, grown, and maintained using DMEM/F12 supplemented with 10% FBS and 200 μg/ml of Hygromycin B (Invitrogen). The presence of PCPE2 mRNA in CHO-PCPE2 cell lysates was determined by using a QuantiGene 2.0 bDNA assay (Panomics, Fremont, CA).
Immunofluorescent staining of CHO and CHO PCPE2
Fluorescence microscopy of CHO and CHO-PCPE2 cells was carried out using cells grown to 50–70% confluency in chamber culture slides (BD Biosciences, San Jose, CA) and maintained in DMEM/F12 medium containing 10% FBS. After removing the culture media, cells were washed twice with PBS and then fixed for 5 min in 100% methanol, washed five times with PBS, and blocked for 60 min at room temperature in PBS containing 10% FBS. The blocking solution was removed and an anti V5-FITC conjugated antibody (Invitrogen) in PBS/10% FBS was added overnight at a 1:500 dilution in the dark at 4°C. Finally cells were washed twice with PBS and observed under fluorescent microscopy.
In vitro pro-collagen I cleavage assays
Confluent human neonatal foreskin fibroblasts (Hs68, ATCC CRL 1635), grown in DMEM containing 10% FBS and 1 mM glutamine were used for the preparation of procollagen I. Confluent cells were washed three times with PBS and then incubated for 24 h in serum-free DMEM containing 1 mg/ml Dextran (Mr-500,000) and 30 μM ZnSO4. Cell debris was removed by centrifugation (1000 g for 5 min), and then cell culture supernatants were concentrated ten times using an Amicon Ultra Centrifugal filter (10 kDa molecular weight cut off) (Millipore). Pro-collagen (300 μg/ml) was incubated for 2 h at 37°C in the presence or absence of PCPE2 (150 μg/ml) with increasing concentrations of BMP-1 (0.3 μg/ml to 3.0 μg/ml) in a total reaction volume of 50 μl. Analysis of the collagen cleavage products was studied by SDS-PAGE followed by Western blotting using an anti human pro-collagen I antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
HepG2 culture
Human liver-derived HepG2 cells (ATCC HB 8065), which secrete apoAI constitutively, were grown to near confluence in T150 flasks in the presence of DMEM medium supplemented with 10% FBS. Cells were washed twice with PBS and cultured in serum-free DMEM medium supplemented with 1 mg/ml Dextran (Mr-500,000) overnight at 37°C. Under these culture conditions, more than 85% of the apoAI present in the culture supernatant is in the pro form. Cell culture supernatants were concentrated ten times using Amicon Ultra Centrifugal Filters (10 kDa molecular weight cut off). The pro-apoAI was separated from the mature apoAI in a NuPAGE 12% Bis-Tris gel run for 110 min at 200 v. MES-SDS running buffer (Invitrogen) was used to ensure adequate resolution and separation of pro-apoAI from its mature form. Proteins were transferred onto nitrocellulose membranes (Invitrogen) and both the pro-apoAI and apoAI were visualized following incubation with a 1:1000 dilution of a goat anti-human apoAI antibody (Millipore, Bedford, MA) overnight at 4°C and a donkey anti-goat HRP. The effect of BMP-1 (R and D Systems, Minneapolis, MN) and PCPE2 on the processing of pro-apoAI was studied by incubating aliquots of the culture media containing pro-apoAI with various concentration of BMP-1 (R and D systems) and PCPE2 for 2 h at 37°C followed by gradient gel electrophoresis and immunoblotting.
Isolation of plasma lipoproteins by FPLC
Plasma was isolated from blood drawn from healthy human volunteers and spun at 3000 rpm for 15 min at 4°C. Individual lipoprotein subclasses were isolated from 500 μl plasma using two tandem Superose 6 columns (Pharmacia LKB Biotechnology, Piscataway, NJ). Cholesterol was determined using enzymatic colorimetric assays (Wako Biochemical, Osaka, Japan). ApoAI was determined by turbidimetry (Roche, Indianapolis, IN) using the Hitachi 912 Clinical Analyzer (Roche).
Immunoprecipitation of PCPE2, BMP-1, and apoAI
PCPE2 was immunoprecipitated by incubating samples containing PCPE2 with a 1:100 dilution of a commercially available antibody to human PCPE2 (Sigma, Saint Louis, MI) bound to protein G beads. The immunoprecipitation was achieved by incubating with anti-PCPE2 antibody bound to protein G beads in a rotating shaker at 4°C overnight. Immunoprecipates were pelleted by centrifugation and then washed four times with a solution containing 50 mM Tris pH 7.5, 150 mM NaCl containing 0.1% Tween 20 (TBS-TW 20). Loading buffer was added to the precipitates, heated at 80°C for 10 min, and then run on a NuPAGE 4–12% SDS-gradient gel using a MOPS-SDS running buffer (Invitrogen) at 200 v for 60 min. PCPE2 was transferred to a nitrocellulose membrane and probed with an anti-human PCPE2 antibody and a goat anti-rabbit HRP as a secondary antibody. Lumi light plus western blotting substrate was used for signal detection. The association between apoAI and either BMP-1 or PCPE2 was examined by immunoprecipitation. In these experiments, 1 μg of apoAI was incubated overnight with a 1:100 dilution of a goat anti-human apoAI antibody bound to protein G beads. Beads containing apoAI bound to its antibody were washed with a solution containing TBS-TW20 and then incubated overnight at 4°C in PBS containing either 1 μg of BMP-1 or PCPE2. Anti-human PCPE2 or goat anti-human BMP-1 polyclonal antibodies were used to determine PCPE2 or BMP-1 association to pro-apoAI. The association of BMP-1 to either apoAI or PCPE2 was studied using similar conditions to those described for apoAI and PCPE2. In these experiments, beads containing BMP-1 bound to goat anti-human BMP-1 polyclonal antibodies were incubated with either apoAI or PCPE2 as described above. The association between PCPE-2, BMP-1, and apoAI was examined using similar conditions to those described above, followed by electrophoresis and Western blotting using an antibody to the V5 tag in PCPE2.
Surface Plasmon Resonance
Surface Plasmon Resonance was used to study the interactions between apoAI, BMP-1, and PCPE2 on a BIAcore 2000 instrument (BIAcore 2000, Upsala, Sweden). Molecular interactions were studied on a CM5 sensor chip. Changes in refractive indices between two media, glass and carboxymethylated dextran, caused by the interaction of molecules to the dextran side of the sensor chip were measured and reported as changes in arbitrary reflectance units (RU) as detailed in the manufacturer's application notes. All reagents used were purchased from BIAcore Inc., Upsala, Sweden, unless otherwise noted. The carboxymethylated dextran surface of a flow cell on a sensor chip was activated by derivatization with 0.05 M N-hydroxysuccinimide mediated by 0.2 M N-ethyl-N′-(dimethylaminopropyl) carbodiimide for 7 min. Goat anti-human apoAI polyclonal antibody and rabbit anti-human BMP-1 polyclonal antibody at a concentration of 10 μg/ml, in 10 mM Na acetate pH 3.5, was injected into flow cell 2 and 3, respectively, at a rate of 5 μl/min at 20°C and covalently immobilized to the flow cell surface. Deactivation of unreacted N-hydroxysuccinimide esters was performed using 1 M ethanolamine hydrochloride, pH 8.5. Following immobilization, the flow cells were cleaned of any unreacted or poorly bound material with five regeneration injections of 50 mM NaOH (Sigma, St Louis, MI) until a stable baseline is achieved. Flow cell 2, anti-apoAI surface, measured approximately 4630 RUs following surface preparation and flow cell 3, anti-BMP-1 surface, measured approximately 1409 RUs. For flow cell 1, the activated blank surface, 10 mM Na acetate buffer was injected during immobilization in place of antigen.
Purified human apoAI, PCPE2, and BMP-1 were prepared as described above. A 10 μg/ml dilution of each was prepared in running buffer [0.01M HEPES, pH7.4, 0.15M NaCl, 3 mM EDTA, 0.005% polysorbate 20 (v/v)]. The flow rate was set at 5 μl/min, and 25 μl of each sample was injected over the sensor chip with a regeneration injection of 5 μl of 50 mM NaOH between each experimental series. Dissociation time between each injection was 5 min. The data was analyzed using BIAevaluation 3.0 global fit software using a Langmuire 1:1 binding model. Background values as measured on the Activated Blank surface were subtracted from the experimental surface values. Calculation of concentration from RU was achieved by using the conversion recommended by the manufacturer (M = [(RU corrected for background/1000)/mol wt of species] × 8.5).
RESULTS
PCPE2 polymorphisms associate with plasma HDL levels
In a previous screen of 81 candidate genes for association with HDL-C, only CETP was found to be significant after correction for multiple testing (5), but several other genes showed a trend toward an association including PCPE2. This study was carried out by pooling DNA from 666 individuals with extremes of HDL-C from the ACCESS trial (16) and then individually genotyping the most significant SNPs in 832 individuals (5). After individual genotyping, rs16852856 located 5′ to the PCPE2 gene was found to be associated with HDL-C with an uncorrected significance P = 0.0033. To further characterize the potential for a genetic association with HDL-C, results from a whole-genome scan using a distinct population were also surveyed. 1984 individuals from the TNT trial (17) were genotyped for 291,988 SNPs. Again, the only SNP with genome-wide significance for association with HDL-C was found in the CETP gene (16). However, as with the ACCESS study, there was a trend for an association with a PCPE2 SNP, although it was not significant after correction for multiple testing via the very stringent whole-genome criteria. The rs6440116 SNP in PCPE2 (intron 2) was associated with HDL-C with P = 0.0012 prior to correction for multiple testing. A third population was then examined for 24 SNPs in and near PCPE2 to determine whether the previous suggestive results could be confirmed. Over 1500 individuals at the high and low extremes of HDL-C were selected from the University of California at San Francisco (UCSF) Genomic Resource in Arteriosclerosis (GRA). Individuals of European ancestry were used in all studies because of the availability of larger numbers within these cohorts. The positions of the 24 SNPs across 70 kB in and around the PCPE2 gene are shown in Table 1. The linkage disequilibrium (LD) among these SNPs was low with only a few showing r2 greater than 0.8 among all combinations. Again, there were suggestive associations of the PCPE2 genotype with HDL-C but none that would withstand correction for multiple testing. SNPs rs11712967 and rs11713111 in intron 5 were associated with HDL-C (P < 0.04), and these SNPs were in high LD with each other. Three other nearby SNPs, rs11712986, rs9680986, and rs11720678, were not as well associated (P < 0.07). Two of these SNPs, rs11712967 and rs11712986, were also tested in the complete TNT cohort as a continuous trait, but neither showed a significant association with HDL-C, demonstrating that, if PCPE2 really plays a role in HDL-C levels, its genetic effect is not sufficiently strong enough to generate consistently reproducible findings as has been found with CETP. A weak signal in a genetic study does not necessarily mean that a gene is not involved in a particular function but it could point to the fact that there are no genetic variants with strong effects on function or expression. Clearly, the equivocal nature of these results means that additional supportive evidence is required to confirm a functional relationship between PCPE2 and HDL-C.
TABLE 1.
List of SNPs in and around the PCPE2 gene examined
| Name | P | Position | MAF | Location |
|---|---|---|---|---|
| rs1871349 | 49030555 | 0.206 | 3′ | |
| rs13060227 | 49034635 | 0.031 | intron 8 | |
| rs17554211 | 49037595 | 0.125 | exon 7 | |
| rs9846406 | 49037829 | 0.222 | intron 6 | |
| rs13089614 | 49039189 | 0.075 | intron 6 | |
| rs2608069 | 49040793 | 0.098 | intron 6 | |
| rs11712986 | 0.055 | 49042408 | 0.281 | intron 6 |
| rs10513171 | 49046244 | 0.05 | intron 5 | |
| rs9680986 | 0.062 | 49046996 | 0.207 | intron 5 |
| rs6804096 | 49047778 | 0.43 | intron 5 | |
| rs11720678 | 0.070 | 49050992 | 0.218 | intron 5 |
| rs11712967 | 0.037 | 49051501 | 0.233 | intron 5 |
| rs11713111 | 0.035 | 49051736 | 0.218 | intron 5 |
| rs3816690 | 49062176 | 0.054 | intron 3 | |
| rs10935472 | 49073950 | 0.373 | intron 2 | |
| rs920466 | 49075230 | 0.364 | intron 2 | |
| rs7624656 | 49080948 | 0.108 | intron 2 | |
| rs6788089 | 49089101 | 0.133 | intron 2 | |
| rs7637508 | 49094431 | 0.076 | intron 2 | |
| rs2707985 | 49094966 | 0.213 | intron 2 | |
| rs6794287 | 49097812 | 0.33 | intron 2 | |
| rs7639314 | 49098256 | 0.229 | intron 2 | |
| rs2707982 | 49099622 | 0.383 | intron 2 | |
| rs2248811 | 49102088 | 0.22 | intron 1 |
MAF, minor allele frequency; PCPE2, procollagen C-proteinase enhancer-2; SNP, single-nucleotide polymorphism.
PCPE2 shares sequence homology with proteins containing CUB domains
To better understand the potential role of PCPE2 in regulating HDL-C, its protein sequence was aligned against known protein coding sequences. In addition to high homology to its family member PCPE1, homology with PCPE2 was found with several proteins containing CUB domains including the protein encoded by the Cubulin gene, BMP-1, and other members of the astacin family (14). The two CUB domains near the N terminus are the most conserved regions of the protein with the C-terminal NTR domain less well conserved. The CUB domains span exons 3–5, the region where most of the associated SNPs are found, although there is no obvious functional connection to any of the SNPs. Although the role of the CUB domains is largely unexplored, a number of proteins containing these domains have been shown to form multimers and play a role in a wide range of biological functions including complement activation (18), inflammation (19), receptor-mediated endocytosis (20), and tumor suppression (21). Cubulin and Megalin; two proteins rich in CUB domains, have been previously shown to bind apoAI and facilitate the endocytosis of HDL (22) suggesting that PCPE2 might also interact with apoAI and participate in the regulation of HDL-C levels.
Cloning and expression of human PCPE2
To examine the biological importance of PCPE2 in HDL, a full-length human PCPE2 cDNA was cloned and expressed in ChoFlp-in (CHO) cells as described under Materials and Methods. Whereas no detectable levels of PCPE2 mRNA were present in control CHO cells, cells transfected with a full-length PCPE2 cDNA demonstrated the presence of a large number of PCPE2 transcripts (Fig. 1A). The study of the cellular distribution of PCPE2 was greatly facilitated by the use of a V5 tag on the 3′terminus. While minimal signal was observed in untransfected cells (Fig. 1B insert), cells expressing PCPE2 displayed a prominent fluorescence in their cytoplasm, suggesting that PCPE2 could, at least in part, localize within the cytoplasm of CHO cells. PCPE2, unlike PCPE1, contains a RGD (Arg-Gly-Asp) site within residues 149–151 (12) which could mediate binding to intracellular proteins and/or integrins (23). To examine whether PCPE2 is secreted, cell lysates and supernatants from CHO-PCPE2 cells were immunoprecipitated and subjected to SDS-PAGE followed by immunobloting using anti-V5 antibodies. Fig. 1C demonstrates the presence of PCPE2 in cell extracts as well as in culture supernatants. PCPE2 migrates on SDS-polyacrylamide gels under reducing conditions with apparent molecular masses of 48 kDa and 50 kDa. These molecular masses are consistent with those reported in the literature but somewhat greater than the predicted molecular mass based on its amino acid sequence (12). The apparent discrepancy might be explained by the presence of the C-terminal V5-His tag or differences in glycosylation within the PCOLCE2 gene product, but this remains to be explored.
Fig. 1.
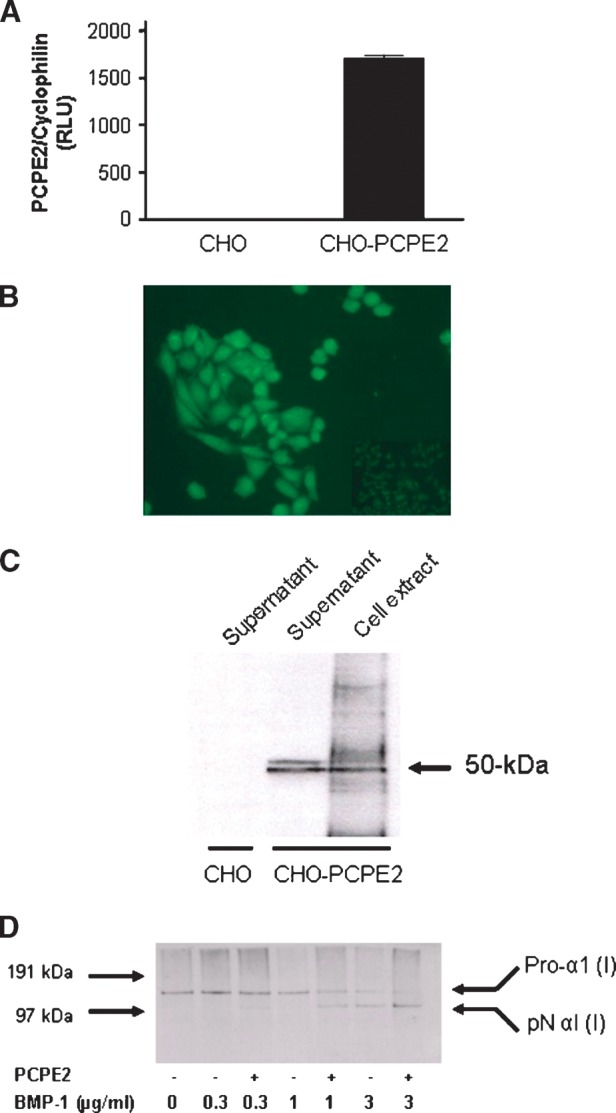
Expression of human PCPE2 in CHO cells. A: CHO and CHO-PCPE2 cells were grown to confluency. PCPE2 mRNA was determined in cell lysates as described under Materials and Methods. B: Fluorescence microscopy of CHO and CHO-PCPE2 cells was carried out in cells grown to 50–70% confluency in chamber culture slides maintained in DMEM/F12 medium. CHO cells are shown as an insert. C: CHO and CHO-PCPE2 cells were grown in DMEM/F12 medium as described under Materials and Methods. Cell lysates and cell culture supernatants were fractionated by electrophoresis and visualized by Western blotting using an antibody against the V5 tag. The molecular mass (in kDa) of PCPE2 is indicated. D: In vitro processing of pro-collagen by BMP-1 and PCPE2. Confluent human neonatal foreskin fibroblasts grown in DMEM containing 10% FBS and 1 mM glutamine were used for the preparation of pro-collagen I. Pro-collagen (300 μg/ml) was incubated for 2 h at 37°C in the presence or absence of PCPE2 (150 μg/ml) and increasing concentrations of BMP-1. Analysis of the collagen cleavage products was studied by SDS-PAGE followed by Western blotting using an anti human pro-collagen I antibody.
To determine whether PCPE2 retains the pro-collagen C-proteinase enhancer activity described in the literature (13), an in vitro pro-collagen cleavage assay was developed and optimized. PCPE2 was incubated with procollagen I in the presence or absence of increasing amounts of BMP-1. Conversion of the Pro-α1 (I) to pNα1 (I) was demonstrated by SDS-PAGE followed by immunobloting using an anti-collagen I antibody (Fig. 1D), indicating that the recombinant PCPE2 retains its intrinsic enhancer activity and, therefore, is suitable for testing its biologic activity.
PCPE2 enhances the BMP-1 cleavage and maturation of pro-apoAI
The sequence similarities within the CUB domains of PCPE2, PCPE1, cubulin, and BMP-1 suggest that these proteins share common folding motifs. In addition, the recent finding identifying BMP-1 as the key enzyme responsible for the cleavage of pro-apoAI into its mature form (15) raises the intriguing possibility that PCPE2 alone or in combination with BMP-1 participates in the processing of pro-apoAI and, therefore, influences the biogenesis of HDL. To examine this possibility, pro-apoAI was prepared from HepG2 cell supernatants and incubated at 37°C in the presence of either BMP-1 or PCPE2. Conversion of pro-apoAI to apoAI was determined by 12% SDS-PAGE followed by immunoblotting using a commercially available antibody to apoAI. While BMP-1 fully cleaves pro-apoAI and generates its mature form as previously described (15), no detectable conversion is observed when pro-apoAI is incubated in the presence of PCPE2 (Fig. 2A). This finding indicates that PCPE2 has no ability to induce the conversion of pro-apoAI into its mature apoAI form. However, when BMP-1 is incubated in the presence of PCPE2, the conversion of pro-apoAI to apoAI is greatly increased (Fig. 2B), suggesting that the presence of PCPE2 enhances the ability of BMP-1 to cleave pro-apoAI. This effect appears to depend on the intrinsic PCPE2 enhancer activity and is proportional to the amount of BMP-1 present in the incubation medium (Fig. 2B). The conversion of pro-apoAI into apoAI was increased up to 2-fold (Fig. 2C).
Fig. 2.

Pro-apoAI processing by BMP-1 and PCPE2. A: Pro-apoAI was incubated for 2 h at 37°C in the presence of either BMP-1 or PCPE2 followed by SDS-PAGE and immunoblotting. 20 kDa and 30 kDa molecular weigh standards are shown in lane 4. B: PCPE2 enhancing of BMP-1 activity was shown by incubating pro-apoAI in the presence or absence of PCPE2 and increasing concentrations of BMP-1 for 2 h at 37°C. C: The ability of PCPE2 to enhance the processing of pro-apoAI by BMP-1 was assessed by determining the amount of apoAI formed following pro-apoAI incubation with either BMP-1 alone or in combination with PCPE2. ApoAI levels were determined by scanning the gels using a Lumi-Imager F1 Workstation. Open squares: pro-apoAI incubated in the absence of PCPE2; Closed squares: pro-apoAI incubated in the presence of PCPE2.
BMP-1, PCPE2, and pro-apoAI interact to form a ternary complex
The interaction among BMP-1, pro-apoAI, and PCPE2 was studied using a BIAcore instrument that relies on Surface Plasmon Resonance to measure molecular interactions on a sensor chip. BMP-1 or pro-apoAI was captured on sensor chips using either anti–BMP-1 or anti-apoAI polyclonal antibodies. The interactions among the various proteins, their stoichiometry, and dissociation rates were determined as described under Materials and Methods. The injection of pro-apoAI over the sensor chip containing BMP-1 led to the formation of BMP-1/pro-apoAI complexes, indicating that the cleavage of pro-apoAI by BMP-1 requires the physical interaction between these two proteins (Fig. 3A). A significant change in the refractive index was observed when PCPE2 was injected over the BMP-1/pro-apoAI complex, indicating that PCPE2 interacts and binds to BMP-1/pro-apoAI complexes to form a ternary pro-apoAI/BMP-1/PCPE2 complex. The relatively slow dissociation off rate of PCPE2 from the BMP-1/pro-apoAI complex (Kd = 7.65 × 10−4 /s) appears to indicate that this ternary structure is quite stable, and its formation begins by the binding of pro-apoAI to BMP-1 followed by the association of PCPE2 to pro-apoAI/BMP-1. Various permutations were examined to determine the sequence of events involved in the molecular interaction among these three proteins. Although PCPE2 binds to BMP-1 (Fig. 3B) with a similar stoichiometry and affinity to that observed for pro-apoAI, the subsequent binding of pro-apoAI to the PCPE2/BMP-1 complex appears to be less favorable as demonstrated by the faster dissociation rate [Kd = 1.58 × 10−3 /s versus 7.65 × 10−4 /s (Fig. 3B)]. Interestingly, PCPE2 interacts with pro-apoAI at a 1:1 stoichiometry and with high affinity (Kd = 1.85 × 10−4 /s). Very little, if any, BMP-1 binds to the pro-apoAI/PCPE2 complex, suggesting that the interaction between PCPE2 and pro-apoAI likely hinders the binding of BMP-1 to the PCPE2/pro-apoAI complex (Fig. 3C). It is plausible that PCPE2 functions as a negative regulator of pro-apoAI processing when tightly bound to pro-apoAI. It is possible that the binding of pro-apoAI to its antibody on the sensor chip blocks a structural domain within the pro-apoAI that is required for its interaction to BMP-1, although this seems unlikely given that a direct association between BMP-1 and pro-apoAI can be demonstrated when BMP-1 is captured to the sensor chip (data not shown). Although no data are available at this time on the structural domains within the apoAI enabling its association to either BMP-1 or PCPE2, immunoprecipation studies using either the pro-apoAI or its mature form suggest that the N-terminal hexapeptide present in the pro form of apoAI does not signficantly influence binding to either BMP-1 or PCPE2 (data not shown). Association among BMP-1, PCPE2, and pro-apoAI was also demonstrated by immunoprecipitation as shown in Fig. 4, further supporting the physiological relevance of these interactions and findings from the BIAcore experiments.
Fig. 3.

Interaction among BMP-1, PCPE2 and pro-apoAI. The interaction among BMP-1, PCPE2 and pro-apoAI was studied by Surface Plasmon Resonance as described under Materials and Methods. A: BMP-1 was captured to sensor chips using a BMP-1antibody followed by pro-apoAI injection, BMP-1/pro-apoAI complex formation, and subsequent PCPE2 injection. B: PCPE2 was injected over captured BMP-1 followed by injection of pro-apoAI. C: PCPE2/pro-apoAI association was measured by injecting PCPE2 over pro-apoAI captured to its antibody. BMP-1 was then injected and its binding to the PCPE2/pro-apoAI complex was measured. Dissociation rates were measured at 10 s and 20 s intervals (open and solid bar graphs, respectively). Binding is expressed as μM and is shown above each bar graph. Dissociation constants (Kd) for each of the complexes are shown above each figure.
Fig. 4.

Immunoprecipitation of BMP-1/pro-apoAI/PCPE2 complexes. Association between BMP-1, pro-apoAI, and PCPE2 was demonstrated by immunoprecipitation. Immunoprecipitates were run in a NuPAGE 4–12% SDS gel, transferred to nitrocellulose membranes, and immunobloted using an anti-V5 antibody as described under Materials and Methods. 1: Beads containing anti-apoAI antibody were incubated in the presence of BMP-1, washed, and then incubated with PCPE2. 2: Beads containing pro-apoAI bound to its antibody were incubated with PCPE2, washed, and then incubated with BMP-1. 3: Beads containing pro-apoAI bound to its antibody were incubated with BMP-1, washed, and then incubated with PCPE2. 4: Beads containing BMP-1 bound to its antibody were incubated with pro-apoAI, washed, and then incubated with PCPE2. 5: Recombinant PCPE2.
PCPE2 is present in human plasma associated with HDL
To examine the presence and distribution of PCPE2 in human plasma, blood was collected from fasted, normolipidemic individuals. Plasma was fractionated by fast-protein liquid chromatography (FPLC), and individual lipoproteins were identified by measuring the amount of cholesterol and apoAI in the individual fractions. Aliquots from each of the fractions were immunoprecipitated using an anti-PCPE2 antibody and subsequently separated using SDS-PAGE as described under Materials and Methods. As shown in Fig. 5, PCPE2 coelutes with HDL in normolipemic, fasted human plasma. Since the fractionation of plasma by FPLC does not lead to the isolation of pure lipoprotein fractions but rather a mix of lipoproteins and unassociated proteins, a further experiment was conducted to provide additional, supporting evidence for the presence of PCPE2 on HDL. FPLC fractions containing HDL were incubated with anti-human apoAI antibody to immunoprecipitate HDL. HDL proteins were then separated by SDS-PAGE electrophoresis, transferred to a nitrocellulose membrane and probed with an anti-human PCPE2 antibody as described under Materials and Methods. Under these experimental conditions, PCPE2 was present in all HDL fractions, further demonstrating that PCPE2 is present and resides on HDL in plasma from fasted and normolipidemic humans (data not shown). Additional studies will be required to assess whether under various conditions and metabolic settings, PCPE2 remains in HDL or is also distributed among VLDL and LDL fractions.
Fig. 5.

Distribution of PCPE2 in human plasma. Human plasma was fractionated using Superose 6 columns as described under Materials and Methods. Cholesterol and apoAI were determined and plotted as a function of column fraction. The position at which known lipoproteins eluted from the column is indicated. The presence of PCPE2 was determined by immunoprecipitation, SDS-PAGE, and immunoblotting. Molecular weigh standards and recombinant PCPE2 were included to confirm the location of PCPE2. The figure shows a representative elution profile from three independent experiments.
DISCUSSION
ApoAI is the main protein moiety on HDL and a key element in the reverse cholesterol transport pathway. Its biological significance is demonstrated by the inverse relationship observed between plasma HDL levels and atherosclerosis (1, 2). The liver and small intestine are the main sites of HDL synthesis in both mammals and birds (24, 25). Translation of apoAI mRNA has shown that the protein is synthesized as a large precursor containing a 24-amino acid NH2-terminal extension, consisting of a signal sequence (18 amino acids) and a prosegment composed of 6 amino acids: RHFWQQ (26). This hexapeptide is unusual as it ends with paired glutamine residues, suggesting a proteolytic cleavage distinct from that occurring intracellularly on most prosegments. While the signal peptide is released cotranslationally, the hexapeptide is cleaved after secretion (27). The functional significance of the apoAI prosegment is unclear. It has been suggested that its deletion reduces the cotranslational translocation and processing efficiency but does not affect the fidelity of the signal peptidase cleavage (28). This suggests that the hexapeptide helps the nascent preproapoAI to assume an adequate conformation to efficiently translocate through the endoplasmic reticulum membrane and enter the secretory pathway. Although several reports have suggested that a metal-dependent protease is implicated in the removal of the hexapeptide (25, 29, 30), it was not until very recently that Chau, Fielding, and Fielding (15) reignited interest in this area of research by suggesting that BMP-1 is responsible for the cleavage and conversion of pro-apoAI to its mature form.
PCPE2 was initially highlighted as being involved with HDL-C (5). However, the association could not withstand correction for multiple testing. Studies we performed in two additional populations also showed highly suggestive associations, but the signal was not strong enough to be convincing, especially for a gene with no known connections to HDL-C or lipids. It became clear that genetic studies on their own would not answer the question of whether PCPE2 truly played a role in HDL-C modulation and, especially, what that role might be. A weak genetic signal does not necessarily mean a minor regulatory role as many genes have no common variants that generate large effects on function or expression. For example, HMG-CoA Reductase (HMGCR); the target enzyme of the most widely used class of drugs for the treatment of cardiovascular disease; shows weak and sporadic association with LDL-C and statin response (9, 31–33). Nonetheless, biochemical and other data are convincing on the major influence that HMGCR plays on the regulation of LDL-C levels. Thus, in what is likely to become a more common paradigm, biochemical and functional studies on a gene will have to be undertaken to complement the genetic findings to determine whether weak genetic effects have any functional relevance.
PCPE2 was initially identified by EST sequencing in the glaucoma candidate gene region on 3q21-q24 in an effort to identify the gene responsible for primary open-angle glaucoma (12). No coding sequence mutations were detected, and PCPE2 was no longer considered as the primary open-angle glaucoma candidate gene, leaving its biological function unknown. Nevertheless, the striking similarity in the predicted PCPE2 amino acid sequence with those published for the human and mouse PCOLCE1 (34, 35) suggests that PCPE2 participates in protein-protein interactions with other proteins containing CUB domains and may regulate key physiological processes. Interestingly, our studies demonstrate that recombinant PCPE2 made by CHO cells retains its intrinsic enhancing activity and modulates the processing of pro-apoAI by enhancing the activity of BMP-1. We have studied the interaction among pro-apoAI, BMP-1, and PCPE2, demonstrating by immunoprecipitation and Surface Plasmon Resonance that BMP-1, pro-apoAI, and PCPE2 interact with each other and form a ternary pro-apoAI/BMP-1/PCPE2 complex. The most favorable sequence of events begins with the association between pro-apoAI and BMP-1, followed by the binding of PCPE2, which further stabilizes the complex. Although our experimental conditions also show the formation of a BMP-1/PCPE2 complex followed by the subsequent binding of pro-apoAI, the faster dissociation rate between these two proteins appears to indicate the formation of a less stable complex. Moreover, our studies indicate that when PCPE2 associates to pro-apoAI, very little, if any, BMP-1 binds to the pro-apoAI/PCPE2 complex, suggesting that the interaction between PCPE2 and pro-apoAI hinders the subsequent binding of BMP-1. It is tempting to speculate that PCPE2 plays a chaperone-like function in the plasma compartment and thereby regulates the processing of pro-apoAI and HDL synthesis. While its association to BMP-1 will result in the enhancing of pro-apoAI processing and therefore act as an activator, binding of PCPE2 to pro-apoAI could hinder the binding to BMP-1 and, hence, function as a repressor. This potential dual role of PCPE2 might depend on its concentration and will need to be explored and examined in greater detail. The findings from our studies do not reveal, however, if additional proteins with similar CUB domains participate in concert with PCPE2 and BMP-1 in the formation of an optimal protein scaffold that regulates the processing of apoAI.
The physiological relevance of the proteolytic maturation of pro-apoAI is unclear. Pro-apoAI, which represents 4–8% of circulating apoAI (36), is known to be biologically active, and its structure and stability are essentially identical to those for mature apoAI (37, 38). In addition, the lipid binding properties (37), LCAT activation (38), and cholesterol efflux (39) of the pro-apoAI and mature apoAI are indistinguishable in vitro, suggesting that the removal of the prosegment in plasma is not a prerequisite for full expression of the known functions of apoAI. More recently, Chau, Fielding, and Fielding (15) documented that the in vitro cleavage of the pro-apoAI segment accelerated the conversion of a 2.6 nm globular pro-apoAI precursor to its 3.6 nm product. The authors proposed that the removal of the prosegment precedes the conversion of apoAI to its lipid-binding form because the formation of the 3.6 nm product depends on ABCA1. Our studies indicate that PCPE2 is localized in HDL, suggesting that the cleavage of the propeptide occurs after the binding of apoAI to lipids. Additional studies, however, will be required to compare the kinetics of proteolytic cleavage for the lipid-free and lipid-bound pro-apoAI in order to gain a better understanding of the events of apoAI maturation and assembly with lipids in plasma. The role of PCPE2 in the regulation of plasma HDL levels will have to wait for the identification of a loss of function mutation in humans or genetic modification in animal models.
Taken together, the findings from these studies implicate a new player in the regulation of apoAI post translational processing. The increasing availability of large amounts of genetic data for many traits of medical interest is providing a rich source of new potential targets for medical intervention, but many of these genes are poorly understood and the genetic effects may not be strong enough to warrant a drug discovery program. By combining weak genetic association data with sequence conservation information, we were able to guide biochemical experiments to follow a potential functional role for a previously poorly characterized gene with no known role in lipids. Though this work only touches on one aspect of PCPE2's potential roles, our collective information opens a new avenue to the study of mechanisms involved in the regulation of HDL levels and cardiovascular disease and exemplifies what will have to be done in the future to generate practical information from weak genetic data.
Acknowledgments
The authors thank the Fondation Leducq for their assistance in the collection of the GRA cohort used in the study.
Abbreviations
ApoAI, apolipoprotein AI
BMP-1, bone morphogenetic protein-1
CETP, cholesteryl ester transfer protein
CHD, coronary heart disease
CUB, complement C1r/C1s, Uegf, Bmp1
EST, expressed sequence tag
FPLC, fast-protein liquid chromatography
HuAI, transgenic mice expressing human apoAI
PCPE2, procollagen C-proteinase enhancer-2
SNP, single-nucleotide polymorphism
Published, JLR Papers in Press, February 23, 2009.
References
- 1.Robins S. J., D. Collins, J. T. Wittes, V. Papademetriou, P. C. Deedwania, E. J. Schaefer, J. R. McNamara, M. L. Kashyap, J. M. Hershman, L. F. Wexler, et al. VA-HIT Study Group. 2001. Relation of gemfibrozil treatment and lipid levels with major coronary events. VA-HIT: a randomized controlled trial. JAMA. 285 1585–1591. [DOI] [PubMed] [Google Scholar]
- 2.Manninen V., M. O. Elo, M. H. Frick, K. Haapa, O. P. Heinonen, P. Heinsalmi, P. Helo, J. K. Huttunen, P. Kaitaniemi, P. Koskinen, et al. 1988. Lipid alterations and decline in the incidence of coronary heart disease in the Helsinki Heart Study. JAMA. 260 641–651. [PubMed] [Google Scholar]
- 3.Tall A. R., L. Yvan-Charvet, N. Terasaka, T. Pagler, and N. Wang. 2008. ABC transporters and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 7 365–375. [DOI] [PubMed] [Google Scholar]
- 4.Rader D. J. 2006. Molecular regulation of HDL metabolism and function: implications for novel therapies. J. Clin. Invest. 116 3090–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hinds D. A., A. B. Seymour, L. K. Durham, P. Banerjee, D. G. Milos, D. R. Cox, J. F. Thompson, and K. A. Frazer. 2004. Application of pooled genotyping to scan candidate regions for association with HDL cholesterol levels. Hum. Genomics. 1 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, Novartis Institutes of BioMedical Research, R. Saxena, B. F. Voight, V. Lyssenko, N. P. Burtt, P. I. de Bakker, H. Chen, J. J. Roix, et al. 2007. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 316: 1331–1336. [DOI] [PubMed]
- 7.Kooner J. S., J. C. Chambers, C. A. Aguilar-Salinas, D. A. Hinds, C. L. Hyde, G. Warnes, F. J. Gomez-Perez, D. R. Cox, J. Scott, P. M. Milos, et al. 2008. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat. Genet. 40 149–151. [DOI] [PubMed] [Google Scholar]
- 8.The Wellcome Trust Case Control Consortium. 2007. Genome wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447: 661–678. [DOI] [PMC free article] [PubMed]
- 9.Thompson, J. F., C. L. Hyde, L. S. Wood, S. A. Paciga, D. A. Hinds, D. R. Cox, G. K. Hovingh, and J. J. P. Kastelein. 2009. Comprehensive whole genome and candidate gene analysis for response to statin therapy in TNT cohort. Circ. Cardiovasc. Genet. Epub ahead of print. February 12, 2009; doi:10.1161/CIRCGENETICS.108.818062. [DOI] [PubMed]
- 10.Willer C. J., S. Sanna, A. U. Jackson, A. Scuteri, L. L. Bonnycastle, R. Clarke, S. C. Heath, N. J. Timpson, S. S. Najjar, H. M. Stringham, et al. 2008. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 40 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kathiresan S., O. Melander, C. Guiducci, A. Surti, N. P. Burtt, M. J. Rieder, G. M. Cooper, C. Roos, B. F. Voight, A. S. Havulinna, et al. 2008. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 40 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu H., T. A. Acott, and M. K. Wirtz. 2000. Identification and expression of a novel type I procollagen C-proteinase enhancer protein gene from the glaucoma candidate region on 3q21-q24. Genomics. 66 264–273. [DOI] [PubMed] [Google Scholar]
- 13.Steiglitz B. M., D. R. Keene, and D. S. Greenspan. 2002. PCOLCE2 encodes a functional procollagen C-proteinase enhancer (PCPE2) that is a collagen-binding protein differing in distribution of expression and post-translational modification from the previously described PCPE1. J. Biol. Chem. 277 49820–49830. [DOI] [PubMed] [Google Scholar]
- 14.Blanc G., B. Font, D. Eichenberger, C. Moreau, S. Ricard-Blum, D. J. S. Hulmes, and C. Moali. 2007. Insights into how CUB domains can exert specific functions while sharing a common fold. J. Biol. Chem. 282 16924–16933. [DOI] [PubMed] [Google Scholar]
- 15.Chau P., P. E. Fielding, and C. J. Fielding. 2007. Bone morphogenetic protein-1 cleaves human proapolipoprotein A1 and regulates its activation for lipid binding. Biochemistry. 46 8445–8450. [DOI] [PubMed] [Google Scholar]
- 16.Ballantyne C. M., T. C. Andrews, J. A. Hsia, J. H. Kramer, and C. Shear. 2001. Correlation of non-high-density lipoprotein cholesterol with apolipoprotein B: effect of 5 hydroxymethylglutaryl coenzyme A reductase inhibitors on non–high-density lipoprotein cholesterol levels. Am. J. Cardiol. 88 265–269. [DOI] [PubMed] [Google Scholar]
- 17.LaRosa J. C., S. M. Grundy, D. D. Waters, C. Shear, P. Barter, J. C. Fruchart, A. M. Gotto, H. Greten, J. J. Kastelein, J. Shepherd, N. K. Wenger, et al. 2005. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N. Engl. J. Med. 352 1425–1435. [DOI] [PubMed] [Google Scholar]
- 18.Gaboriaud C., N. M. Thielens, L. A. Gregory, V. Rossi, J. C. Fontecilla-Camps, and G. J. Arlaud. 2004. Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends Immunol. 25 368–373. [DOI] [PubMed] [Google Scholar]
- 19.Milner C. M., and A. D. Days. 2003. TSG-6: a multifunctional protein associated with inflammation. J. Cell Sci. 116 1863–1873. [DOI] [PubMed] [Google Scholar]
- 20.Sugiyama T., H. Kumagai, Y. Morikawa, Y. Wada, A. Sugiyama, K. Yasuda, N. Yokoi, S. Tamura, Y. Kojima, T. Nosaka, et al. 2000. A novel low density lipoprotein receptor-related protein mediating cellular uptake of apolipoprotein E-enriched β-VLDL in vitro. Biochemistry. 39 15817–15825. [DOI] [PubMed] [Google Scholar]
- 21.Hooi C. F., C. Blancher, W. Qiu, I. M. Revet, L. H. Williams, M. L. Ciavarella, R. L. Anderson, E. W. Thompson, A. Connor, W. A. Phillips, et al. 2006. ST7-mediated suppression of tumorigenicity of prostate cancer cells is characterized by remodeling of the extracellular matrix. Oncogene. 25 3924–3933. [DOI] [PubMed] [Google Scholar]
- 22.Kozyraki R., J. Fyfe, M. Kristiansen, C. Gerdes, C. Jacobsen, S. Cui, E. I. Christensen, M. Aminoff, A. de la Chapelle, R. Krahe, et al. 1999. The intrinsic factor-vitamin B12 receptor, cubilin, is a high-affinity apolipoprotein A-I receptor facilitating endocytosis of high-density lipoprotein. Nat. Med. 5 656–661. [DOI] [PubMed] [Google Scholar]
- 23.Ruoslahti E. 1996. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 12 697–715. [DOI] [PubMed] [Google Scholar]
- 24.Sliwkowski M. B., and H. G. Windmueller. 1984. Rat liver and small intestine produce proapolipoprotein A-I which is slowly processed to apolipoprotein A-I in the circulation. J. Biol. Chem. 259 6459–6465. [PubMed] [Google Scholar]
- 25.Rajavashisth T. B., P. A. Dawson, D. L. Williams, J. E. Shackelford, H. Lebherz, and A. J. Lusis. 1987. Structure, evolution and regulation of chicken apolipoprotein A-I. J. Biol. Chem. 262 7058–7065. [PubMed] [Google Scholar]
- 26.Zannis V. I., S. K. Karathanasis, H. T. Keutmann, G. Goldberger, and J. L. Breslow. 1983. Intracellular and extracellular processing of human apolipoprotein A-I: secreted apolipoprotein A-I isoprotein2 is a propeptide. Proc. Natl. Acad. Sci. USA. 80 2574–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gordon J. I., H. F. Sims, S. R. Lentz, C. Edelstein, A. M. Scanu, and A. W. Strauss. 1983. Proteolytic processing of human preproapolipoprotein A-I. A proposed defect in the conversion of pro A-I to A-I in Tangier's disease. J. Biol. Chem. 258 4037–4044. [PubMed] [Google Scholar]
- 28.Folz R. J., and J. I. Gordon. 1987. The effects of deleting the propeptide from human preproapolipoprotein A-I on co-translational translocation and signal peptidase processing. J. Biol. Chem. 262 17221–17230. [PubMed] [Google Scholar]
- 29.Edelstein C., J. I. Gordon, K. Toscas, H. F. Sims, A. W. Strauss, and A. M. Scanu. 1983. In vitro conversion of proapoprotein A-I to apoprotein A-I. Partial characterization of an extracellular enzyme activity. J. Biol. Chem. 258 11430–11433. [PubMed] [Google Scholar]
- 30.Kooistra T., V. van Hinsbergh, L. Havekes, and H. J. Kempen. 1984. In vitro studies on origin and site of action of enzyme activity responsible for conversion of human proapoprotein A-I into apoprotein A-I. FEBS Lett. 170 109–113. [DOI] [PubMed] [Google Scholar]
- 31.Chasman D. I., D. Posada, L. Subrahmanyan, N. R. Cook, V. P. Stanton, and P. M. Ridker. 2004. Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA. 291 2821–2827. [DOI] [PubMed] [Google Scholar]
- 32.Thompson J. F., M. Man, K. J. Johnson, L. S. Wood, M. E. Lira, D. B. Lloyd, P. Banerjee, P. M. Milos, S. P. Myrand, J. Paulauskis, et al. 2005. An association study of 43 SNPs in 16 candidate genes with atorstatin response. Pharmacogenomics J. 5 352–358. [DOI] [PubMed] [Google Scholar]
- 33.Krauss R. M., L. M. Mangravite, J. D. Smith, M. W. Medina, D. Wang, X. Guo, M. J. Rieder, J. A. Simon, S. B. Hulley, D. Waters, et al. 2008. Variation in the 3-hydroxyl-3-methylglutaryl coenzyme a reductase gene is associated with racial differences in low-density lipoprotein cholesterol response to simvastatin treatment. Circulation. 117 1537–1544. [DOI] [PubMed] [Google Scholar]
- 34.Lecain E., D. Zelenika, M-C. Laine, T. Rhyner, and B. Pessac. 1991. Isolation of a novel cDNA corresponding to a transcript expressed in thechorid plexus and leptomeninges. J. Neurochem. 56 2133–2138. [DOI] [PubMed] [Google Scholar]
- 35.Takahara K., E. Kessler, L. Biniaminov, M. Brusel, R. L. Eddy, S. Jani-Sait, T. B. Shows, and D. S. Greenspan. 1994. Type I procollagen COOH-terminal proteinase enhancer protein: identification, primary structure, and chromosomal localization of the cognate human gene (PCOLCE). J. Biol. Chem. 269 26280–26285. [PubMed] [Google Scholar]
- 36.Barkia A., C. Martin, P. Puchois, J. C. Gesquiere, C. Cachera, A. Tartar, and J. C. Fruchart. 1988. Enzyme-linked immunosorbent assay for human proapolipoprotein A-I using specific antibodies against synthetic peptide. J. Lipid Res. 29 77–84. [PubMed] [Google Scholar]
- 37.McGuire K. A., W. S. Davidson, and A. Jonas. 1996. High yield overexpression and characterization of human recombinant proapolipoprotein A-I. J. Lipid Res. 37 1519–1528. [PubMed] [Google Scholar]
- 38.Huang W., A. Matsunaga, W. Li, H. Han, A. Hoang, M. Kugi, T. Koga, D. Sviridov, N. Fidge, and J. Sasaki. 2001. Recombinant proapoA-I (Lys107del) shows impaired lipid binding associated with reduced binding to plasma high density lipoprotein. Atherosclerosis. 159 85–91. [DOI] [PubMed] [Google Scholar]
- 39.Okuhira K., M. Tsujita, Y. Yamauchi, S. Abe-Dohmae, K. Kato, T. Handa, and S. Yokoyama. 2004. Potential involvement of dissociated apoA-I in the ABCA1-dependent cellular lipid release by HDL. J. Lipid Res. 45 645–652. [DOI] [PubMed] [Google Scholar]