

Abstract
Apolipoprotein B (apoB) is a nonexchangeable apolipoprotein. During lipoprotein assembly, it recruits phospholipids and triacylglycerols (TAG) into TAG-rich lipoprotein particles. It remains bound to secreted lipoproteins during lipid metabolism in plasma. The β1 region (residues 827–1880) of apoB has a high amphipathic β strand (AβS) content and is proposed to be one region anchoring apoB to lipoproteins. The AβS-rich region between apoB37 and apoB41 (residues 1694–1880) was cloned, expressed, and purified. The interfacial properties were studied at the triolein/water (TO/W) and air/water (A/W) interfaces. ApoB[37–41] is surface-active and adsorbs to the TO/W interface. After adsorption the unbound apoB[37–41] was removed from the aqueous phase. Adsorbed apoB[37–41] did not desorb and could not be forced off by increasing the surface pressure up to 23 mN/m. ApoB[37–41] adsorbed on the TO/W interface was completely elastic when compressed and expanded by ±13% of its area. On an A/W interface, the apoB[37–41] monolayer became solid when compressed to 4 mN/m pressure indicating extended β-sheet formation. It could be reversibly compressed and expanded between low pressure and its collapse pressure (35 mN/m). Our studies confirm that the AβS structure of apoB[37–41] is a lipid-binding motif that can irreversibly anchor apoB to lipoproteins.
Keywords: adsorption, air/water interface, amphipathic β sheet structure, apoB, compressibility, desorption, elasticity, low-density lipoprotein, oil-drop tensiometry, protein monolayer, triolein/water interface
Apolipoprotein B (apoB) is a large protein (4536 residues) that plays an essential role in the formation of triacylglycerol (TAG)-rich lipoproteins by the intestine, as chylomicrons, or by the liver, as VLDL (1). The N-terminal 48% of apoB (apoB48) in the intestine and the full-length apoB (apoB100) in the liver play fundamental roles in the assembly with lipids, including phosphatidylcholine, TAG, and cholesterol, into a nascent emulsion particle which, after further modification, is secreted and ultimately enters the blood. These particles carry dietary (chylomicrons) or liver (VLDL) TAG through the blood to other tissues where they are acted upon by lipoprotein lipase to serve as a source of energy or for cell membrane and lipid droplet synthesis. ApoB is unique among apolipoproteins and a rare member of the family of proteins that bind irreversibly to lipid droplets (1, 2). Only a few other peptides share this irreversible binding, including the oleosins of oil bodies in seeds (3) and perhaps some viral core proteins such as those in hepatitis C virus (4). Oleosins and virus core proteins have a large hydrophobic, proline-rich region that has been suggested to anchor these peptides permanently to their respective emulsion particles. ApoB is the only nonexchangeable apolipoprotein: it remains with the particle from the time it is formed in the liver or intestine until it is removed and catabolized through receptor-mediated cell uptake (1, 2). All remaining plasma apolipoproteins are exchangeable, moving between different lipoproteins as they circulate and are remodeled in the plasma.
ApoB100 has been shown to bind to TAG droplets and to undergo conformational changes in response to changes in surface pressure (5). While the peptide cannot be completely displaced from the triolein/water (TO/W) interface, parts of the peptide appear to desorb as surface pressure is increased and then to readsorb rapidly as the pressure is reduced (5). Previous studies have shown that a 12 amino acid consensus amphipathic β strand (AβS) or a two-strand amphipathic β sheet modeled from B21 to B41 sequence (the first β sheet region of apoB) binds irreversibly to air/water (A/W), hydrocarbon/water (HC/W), and TO/W interfaces, lowering the interfacial free energy (6). Compression to high surface pressure does not displace the peptide from the interface but simply compresses it. These AβS peptides form an almost completely elastic interface at the A/W, HC/W, and TO/W surfaces. From predictions of secondary structure and studies on consensus AβS peptides, it was suggested that the β1 (residues 827–1880) and β2 (residues 2571–4000) domains of apoB100 are the anchors that prevent apoB from leaving the particle once it is assembled (5–8). ApoB48 of course has only the β1 putative anchoring domain.
Based on a consensus of several secondary structure modeling algorithms, the sequence of apoB from B21 to B41 (residues 968–1880) was originally suggested to be a relatively continuous large, amphipathic β sheet roughly 50–60 Å in width and about 200 Å long (8). It is known that the region between B19.5 and B22 can initiate the formation of lipoprotein particles that contain phospholipids and some TAGs in various cell lines (9–16). In C-127 cells containing very low levels of the catalyst microsomal triglyceride transfer protein (MTP), lipoproteins secreted from cells transfected with N-terminal fragments of apoB show an abrupt increase in TAG secretion when the secreted apoB polypeptide is longer than the N-terminal B32. The number of TAG molecules increases from 25 in secreted B32 particles to 113 in B37 particles and to 191 in B41 particles (12). B37 and B41 nascent particles are quasi-spherical with a TAG core clearly observable by cryoelectron microscopy. Thus, this part of the β1 region of apoB (B32–B41) appears to favor the recruitment of TAG, presumably by binding nascent TAG as the sequence translocates through the endoplasmic reticulum. Having shown that small AβS bind irreversibly, we needed to show that an actual sequence from apoB, predicted to be rich in AβS and associated with the secretion of TAG, has properties similar to the small consensus peptides (6). We cloned, expressed, and purified a polypeptide from the β strand–rich region between apoB37 and 41 (amino acids 1694–1880) and then studied its behavior at the oil/water and A/W interfaces. This region contains 10 AβS of 11–15 amino acids (Fig. 1) and several shorter strands (7, 8). The total region is at least 70% β strand. Studies of this sequence show that it is irreversibly bound to the TAG/water interface; it cannot be pushed off by Π up to 24 mN/m; and it is almost completely elastic, therefore a potential anchoring region of apoB to triaclyglycerol-rich lipoprotein. This study provides substantial validity to the previous speculations, derived from small peptide studies, suggesting that AβS regions of apoB serve to anchor apoB48 and apoB100 to their lipoproteins.
Fig. 1.

Extended putative amphipathic β strands (AβS) in the sequence of apoB[37–41] peptide (residue 1694–1880). Residues in boxes are the hydrophobic side chains of AβSs. Several shorter strands are not included.
MATERIALS AND METHODS
Subcloning and expression of apoB1694–1880
A fragment encompassing codons for apoB1694–1880 (the region from B37 to B41, apoB[37–41]) from the apoB48 cDNA (17) was subcloned using primers containing NcoI and HindIII restriction sites for the in-frame insertion of apoB sequence immediately downstream of a His6-Ser-Ser- tag sequence in a modified pET30a plasmid (pET30a_sHT) (pETB37sense: 5′-TCTAAGGCCATGGTCGACAGCAAAAACATTTTC-3′; pETB41anti: 5′-CGGCCGCAAGCTTAGATGGTCATGGTAAACGG-3′). Following amplification of the fragment using Vent DNA polymerase (New England Biolabs, Boston), the amplified fragment was digested with NcoI and HindIII and ligated to pET30a_sHT which had been digested with the same enzymes. Transformed E. coli (BL21) were selected on kanamycin (30 μg/ml) and screened for insert.
Bacteria strain BL21(DE3) was transformed with pETB3741 plasmid, and a single colony was selected and transferred to 3 mls of LB-kanamycin broth and grown at 37°C to A600 of approximately 1.0. The bacteria were collected by centrifugation and transferred to 100 ml of fresh LB-kanamycin broth and grown to A600 of approximately 1.0. The vigorous culture was finally transferred to 900 mls of LB-kanamycin and grown to an A600 of 0.6–0.8. Isopropyl-thioglucoside (IPTG) was added to a final concentration of 0.3 mM, and the incubation was continued for 2.5 h at 37°C. The bacteria were then collected by centrifugation, and the pellet was stored at −80°C.
Purification of apoB1694–1880
To purify the apoB1694–1880 fragment, the bacteria were thawed on ice and suspended in 20 ml of 50 mM sodium phosphate, pH 8.0, 150 mM NaCl containing one-half of a protease inhibitor tablet (Complete®, Roche) and 5 ml of lysozyme solution (4 mg/ml). The suspension was then sonicated (6 × 10 s), and the inclusion bodies containing the apoB protein were collected by centrifugation (30 min, 10000 rpm, SS34 rotor). Inclusion bodies were solubilized in 25 ml of 50 mM sodium phosphate, pH 8.0, 150 mM NaCl, 8 M urea, and protease inhibitors. After solubilization, the solution was mixed with a 50% (v/v) suspension of Ni-NTA His-Bind resin (Novagen) for 1 h. The resin was poured into a disposable chromatography column and washed successively with 50 mM sodium phosphate, 8 M urea at pH 8.0 (10 ml), and then at pH 5.9 (20 ml). ApoB1694–1880 was then eluted from the column with 10 ml of 50 mM sodium phosphate, pH 4.3, 8 M urea and dialyzed against 2 l of 20 mM sodium phosphate, pH 3.0, 50 mM NaCl. For storage and transport, the protein was dialyzed against 5 mM acetic acid and lyophilized. The purification profile is shown in Fig. 2.
Fig. 2.

Purification of histidine-tagged apoB[37–41] from BL21(DE3) inclusion bodies. The left panel is an SDS-PAGE and the right panel is a western blot of purification of his-tagged apoB[37–41]. Aliquots of each fraction were resolved on 10% SDS-PAGE gels and revealed by Coomassie Blue R250 staining or western blot analysis for the apoB epitope 1C4. Lane 1 = Total (non-induced); Lane 2 = Total (IPTG induced); Lane 3 = Sonication supernatant; Lane 4 = Sonication pellet (inclusion bodies); Lane 5 = Urea soluble pellet; Lane 6 = Ni-NTA pH 4.3 eluate.
Circular dichroism and crosslinking studies
Circular dichroism (CD) spectra were collected between 190 and 260 nm on an OLIS DSM CD spectrophotometer at 25°C in 20 mM phosphate buffer containing 50 mM NaCl. In pH experiments, the pH was adjusted by changing the ratios of monobasic and dibasic sodium phosphate and verified by pH measurements. Crosslinking studies were performed on 84 μM samples at low pH after overnight incubation as previously described (18).
Oil-drop tensiometry and interfacial tension (γ) measurement
The interfacial tension of the TO/W interface in the presence of apoB[37–41] peptide in the aqueous phase was measured with an I. T. CONCEPT (Longessaigne, France) Tracker oil-drop tensiometer (19). An aliquot of peptide stock (approximately 0.1–0.6 mg/ml apoB[37–41] dissolved in 2 mM pH 3.0 phosphate buffer) was added to the aqueous phase (2 mM pH 4.85 phosphate buffer) to obtain a peptide concentrations ranging from 2.6 × 10−8 M to 2.2 × 10−7 M. At these concentrations and pH 4.85, apoB[37–41] peptide is soluble. A 16 μl triolein drop was formed in the aqueous phase and the interfacial tension (γ) was recorded continuously until it approached an equilibrium level. The surface pressure Π was obtained from γ of the interface without peptide (γTO) minus γ of the interface with peptide (γpep) (i.e., Π = γTO − γpep). All experiments were carried out at 25 ± 0.1°C in a thermostated system. All the peptide stock concentrations were determined by Lowry protein assay. Triolein (greater than 99% pure) was purchased from NU-CHEK PREP, INC. (Elysian, MN);its interfacial tension against buffer was 32 mN/m. All other reagents were of analytical grade. KCl was heated to 600°C for 6 h to remove all organic contaminants before use.
Buffer exchange procedure
While doing the surface tension measurements, we exchanged the aqueous phase buffer (6 ml) containing the peptide with buffer without peptide by continuously removing the aqueous phase from the surface and infusing new buffer continuously near the bottom of the stirred cuvette. Usually approximately 150 ml buffer was exchanged. This exchange volume removed greater than 99% of the peptide in the aqueous phase. If peptide desorbs into the aqueous phase during or after exchange, then surface concentration will fall and γ will rise.
Instant compression and expansion of the interfaces
Once γ approached an equilibrium level, the oil drop (16 μl) was compressed by rapidly decreasing the volume by as little as 6% (1 μl) or as much as 25% (4 μl). In special cases the volume was decreased by 50% (8 μl). The sudden decrease in drop volume (V) instantaneously decreased the drop surface area (A) and resulted in a sudden compression causing γ to drop abruptly to a certain level, γ0, and generated an instant surface pressure, Π0 = γTO − γ0, where γTO is the surface tension of pure triolein (32 mN/m). The reduced volume was held for several minutes, and γ was continuously recorded. If it rose back toward γeq then ΠMAX, the maximum Π allowed without causing displacement of part of the peptide from the surface was estimated (5). Then the surface was expanded by increasing the drop volume back to the original volume (16 μl) and then held constant for several minutes. This procedure was done both before buffer exchange and after buffer exchange. In either case γ will fall after compression. If peptide molecules readily desorbed from the surface, γ would rise back toward an equilibrium value (the desorption curve). If they do not desorb, γ would remain at a relatively low level. When the compressed surface is re-expanded to the initial area, the change in surface tension may depend on whether there is an aqueous concentration of peptide or whether the peptide has been removed. Before peptide removal, if compression caused desorption of the peptide, then re-expansion will provide space for peptide in the aqueous phase to readsorb to the surface. The kinetics of readsorption will be slow, similar to the kinetics of the original adsorption from the aqueous solution. If the peptide compresses but does not desorb, the peptide should rapidly return to γeq on re-expansion. After the peptide has been removed from the aqueous phase, re-expansion after compression could follow two paths. If peptide has been desorbed, the surface tension will rise to a higher value than the original equilibrium γ and remain constant at the high value, because in the absence of peptide in the aqueous phase no readsorption can occur. If, on the other hand, peptide was not desorbed but merely compressed, the peptide will simply re-expand to conform to the larger area, and surface tension will rise back to the original equilibrium value. A third possible change might occur at high pressure where the peptide is compressed into a small area and partly leaves the surface to refold into a denatured conformation. In this case, the peptide might occupy a smaller area and on re-expansion appear to have been lost or very slowly respread, gradually decreasing γ back toward equilibrium.
Oscillation of the interface and the elasticity analysis
Oscillations were carried out at the equilibrium γ (γe). The drop volume (16 μl) was sinusoidally oscillated at varied amplitudes (6%–25%) and periods (8–128 s) after γ reached an equilibrium level (γe). The area (A) and γ changes were followed as the volume (V) oscillated. In the elasticity analysis, the interfacial elasticity modulus ɛ (ɛ = dγ/d ln A), the phase angle φ between compression and expansion, and the elasticity real part ɛ′ and the elasticity imaginary part ɛ″ were obtained (ɛ′ = |ɛ|cosφ, ɛ″ = |ɛ|sinφ) (20, 21).
Slow compression and expansion of the interface
A 16μl drop was formed in a solution containing 1.3 × 10−7 M of peptide. After reaching γeq (approximately 16 mN/m), the peptide solution was exchanged with peptide-free buffer. The drop was then compressed at a rate of 0.02 μl/s to a certain volume. The compressed volume was held for several minutes and then was re-expanded at a rate of 0.02 μl/s to the original volume. After remaining at 16 μl for several minutes, the compression was repeated but at a larger total compression. Compressions were made at 1, 2, 3, 4, 5, 6, and 8 μl giving surface decreases in area of approximately 5, 8, 13, 17, 22, 27, and 34%. Area, volume, and γ were continuously monitored. After the final compression, the area was re-expanded, and γ was monitored for 20 h.
Langmuir balance studies at the A/W interface
A solution of apoB[37–41] peptide (45 μg/ml) in 30% w/v isopropanol/2 mM phosphate buffer (pH 3.0) was spread slowly (approximately 50 μl/min) on a clean surface of 3.5 M KCl 10 mM pH 7.4 PBS buffer at 25°C according to the techniques of Phillips and Krebs (22). Monolayers of 9 μg apoB[37–41] were compressed at 5 mN/m per minute on a KSV 5000 mini trough of a Langmuir/Pockles surface balance (Helsinki, Finland) and Π-A curves were obtained. To check the reversibility of the Π/A isotherms of the apoB[37–41] monolayer, the monolayer was first compressed to certain Π (15, 30, or 40 mN/m) and then expanded at 5 mN/m per min to reach a Π lower than 1 mN/m, respectively. The state of the monolayer (liquid, condensed viscous, or solid phase) was detected by putting talc powder on the surface, then directing a fine jet of air and directly observing the motion of talc particles (23, 24). In the liquid state, the talc particles move rapidly and freely; in the condensed viscous state, they move slowly; and in the solid state, they are nearly stationary.
RESULTS
Characterization of the apoB[37–41] peptide
The apoB[37–41] fragment was soluble up to 8.4 × 10−5 M at low pH and present in solution primarily as monomers although some aggregates were seen in crosslinking experiments. The pH was raised in steps and the far UV CD spectrum was recorded as a function of pH (Fig. 3A). The loss of solubility appeared to coincide with the loss of the characteristic β secondary structure features between 210 and 217 nm. When the molar ellipticity at 214 nm was plotted against pH (Fig. 3B), the apparent loss of features of β-structure were evident above pH 5.5. This apparent loss secondary structure is due to the loss of protein from solution as precipitation occurs. Therefore, subsequent studies were performed at low concentrations of 2.2 × 10−7 M or lower and at pH of 4.85 where the protein is soluble and probably predominantly monomeric.
Fig. 3.
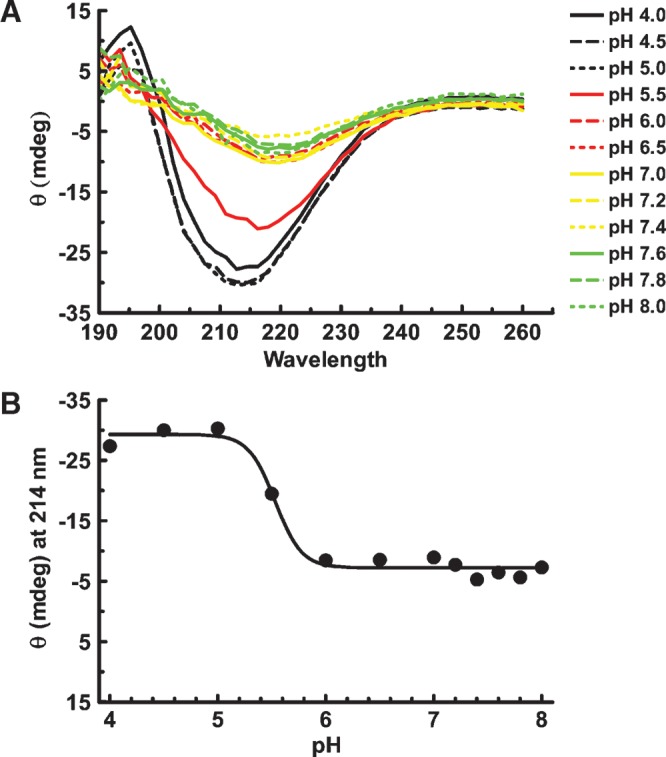
pH titration of soluble apoB[37–41] fragment. A: ApoB[37–41] protein was diluted to 14 μM (0.3 mg/ml) in 20 mM phosphate buffer containing 50 mM NaCl at the indicated pH. After incubation overnight at 4°C, the far UV spectrum was collected on an Olis DSM 17 CD spectrophotometer. Compton calculations of secondary structure contributions were performed at pH 3.0 using the instrument software, revealing 15.6% α-helix, 28.1% β-sheet, 24.6% β-turn, and 34.4% other structure. B: Ellipticity (θ) at 214 nm is plotted as a function of pH. Evidence of apoB structure greatly diminished above pH 5.5 due to precipitation of apoB[37–41] at higher pH.
Adsorption of apoB[37–41] onto the TO/W interface and the compression and expansion of the interface with apoB[37–41] peptide in the aqueous phase before buffer exchange
Concentrations of peptide from 2.6 × 10−8 to 2.2 × 10−7 M in the aqueous phase were studied. The lowest concentrations lowered γ to only 30 mN/m (2.6 × 10−8 M) and 21.3 mN/m (5.2 × 10−8 M). In the latter case, equilibration was carried out for 12,000 s. Sixteen individual experiments were carried out at concentrations from 7.9 × 10−8 to 2.2 × 10−7; the mean γeq was 16.3 ± 1.1 mN/m. There was no significant difference between γeq and concentration in these experiments. Eight experiments were carried out at 8.6 × 10−8 M (γeq= 16.2 ± 0.8 mN/m). Most of the data shown were from these experiments except as noted.
Figure 4A shows a typical adsorption curve (0–7200 s) of apoB[37–41] at TO/W interface and the instant compression and expansion of the interface after γ reached an equilibrium level (after 7200 s). The peptide concentration in the aqueous phase is 8.6 × 10−8 M. In the top figure, the left side (0–7200 s) shows that apoB[37–41] peptide is surface-active and lowered the surface tension (γ) of the TO/W interface (32 mN/m) to reach an equilibrium level (γeq) in about 7200 s. The right side (after 7200 s) shows an example of the tension-time curve of instant compression and re-expansion of the interface with 8.6 × 10−8 M apoB[37–41] peptide in the aqueous phase. After γ approached an equilibrium level, the 16 μl triolein droplet was compressed by decreasing the volume by 1 μl, 2 μl, and 4 μl, respectively. Each instant compression made γ decrease and then remain almost constant at a lower level for several minutes. A slight trend to rise was present with the larger compressions, but the level did not return to equilibrium. The drop was then expanded to 16 μl, and every expansion made the γ increase immediately back or close to the equilibrium value. At 1 μl compression, the γ dropped from γeq to 14.7 mN/m and stayed between 14.7 and 14.8 mN/m while holding that compressed volume. No net change in tension was observed at the compressed volume, suggesting that there was no desorption of the peptide from the surface. When the surface was expanded back to the original volume of 16 μl, γ rapidly returned to the γeq. If peptide had desorbed on compression, re-expansion would have elevated γ above the equilibrium level, followed by a decrease in tension to γeq as the peptide readsorbed. At 2 μl and 4 μl compressions, γ dropped instantaneously from the γeq to 11.6 mN/m and 8.7 mN/m and rose slightly to 12.0 mN/m and 9.2 mN/m, respectively, while holding the compressed volume. On expansion, the drop volume was returned to 16 μl, and γ rapidly returned to 17.6 mN/m and 18.0 mN/m, respectively. These values were slightly higher than equilibrium but relaxed back to the equilibrium γ rapidly. In several similar experiments the changes at 2 μl compression were not always seen but at 4 μl compression, four of five curves showed the higher γ after re-expansion and slow relaxation back to γeq. The results from several experiments suggested that ΠMAX was about 16.0 mN/m. Therefore, once apoB[37–41] adsorbs onto the TO/W interface, it cannot be fully displaced from the interface, but a small fraction of the sequence is desorbed above the Π = 16 mN/m. Other experiments (not shown) showed that large compressions produced a large decrease in γ as low as 4 mN/m (Π = 28) which slowly relaxed over several minutes to about 6 or 7mN/m. When the area was returned to precompression level, γ was much higher (up to 28 mN/m), suggesting that something had been displaced from the surface. During the immediate post-expansion period, there was a sudden small, rapid decrease in γ suggestive of something rapidly snapping back on, followed by a very slow decrease back toward γeq. Because of the confounding variable of an aqueous phase containing protein which might adsorb to open parts of the surface after rapid re-expansion, we decided to circumvent this problem by first adsorbing the peptide, exchanging it, and then looking at larger compression.
Fig. 4.


Examples of the interfacial tension (γ) and the area versus time curves for apoB[37–41] peptide at TO/W interface and the instant compression and re-expansion measurement before and after buffer exchange procedure. A: The instant compression and re-expansion was done after the tension approaching an equilibrium level without buffer exchange. A 16 μl triolein drop was formed in 2 mM pH 4.85 phosphate buffer. The concentration of apoB[37–41] peptide was 8.6 × 10−8 M in the aqueous phase. When γ reached the equilibrium level in about 7200 s, the 16 μl triolein drop was compressed by decreasing the volume by 1, 2, and 4 μl (corresponding to 2–16% changes in area), and then after several minutes re-expanded to the original volume 16 μl, respectively. B: The instant compression and re-expansion was done after the tension approaching an equilibrium level and after the buffer exchange. A 16 μl triolein drop was formed in 2 mM pH 4.85 phosphate buffer. The concentration of apoB[37–41] peptide was 1.3 × 10−7 M in the aqueous phase before buffer exchange. After approaching the equilibrium level, 150 ml buffer was exchanged (shown as the first bar in the figure). The peptide was virtually absent after the exchange. Then the 16 μl triolein drop was compressed by decreasing the volume by 1, 2, 4, and 8 μl (corresponding to 2–33% changes in area) and after several minutes re-expanded to the original volume 16 μl, respectively. Then the tension was lowered back to the equilibrium level by decreasing the volume at a rate of 0.02 μl/s, the second buffer exchange was carried out while the decreased volume was held (shown as the second bar in the figure). Then the volume was increased back to 16ul at a rate of 0.02 μl/s. Two 8 μl compressions and re-expansions were then made. All the measurements were carried out at 25 ± 0.1°C.
Buffer exchange and the compression and expansion of the interface without apoB[37–41] peptide in the aqueous phase
Figure 4B shows an example of the tension time curve for the buffer exchange procedure and the instant compression and expansion of the interface after buffer exchange. A second buffer exchange was carried out, and large compressions were repeated. When γ reached the equilibrium level, the aqueous phase pH 4.85 2 mM phosphate buffer containing 1.3 × 10−7M apoB[37–41] peptide was replaced continuously by pure pH 4.85 2 mM phosphate buffer with approximately 150 ml buffer being exchanged.
Compression at 4 μl shows only a very small quick rise on compression and a very small fall on re-expansion. (The data at 4 μl compression and re-expansion vary from experiment to experiment. Most show a small increase in post-expansion γ, but about 20% of the time γ simply returns to γeq. Part of this is mechanical; the actual starting drop volume differs some and the desired decrease in volume within each experiment is slightly imprecise. Also, as shown later, the 4 μl change is near a critical point for displacement of some peptides on compression.) Compression of 8 μl lowers γ to approximately 4 mN/m (π = 28), and after re-expansion, a rapid snapback is seen which plateaus rapidly to a higher γ (approximately 20 mN/m). Repeated compression of 8 μl produced on decompression an even higher γ with a rapid snapback to a higher plateau value. This indicated that the compression forced something off of the interface, and when the interface was re-expanded, a small fraction of that snaps back on (the rapid fall in γ) but a larger fraction is either lost into the solution or re-expands very slowly. To test for the latter possibility, after the two large compressions to 8 μl, the area was adjusted to return γ to the precompression value of approximately 17 mN/m. Note that the area was reduced from approximately 30 to 28 mm2, showing that ±6% of the originally adsorbed peptide was displaced from the surface by the 8 μl compressions. A second exchange was then carried out with no changes in γ, indicating that the polypeptide did not desorb during this period. Repeat 8 μl compression after this washout produced curves similar to those before the second washout but with a slightly increasing re-expansion of γ to approximately 22 mN/m. This suggests that with each compression of γ to less than 5 mN/m some peptide is displaced from the surface.
The compression and expansion experiments showed similar trends to those before buffer exchange (compare Fig. 4A and B). When the surface was compressed by decreasing the drop volume from 16 μl to about 15 μl, 14 μl and 12 μl (i.e., 2–16% change in area, respectively), the equilibrium γ dropped instantly and showed either no change (1 μl or 2 μl compression) or only a slight change (4 μl compression) while holding the compressed volume. When the surface was expanded to the original volume (16 μl), γ rose quickly to either the equilibrium γ (1 μl or 2 μl expansion) or a slightly higher γ and then relaxed back to the equilibrium (4 μl expansion). Therefore, even without the apoB[37–41] polypeptide in the aqueous phase, the bound apoB[37–41] peptide cannot be fully pushed off the surface by small compressions. The major changes in γ seen following the largest compression expansion are examined in the next sections.
To examine the effect of pH, we carried out experiments at pH7.4. The peptide was allowed to adsorb from pH4.85 and then exchanged at pH4.85 so that precipitation would not occur during change. After a full exchange finished, a second exchange was carried out using pH7.4 buffer to change the pH of the solution to 7.4. The only peptide remaining was that bound to the interface. A series of compressions of 1, 2, 4, and 8 μl were made, and the results are substantially the same as pH4.85 (Fig. 4B). This was repeated and the results were substantially the same (data not shown). These experiments indicate that once the peptide is adsorbed, a change in pH from 4.85 to 7.4 does not change its surface behavior.
Slow compression and re-expansion
We carried out a series of slow compressions where the volume was changed at 0.02 μl/s so that a Π/A curve could be produced (Fig. 5). In these slow compressions, the rapid snapback seen in rapid re-expansion is largely lost in the slow re-expansion phase. Compression/re-expansion cycles were done at volume changes of 1, 2, 3, 4, 5, 6, and 8 μl (Fig. 5A). The percentage decrease in areas of the compressed film is plotted against the peak γ following reexpansion (Fig. 5B). This showed that there was little change in the post-expansion γ when compressions of up to and including 3 μl were used. However, a small change occurred at 4 μl, and proportionally larger changes occurred at each higher compression. This plot shows that there is a threshold for the displacement of protein from the surface at between 3 and 4 μl (or around 15% reduction in area). The threshold Π necessary to produce this displacement is about 23 mN/m corresponding to γ of approximately 9 mN/m (Fig. 5A).
Fig. 5.


Slow compression and expansion procedure after buffer exchange. A: The tension and area versus time curves for the slower compression and expansion following the adsorption and the buffer exchange. A 16 μl triolein drop was formed in pH 4.85 2 mM phosphate buffer. The concentration of apoB[37–41] in the aqueous phase was 1.3 × 10−7 M before buffer exchange. 150 ml phosphate buffer was exchanged (shown as the bar in the figure). Then the drop was compressed by decreasing the drop volume by −1, −2, −3, −4, −5, −6, and −8 μl at a rate of 0.02 μl/s to different level. After the compressed volume was held for several minutes, the drop was re-expanded by increasing the drop volume to the original 16 μl at the same rate respectively. Then the tension was followed over 20 h period (shown as the break in the figure) while the tension dropped slowly from 23 mN/m to 19.7 mN/m. B: The plot of the instant tension produced after the slow re-expansion versus the percentage decrease in area of each corresponding slow compression derived from curves in the left panel.
The slow ramping experiment generates Π/A curves indicated in Figs. 6A and 6B. The Π/A curves for ΔV of 1, 2, 3, and 4 μl fall on virtually the same line with no significant hysteresis on compression and expansion (Fig. 6A). However, compression from 5, 6, or 8 μl (Fig. 6B) shows significant hysteresis with the area at a given Π on compression being greater than that on reexpansion. Further, it showed that the greater the compression, the greater the decrease in area on reexpansion. The slope is fairly constant up to a Π of approximately 20 mN/m, then becomes progressively less. This indicates that compressibility increases when the peptide begins to be displaced from the surface. In other experiments (not shown) to test if high pressure would allow peptide to “wash off” of the surface, the area was decreased to reach a surface pressure of about 27 mN/m and then a buffer exchange was carried out. The γ stayed absolutely constant during the exchange period, indicating that the peptide did not leave the surface even at this high pressure. When the area was returned to the normal level, the γ increased to the precompression level, indicating that the surface was relatively stable in its compressed state.
Fig. 6.
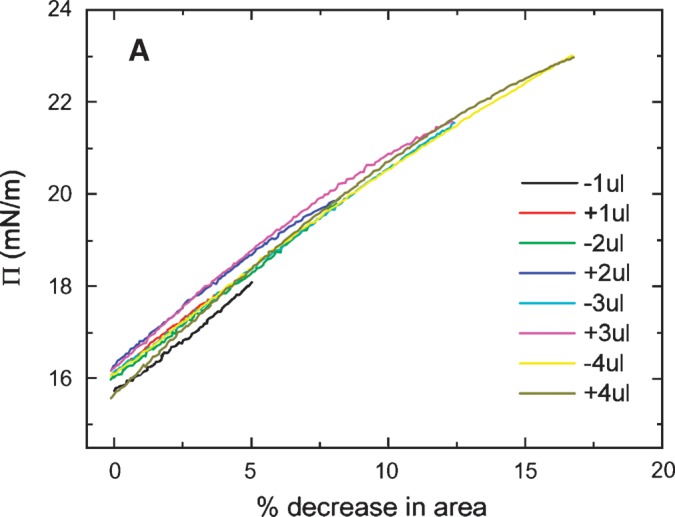
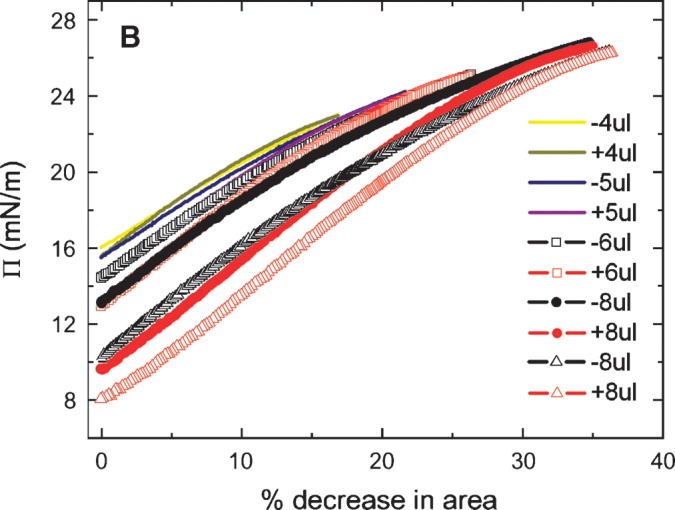
Π versus the % change in area plots of slow compression and expansion derived from the measurement in Fig. 5 (left). A: Plots derived from 1, 2, 3 and 4 μl compression and expansions. Little hysteresis was shown in the plots indicating an elastic behavior. B: Plots derived from 4, 5, 6, and 8 μl compression and expansion. Plots of compression and expansion greater than 4 μl showed hysteresis indicating loss of protein at the interface.
To assess the state of the peptide at the interface after large compression, the surface was reexpanded to 30 mm2, and γ was followed for 20 h to see if there was a continued fall of γ with time (Fig. 5A, right). Over the 20 h equilibration period, γ gradually fell from approximately 23 to 19.7 mN/m, suggesting that the compressed peptide that was partly displaced from the surface very gradually respreads onto the surface. We suspect that the highly compressed peptide forms some multilayered or denatured conformation that with adequate time (days) might reform the original monolayer present at γeq.
Oscillations and elasticity analysis of apoB[37–41] before and after buffer exchange
Equilibrium oscillation experiments of apoB[37–41] at different amplitudes and periods were carried out before and after buffer exchange (Figs. 7A, 7B). Figure. 7A shows a set of the Π-A curves of apoB[37–41] peptide which were derived from the oscillations at 16 ± 2 μl and 16 ± 4 μl with periods from 8 to 128 s before buffer exchange. The concentration of apoB[37–41] in the aqueous phase was 8.6 × 10−8 M. The elasticity analysis data are listed in Table 1. At every condition we have studied, the Π-A curve shows little hysteresis and the phase angle is insignificantly small (less than 3°) indicating a pure elastic surface. Thus, there is no evidence of exchange of the apoB[37–41] peptide molecule between the surface and the bulk solution during oscillation even though the Π approaches 24 mN/m. This is further evidence that the apoB[37–41] polypeptide does not desorb once bound to the TO/W interface. In comparison with other peptides (the consensus sequence peptide of apoB: P12, P27, and the consensus peptide of apoA-I: CSP, the N- and C-terminal peptides of apoA-I: [1–44]apoA-I and [198-243]apoA-I) and proteins (apoA-1 and apoB) in our previous studies (5, 6, 25, 27), apoB[37–41] shows a intermediate elastic modulus ranging from 57-80 mN/m (Table 1).
Fig. 7.
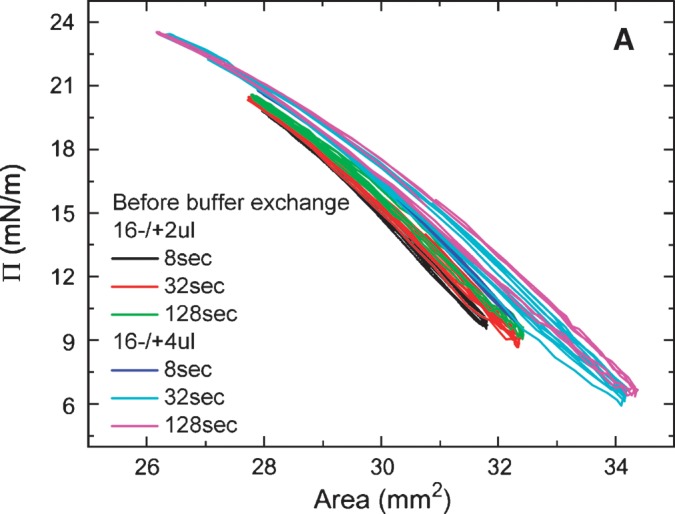
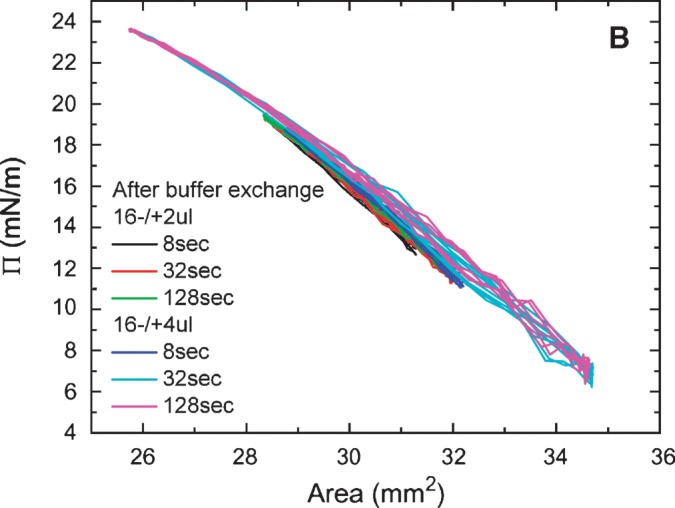
Surface pressure (Π) versus area plots of apoB[37–41] peptide at the TO/W interface derived from the oscillations without (A) and with (B) buffer exchange. A: Without buffer exchange and at equilibrium tension (γeq), a 16 μl triolein drop was oscillated at 16 ± 2 μL, 16 ± 4 μL, and different periods from 8 to 128 s, respectively. The concentration of apoB[37–41] in aqueous phase was 8.6 × 10−8 M. B: After buffer exchange and at the equilibrium tension (γeq), a 16 μl triolein drop was oscillated at 16 ± 2μL, 16 ± 4μL, and different periods from 8 to 128 s, respectively. ApoB[37–41] was 8.6 × 10−8 M in the aqueous phase before buffer exchange and was virtually absent after buffer exchange.
TABLE 1.
Dynamic interfacial properties of apoB[37–41] at TO/W interface
| V+ΔV (μl) (design) | V+ΔV((μl) (actual) | A+ΔA (mm2) | %change in A | Period (s) | Mean γ (mN/m) | ɛ (mN/m) | φ (°) | ɛ′ (mN/m) | ɛ″ (mN/m) | |
|---|---|---|---|---|---|---|---|---|---|---|
| (−) | 16 ± 2 | 16.1 ± 1.6 | 29.9 ± 1.9 | ±6.4 | 8 | 17.1 | 79.6 | 1.6 | 79.5 | 2.2 |
| 16.3 ± 1.9 | 30.0 ± 2.3 | ±7.7 | 32 | 17.3 | 76.0 | 2.2 | 76.0 | 2.9 | ||
| 16.3 ± 1.9 | 30.1 ± 2.3 | ±7.6 | 128 | 17.0 | 73.9 | 2.6 | 73.8 | 3.4 | ||
| 16 ± 4 | 16.3 ± 1.8a | 30.1 ± 2.2 | ±7.3 | 8 | 16.5 | 75.5 | 0.5 | 75.5 | 0.7 | |
| 16.4 ± 3.2 | 30.3 ± 3.9 | ±12.9 | 32 | 16.5 | 66.3 | 2.8 | 66.3 | 3.2 | ||
| 16.4 ± 3.4 | 30.3 ± 4.0 | ±13.2 | 128 | 16.4 | 63.3 | 2.9 | 63.2 | 3.2 | ||
| (+) | 16 ± 2 | 16.3 ± 1.2 | 29.9 ± 1.4 | ±4.7 | 8 | 16.0 | 65.3 | −0.7 | 65.3 | −0.8 |
| 16.5 ± 1.5 | 30.2 ± 1.8 | ±6.0 | 32 | 16.4 | 65.2 | 0.8 | 65.2 | 0.9 | ||
| 16.5 ± 1.5 | 30.2 ± 1.8 | ±6.0 | 128 | 16.4 | 64.1 | 0.4 | 64.1 | 0.4 | ||
| 16 ± 4 | 16.8 ± 1.4a | 30.5 ± 1.7 | ±5.6 | 8 | 16.7 | 66.4 | −0.2 | 66.4 | −0.2 | |
| 16.7 ± 3.7 | 30.3 ± 4.4 | ±14.5 | 32 | 16.4 | 57.8 | 1.2 | 57.8 | 1.2 | ||
| 16.6 ± 3.7 | 30.2 ± 4.4 | ±14.6 | 128 | 16.2 | 56.6 | 1.5 | 56.6 | 1.4 |
All the oscillations were carried out in pH 4.85 phosphate buffer (2 mM) at 25 ± 0.1°C before (−) or after (+) buffer exchange. The concentration of apoB[37–41] in the aqueous phase before buffer exchange was 8.6 × 10−8 M. V, initial drop volume; ΔV, oscillation amplitude; Mean γ, mean interfacial tension of near equilibrium oscillation; ɛ, viscoelastic modulus; φ, viscous phase angle, a phase difference between dγ and dA; ɛ′, elastic component, the real part of modulus; ɛ″, viscous elastic component, the imaginary part of modulus.
Although the apparatus was set to oscillate at 16 ± 4μl for 8 s, the machine delivered less than half the desired amplitude. Thus, these data are like the 16 ± 2μl for 8 s. Significant differences were found between before (−) and after (+) buffer exchange values for ɛ, φ, and ɛ′ by one-way ANOVA.
After the buffer exchange, equilibrium oscillations of the apoB[37–41] TO/W interface were carried out at varied amplitudes and periods. Figure. 7B shows a set of the Π-A curves of apoB[37–41] TO/W interface, which were derived from oscillations at 16 ± 2μl and 16 ± 4 μl with periods from 8 to 128 s There was no apoB[37–41] in the aqueous phase. The elasticity analysis data are listed in Table 1. The Π-A curves at different oscillation frequencies show almost linear curves with little hysteresis and almost no phase angle, indicating a pure elastic surface and quite similar to those in Fig. 7A. However, ɛ and ɛ′ are significantly higher prior to exchange, suggesting that the peptide in the aqueous phase interacts in some way with the surface to modestly increase ɛ′.
ApoB[37–41] monolayer at the A/W interface and compression reversibility
Figure 8 shows (A) the pressure/area (Π-A) isotherms and (B) the reversibility when compressed to different pressures of apoB[37–41] at the A/W interface. In these experiments 9 μg apoB[37–41] peptide (0.045 mg/ml) was spread on a high-salt buffer (3.5 M KCl 10 mM pH 7.4 phosphate buffer) subphase. The 0.045 mg/ml peptide stock was prepared in 30% w/v isopropanol/2 mM pH 3.0 phosphate buffer solutions to help spreading of the peptide on the surface. Talc powder was added on the surface to check the fluidity of the monolayer by direct observation. In Fig. 8A, the Π-A curve lifts off at approximately 4000 Å2 per apoB[37–41] molecule (approximately 21.4 Å2/amino acid), which suggests that apoB[37–41] is flat on the surface. Extrapolation of the steep part of the Π-A curve to the baseline gives a limiting area of approximately 16 Å2/amino acid. The curve then rises steeply and breaks at 35 mN/m (collapse pressure) where apoB[37–41] molecule has an area of approximately 1830 Å2 (approximately 9.8 Å2/aa). The monolayer of apoB[37–41] peptide was liquid (talc can move freely on the surface) when Π was less than 1 mN/m; the monolayer was condensed and viscous (talc can move slowly but the surface became stiff) when Π was between 1 and 4 mN/m; and the monolayer was solid when Π was greater than 4 mN/m.
Fig. 8.


The pressure/area (Π-A) isotherm (A) and the up and down experiments (B) of apoB[37–41] at the A/W interface. ApoB[37–41] peptide (9 μg) in 30% (wt/vol) isopropanol/phosphate buffer at pH 3.0 was spread on a 3.5 M KCl 10 mM pH 7.4 phosphate subphase at 25°C. A: The monolayer was compressed at 5 mN/m per min to produce the Π-A isotherm. Lift-off occurs at 4000 Å2/molecule (21.4 Å2/amino acid) and collapses at 35 mN/m (9.8 Å2/amino acid). The physical state of the surface as a function of Π is indicated. B: The monolayer was compressed and expanded at 5 mN/m per min, first from 0 to 15 mN/m (solid curve), then from 0 to 30 mN/m (dash curve), and finally from 0 to 40 mN/m (dot curve), respectively. The curve compressed to 40 mN/m (above the collapse Π of 35 mN/m), showing that the expansion curve has a smaller area than the compression curve indicating nonreversibility.
The reversibility following compression was studied by re-expanding from 15 mN/m, 30 mN/m, and 40 mN/m, respectively (Fig. 8B). When Π rose from 0 to 15 mN/m and then dropped back to the baseline (solid curve), the up and down Π-A curves were overlapping with each other, indicating the monolayer was reversible at this range, even though the monolayer was already solid above 4 mN/m. When Π rose from 0 to 30 mN/m and then dropped back to the baseline (dashed curve), the up and down Π-A curves were almost identical, indicating the monolayer was reversible in this range as well. However, when Π rose from 0 to 40 mN/m (a Π greater than the collapse pressure of 35 mN/m) and then dropped back to the baseline (dotted curve), the up and down Π-A curves were not similar, indicating the monolayer was not reversible beyond the collapse point. The area of apoB[37–41] on re-expansion from 40 mN/m was always less than that of the original compression. ApoB[37–41] peptide consists of a unique amphipathic β-sheet structure, which can be tightly bound into a solid monolayer but still reversibly compressed and expanded below the collapse pressure. Thus, the amphipathic β-sheet structure is beautifully engineered to serve as the fundamental building block for the nonexchangeable lipid binding motif of apoB.
DISCUSSION
ApoB is a unique nonexchangeable apolipoprotein. The two major characteristics of apoB are strong lipid binding and great conformational flexibility. The former allows apoB to effectively recruit lipids to form stable nascent TAG-rich lipoprotein particles and to be anchored during lipid metabolism (1, 2) from nascent VLDL to LDL. The latter allows the conformational changes of anchored apoB on lipoprotein particles of different sizes and compositions, which promote multiple functions including binding to antibodies, enzymes, receptors, and oxidation products (28–31).
ApoB is rich in AβS and amphipathic α helix (AαH) and can be divided into five distinct super domains: NH2-βα1-β1-α2-β2-α3-COOH (7, 32, 33). The first domain (βα1), from the N-terminal up to apoB20, is an α-helix and β structure mixed region with the homology to MTP and lipovitellin (34–37). This domain can be secreted with little lipid (9, 10) and also as an “HDL” density particle in McA-RH7777 cells (38). In cells transfected with MTP, constructs as short as B19.5 can be secreted with lipid. In vitro, the first 20% of apoB has variable affinity for lipids. ApoB17 was shown to bind to phospholipids (39), phospholipid-TAG emulsions (40), and TAG drops (41). ApoB 19 and 20.1 also bind to TAG drops (42). ApoB 5.9, corresponding to the N-terminal β barrel, does not bind to phospholipids; however, fragments of apoB such as apoB[6–17], [6–13], etc. bind to phospholipids with different kinetics (43–45). The complex region of apoB[6–17] also binds to TAG drops in a complicated fashion (46). Thus the N-terminal 20% of apoB contains many subdomains with phospholipid and/or TAG binding regions which are folded during translocation in the endoplasmic reticulum in ways which allow it to be secreted with only a few molecules of lipid or as a more lipidated particle (38), especially if MTP is highly expressed (13). The second domain (β1) between apoB20 and apoB41 is made up mainly of amphipathic β sheet structure and plays an essential role in recruiting lipids to form TAG-rich nascent lipoprotein particles (9–16). The region from apoB32 to apoB41 has a marked ability to recruit TAG to nascent lipoproteins (12). The third domain (α2) from apoB42 to apoB55 and the fifth domain (α3) from apoB90 to the C-terminal of apoB are rich in AαH, and many of these resemble the AαH of exchangeable apolipoproteins (7). The fourth domain (β2) from apoB55 to apoB90 is rich in AβS, but contains three predicted AαH around the LDL receptor binding site (7). All but domain one bind strongly to LDL based on studies using proteases (47).
It has been suggested that AβS and AαH structure are responsible for the lipid binding properties of apoB and, particularly, AβS may anchor apoB on lipid surfaces (5–7). Small and Atkinson predicted that the region between apoB-21 and apoB-41 contained at least 41 AβS of 11 residues or longer, and their direct binding to TAG was energetically favored (8). These predictions are consistent with the model of LDL presented by Segrest et al. (7).
We have looked at the surface properties of two consensus AβS peptides P12 and P27 (6) modeling the AβS and sheet structure between apoB21 and apoB41. P12 has one consensus β strand sequence of 12 amino acids and P27, a dimer of P12 linked with a β turn (-NGN-) intended to produce a two amphipathic β-strand sheet structure. Both of the AβS peptides bind very strongly to hydrophobic surfaces, including TO/W, dodecane/water (DD/W), and A/W interfaces, to markedly lower the interfacial tension. They cannot be pushed off the surface when the surface area is compressed by approximately 15%. P12 can withstand a pressure of 27 mN/m and P27 can withstand a pressure of 25 mN/m at the TO/W interface without net desorption. Higher pressure could not be tested. Both of the peptides are completely elastic at interfaces when the surface is compressed and expanded by ±15% in area.
Based on several secondary structure prediction algorithms (6–8, 32), apoB[37–41] contains 10 strong amphipathic β strands of 11–15 amino acids and several shorter strands (Fig. 1). Compared with P12 or P27, apoB[37–41] represents a large, continuous β-sheet structure. As expected, apoB[37–41] showed strong lipid binding at the TO/W interface. It lowered the surface tension and stabilized the surface remarkably (Fig. 4, top). When compressed by 2–4 μl, there is a small increase in γ after compression and a rapid snapback on re-expansion consistent with a small part of the sequence being pushed off above a ΠMAX of approximately 16.0 mN/m and rapidly readsorbing on re-expansion. However, most of the bound apoB[37–41] can be compressed by up to approximately 15% in area to a surface pressure of approximately 23 mN/m without desorption (Fig. 4B). This is consistent with the interfacial behavior of P12 and P27 at the TO/W interface (6). Therefore, we think the strong binding of apoB[37–41] to the lipids comes from the major AβS structure. ApoB[37–41] is almost completely elastic on the surface when the surface was compressed and expanded within ±15% change in area (Fig. 7A and Table 1), which indicates that apoB[37–41] binds to the surface, stays firmly bound to the surface, and retains elasticity. When the peptide in the aqueous phase surrounding the triolein droplet is exchanged, the bound apoB[37–41] does not desorb. Upon compression, the bound peptide remains largely bound up to a decrease in area of approximately 15% and within this range of oscillation is fully elastic on the surface (Fig. 7B, Table 1). When apoB[37–41] binds to the interface, the AβSs form a large, well organized β-sheet structure with hydrogen bonds between strands as shown by the formation of a solid interface at the A/W interface at low pressure (approximately 4 mN/m) (Fig. 8A).
When compressed to a high pressure (greater than 24 mN/m) and to an area less than 85% of its area at γeq, a more permanent displacement of the peptide occurs. There is no evidence that the entire peptide is pushed off into the aqueous phase, but rather, that a decrease in area of greater than 15% causes a surface change into a conformationally denatured, perhaps multilayered structure which is essentially irreversible in the short term but may be slowly reversible if given many days to respread (Fig. 5A). This contrasts to full-length apoB which can be compressed to 28 mN/m with a decrease in area of 55% and then snap back rapidly onto the surface of the TO drop on re-expansion to reestablish γeq. This great flexibility must derive from the other parts of apoB, most probably from the large AαH regions of α1 and α2 (7). The ΠMAX of apoB is at about 13 mN/m, and various domains of apoB are progressively displaced by larger compressions (5) so that about half of the whole sequence can be desorbed. The ΠMAX of apoB[37–41] of about 16 mN/m is very limited and probably only represents a low percentage of the sequence that can reversibly desorb. Thus, this domain is much more rigid and inflexible relative to full-length apoB.
Studies on synthetic model AβS peptides at A/W interface suggest a flat, well-organized anti-parallel β-sheet structure on the surface (48–51). A study on a group of designed amphiphilic β-sheet peptides with the sequence Pro-Glu-(Phe-Glu)n-Pro (n = 2–7) by grazing incidence X-ray diffraction (GIXD) showed that those peptides formed stable, ordered 2D monolayer structures at A/W interface (50). Further study on Pro-Glu-(Phe-Glu)5-Pro revealed the elastic-like behavior of the peptide upon compression and expansion along the Π-A isotherm by using in situ GIXD technique (51). Upon compression, Pro-Glu-(Phe-Glu)5-Pro lifted off at about 16 Å2/residue, followed by a steep increase, and collapsed at 25 mN/m with an area of approximately 11.5 Å2/residue. The macroscopic 2D compressibility CM is about 11.3 mN/m beyond the limiting area. When compressed above approximately 17 mN/m, on re-expansion Pro-Glu-(Phe-Glu)5-Pro showed a smaller area per residue than during compression. It was suggested that the ordered β-sheet monolayer may undergo a quasi-reversible compression and expansion cycle at the interface (51). Since the N and C-terminal prolines are aligned and define the long axis of the 2D cell on the interface, d01 spacing is the length of the peptide in the interface, and d02 is the distance between chains. This gives a Pro-Glu-(Phe-Glu)5-Pro β-sheet unit cell of a = 9.5 Å, b = 46 Å, γ = approximately 90°, and a corresponding area of 218.5 Å2 (16.8 Å2/residue). This is typical for a standard β-sheet unit cell of 7 Å × 9.5 Å (Z = 4) = 66 Å2 (or 16.5 Å2/amino acid). The in situ GIXD data showed that the 2D peptide assembly can be compressed up to 37% along the peptide backbone axis, and on re-expansion, the β-sheet structure can be reverted elastically to its original conformation. While the distance between the β strands along the hydrogen bond direction (d2,0) remained unchanged during the compression and expansion cycle, the d1,0 spacing decreased from 46 Å (approximately 3.5 Å/amino acid) at π = 1.4 mN/m to 35.8 Å (approximately 2.8 Å/amino acid) at π = 27.1 mN/m. Thus, the length of the peptide parallel to the surface is shorter by 22%. The authors suggested that beyond the limiting area per molecule where peptides were closely packed, the compression affected the peptide backbone conformation to allow the β-sheet domain to bend out of the surface when compressed to above a baseline “0” pressure without affecting the side-to-side H-bonded dimension. We suggest that, like Pro-Glu-(Phe-Glu)5-Pro (51), apoB37–41 sheets can also bend out of the plane of the surface when compressed.
ApoB[37–41] showed a typical behavior of the β-sheet structure at the A/W interface. It lifts off at approximately 21 Å2/residue (limiting area approximately 16 Å2/residue) followed by a steep increase in the pressure, and collapses at 35 mN/m with an area of 9.8 Å2/residue (Fig. 8A and B), which suggests that apoB[37–41] is flat on the surface at its limiting area of 16 Å/residue. The monolayer became solid when the pressure was greater than 4 mN/m, and it can be reversibly compressed and expanded below the collapse pressure (Fig. 8B). Beyond the collapse pressure, the monolayer showed an irreversible compression and expansion cycle similar to the quasi-reversible cycle noted for Pro-Glu-(Phe-Glu)5-Pro (51). Based on the definition of the compressibility of the peptide monolayer (49), the compressibility of apo[B37–41] at the steep part in the Π-A isotherm is 10.7 mN/m, which is similar to but slightly less than Pro-Glu-(Phe-Glu)5-Pro (11.3 mN/m) (51). Thus, the apoB[37–41] monolayer exhibits β-sheet structure characteristics of a well-organized, solid network on the surface while having compressibility and elasticity.
Compared with the strong lipid binding behavior of AβS structure, the easy desorption and fast readsorption behavior of AαH structure is suggested to correspond to the major conformational changes of apoB (5, 6, 26). The CSP of AαH derived from the exchangeable apolipoproteins resembles some of the AαH sequences in α2 and α3 domains of apoB (26). CSP binds to TO/W interface and lowers the interface tension. On compression it desorbed from the interface above an interfacial pressure of 16 mN/m, and when the surface is expanded back to the original area and the pressure is relaxed, CSP readsorbed onto the surface. It is suggested that AαH, like CSP, are responsible for the major flexibility of apoB when compressed and re-expanded.
The conformational changes of apoB corresponding to the pressure changes are also reflected in the Π-A isotherm of apoB when apoB was spread onto the A/W interface (5). ApoB lifts off at 56000 Å2 per molecule, the pressure rises to a transition at about 23 mN/m with the area of 34000 Å2 per molecule, followed by a gradual increase to 34 mN/m with the area of 20000 Å2 per molecule, and then the pressure rises steeply to about 50 mN/m before an apparent monolayer collapse. At low pressure the conformation of apoB was between fully extended to closely packed with the area between 68000 (assuming 15 Å2 per residue) and 56000 Å2 at liftoff. When the pressure increased to about 23 mN/m with the area of 34000 Å2 per molecule, we speculated that most of the AαH domains were pushed off the surface, leaving AβS bound to the surface. This is consistent with the estimation of Segrest et al. (7) that the total area covered by AβS in β1 and β2 domain of apoB, assuming 16 Å2 per residue, is about 32000 Å2. The region between 34000 and 20000 Å2 may be responsible for the ejection of the rest of the most hydrophobic AαH and the elastic compression of AβS. The total reversible reduction in area of apoB from 56,000 Å2 at liftoff to 20,000 Å2 at 34 mN/m is approximately 64%, demonstrating great flexibility. At 20000 Å2, if only the AβS of β1 and β2 domains remained bound on the A/W surface, the result indicates that AβS in apoB can be compressed to 10 Å2 per residue. This is consistent with the fact that small consensus peptides derived from the first β region of apoB can be compressed on a TO/W interface to 10Å2 per residue (6), while apoB[37–41] can be compressed to 9.8 Å2 per residue before collapse at A/W interface. In other words, at higher pressure only AβS anchors this peptide on the surface, and the elastic, compressible nature of AβS is responsible for the limited flexibility at this point.
From the time TAG-rich lipoprotein particles are secreted into the plasma as chylomicrons or VLDL to the time the metabolized particle is taken up as a chylomicron remnant or an LDL particle, apoB remains anchored on the lipoprotein surface. While the lipoprotein particles undergoing metabolism in plasma change in size and composition, the surface pressure may also change, increasing if surface-active molecules are added to the surface or core molecules are depleted. As a result, the coverage of apoB probably changes also and is accompanied by changes in some of its surface conformation (28–31). It is reasonable to assume that the nascent TAG-rich lipoprotein particles possess a relatively low surface pressure because free cholesterol and exchangeable apolipoproteins are able to rapidly bind to the surface. Increase in such surface-located molecules should increase Π. As lipoprotein lipase acts on the surface and increases the surface molecules (fatty acids and monoacylglycerols) and decreases the core lipids (TAG), the surface pressure should increase progressively. This should force off some of the more weakly bound exchangeable apolipoproteins and apoB amphipathic α helices. As soluble apolipoproteins desorb and the products of lipolysis are removed by albumin, the surface pressure should relax. Thus, we suggest that moderate changes in surface pressure are responsible for major conformational changes of apoB, which give rise to a changing capacity or affinity to bind antibodies, enzymes, and receptors. Further, the strong binding and limited elastic feature of AβS structure may be a driving force for recruiting lipids during lipoprotein assembly in the endoplasmic reticulum. In this work, we show that apoB[37–41], rich in AβS structure, is a minimally flexible subdomain that irreversibly binds to TAG and is probably part of the anchor sequence of apoB that recruits TAG during nascent lipoprotein formation.
Acknowledgments
The authors thank Donna Ross for manuscript preparation.
Abbreviations
AαH, amphipathic α helix
AβS, amphipathic beta strand
ApoB, apolipoprotein B
apoB48, N-terminal 48% of apoB
apoB100, full-length apoB
A/W, air/water
CSP, a consensus sequence peptide of exchangeable apolipoproteins
DD/W, dodecane/water
DPC, dodecylphosphocholine
GIXD, grazing incidence X-ray diffraction
HC/W, hydrocarbon/water
IDL, intermediate density lipoprotein
MTP, microsomal triglyceride transfer protein
TAG, triacylglycerol
TO/W, triolein/water
This work was supported in part by National Institutes of Health USA Grant NIH-NHLBI 2P01 HL26335-21 and the Canadian Institutes of Health Research (CIHR) Grant MOP-67073.
Published, JLR Papers in Press, February 26, 2009.
References
- 1.Kane, J. P., and R. J. Havel. 2001. Disorders of the biogenesis and secretion of lipoproteins containing the B apolipoproteins. In The Metabolic and Molecular Bases of Inherited Disease. C. R. Scriver, A. L. Beaudet, D. Valle, et al., editors. McGraw–Hill, New York. 2717–2752.
- 2.Havel, R. J., and J. P. Kane. 2001. Introduction: structure and metabolism of plasma lipoproteins. In The Metabolic and Molecular Bases of Inherited Disease. C. R. Scriver, A. L. Beaudet, D. Valle, et al., editors. McGraw–Hill, New York. 2705–2716.
- 3.Huang A. H. C. 1996. Oleosins and oil bodies in seeds and other organs. Plant Physiol. 110: 1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hope R. G., D. J. Murphy, and J. McLauchlan. 2002. The domains required to direct core proteins of hepatitis C virus and GB virus-B to lipid droplets share common features with plant oleosin proteins. J. Biol. Chem. 277: 4261–4270. [DOI] [PubMed] [Google Scholar]
- 5.Wang L., M. T. Walsh, and D. M. Small. 2006. Apolipoprotein B is conformationally flexible but anchored at a triolein/water interface: a possible model for lipoprotein surfaces. Proc. Natl. Acad. Sci. USA. 103: 6871–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L., and D. M. Small. 2004. Interfacial properties of amphipathic β strand consensus peptides of apolipoprotein B at oil/water interfaces. J. Lipid Res. 45: 1704–1715. [DOI] [PubMed] [Google Scholar]
- 7.Segrest J. P., M. K. Jones, H. De Loof, and N. Dashti. 2001. Structure of apolipoprotein B-100 in low density lipoproteins. J. Lipid Res. 42: 1346–1367. [PubMed] [Google Scholar]
- 8.Small D. M., and D. Atkinson. 1997. The first beta sheet region of apoB(apoB21–41) is a amphipathic ribbon 50–60Å wide and 200Å long which initiates triglyceride binding and assembly of nascent lipoproteins. Circulation. 96 (8S): I-1. [Google Scholar]
- 9.Yao Z. M., B. D. Blackhart, M. F. Linton, S. M. Taylor, S. G. Young, and B. J. McCarthy. 1991. Expression of carboxyl-terminally truncated forms of human apolipoprotein B in rat hepatoma cells. Evidence that the length of apolipoprotein B has a major effect on the buoyant density of the secreted lipoproteins. J. Biol. Chem. 266: 3300–3308. [PubMed] [Google Scholar]
- 10.Spring D. J., L. W. Chen-Liu, J. E. Chatterton, J. Elovson, and V. N. Schumaker. 1992. Lipoprotein assembly. Apolipoprotein B size determines lipoprotein core circumference. J. Biol. Chem. 267: 14839–14845. [PubMed] [Google Scholar]
- 11.McLeod R. S., Y. Wang, S. Wang, A. Rusiñol, P. Links, and Z. Yao. 1996. Apolipoprotein B sequence requirements for hepatic very low density lipoprotein assembly. Evidence that hydrophobic sequences within apolipoprotein B48 mediate lipid recruitment. J. Biol. Chem. 271: 18445–18455. [DOI] [PubMed] [Google Scholar]
- 12.Carraway M., H. Herscovitz, V. Zannis, and D. M. Small. 2000. Specificity of lipid incorporation is determined by sequences in the N-terminal 37 of apoB. Biochemistry. 39: 9737–9745. [DOI] [PubMed] [Google Scholar]
- 13.Shelness G. S., L. Hou, A. S. Ledford, J. S. Parks, and R. B. Weinberg. 2003. Identification of the lipoprotein initiating domain of apolipoprotein B. J. Biol. Chem. 278: 44702–44707. [DOI] [PubMed] [Google Scholar]
- 14.Manchekar M., P. E. Richardson, T. M. Forte, G. Datta, J. P. Segrest, and N. Dashti. 2004. Apolipoprotein B-containing lipoprotein particle assembly: lipid capacity of the nascent lipoprotein particle. J. Biol. Chem. 279: 39757–39766. [DOI] [PubMed] [Google Scholar]
- 15.Richardson P. E., M. Manchekar, N. Dashti, M. K. Jones, A. Beigneux, S. G. Young, S. C. Harvey, and J. P. Segrest. 2005. Assembly of lipoprotein particles containing apolipoprotein-B: structural model for the nascent lipoprotein particle. Biophys. J. 88: 2789–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dashti N., M. Manchekar, Y. Liu, Z. Sun, and J. P. Segrest. 2007. Microsomal triglyceride transfer protein activity is not required for the initiation of apolipoprotein B-containing lipoprotein assembly in McA-RH7777 cells. J. Biol. Chem. 282: 28597–28608. [DOI] [PubMed] [Google Scholar]
- 17.Hussain M. M., Y. Zhao, R. K. Kancha, B. D. Blackhart, and Z. Yao. 1995. Characterization of recombinant human apoB-48–containing lipoproteins in rat hepatoma McA-RH7777 cells transfected with apoB-48 cDNA: overexpression of apoB-48 decreases synthesis of endogenous apoB-100. Arterioscler. Thromb. Vasc. Biol. 15: 485–494. [DOI] [PubMed] [Google Scholar]
- 18.Ren X., L. Zhao, A. Sivashanmugam, Y. Miao, L. Korando, Z. Yang, C. A. Reardon, G. Getz, C. G. Brouillette, W. G. Jerome, et al. 2005. Engineering mouse apolipoprotein A-I into a monomeric, active protein useful for structural determination. Biochemistry. 44: 14907–14919. [DOI] [PubMed] [Google Scholar]
- 19.Labourdenne S., N. Gaudry-Rolland, S. Letellier, M. Lin, A. Cagna, G. Esposito, R. Verger, and C. Rivière. 1994. The oil-drop tensiometer: potential applications for studying the kinetics of (phopho)lipase action. Chem. Phys. Lipids. 71: 163–173. [Google Scholar]
- 20.Benjamins, J., and E. H. Lucassen-Reynders. 1998. Surface dilational rheology of proteins adsorbed at air/water and oil/water interfaces. In Proteins at Liquid Interfaces. D. Möbius and R. Miller, editors. Elsevier, Amsterdam. 341–384.
- 21.Benjamins J., A. Cagna, and E. H. Lucassen-Reynders. 1996. Viscoelastic properties of triacylglycerol/water interfaces covered by proteins. Coll. Surf. 114: 245–254. [Google Scholar]
- 22.Phillips M. C., and K. E. Krebs. 1986. Studies of apolipoproteins at the air-water interface. Methods Enzymol. 128: 387–403. [DOI] [PubMed] [Google Scholar]
- 23.Fahey D. A., and D. M. Small. 1986. Surface properties of 1,2-dipalmitoyl-3-acyl-sn-glycerols. Biochemistry. 25: 4468–4472. [DOI] [PubMed] [Google Scholar]
- 24.Fahey D. A., and D. M. Small. 1988. Phase behavior of monolayers of 1,2-dipalmitoyl-3-acyl-sn-glycerols. Langmuir. 4: 589–594. [Google Scholar]
- 25.Wang L., D. Atkinson, and D. M. Small. 2003. Interfacial properties of an amphipathic α-helix consensus peptide of exchangeable apolipoproteins at air/water and oil/water interfaces. J. Biol. Chem. 278: 37480–37491. [DOI] [PubMed] [Google Scholar]
- 26.Wang L., D. Atkinson, and D. M. Small. 2005. The interfacial properties of apoA-I and an amphipathic α-helix consensus peptide of exchangeable apolipoproteins at the triolein/water interface. J. Biol. Chem. 280: 4154–4165. [DOI] [PubMed] [Google Scholar]
- 27.Wang L., N. Hua, D. Atkinson, and D. M. Small. 2007. The N-terminal (1-44) and C-terminal (198-243) of apolipoprotein A-I behave differently at the triolein/water interface. Biochemistry. 46: 12140–12151. [DOI] [PubMed] [Google Scholar]
- 28.Kane J. 1991. Plasma lipoproteins and their receptors. Curr. Opin. Struct. Biol. 1: 510–515. [Google Scholar]
- 29.Chen G. C., W. Liu, P. Duchateau, J. Allaart, R. L. Hamilton, C. M. Mendel, K. Lau, D. A. Hardman, P. H. Frost, M. J. Malloy, et al. 1994. Conformational differences in human apolipoprotein B-100 among subspecies of low density lipoproteins (LDL). Association of altered proteolytic accessibility with decreased receptor binding of LDL subspecies from hypertriglyceridemic subjects. J. Biol. Chem. 269: 29121–29128. [PubMed] [Google Scholar]
- 30.McNamara J. R., D. M. Small, Z. Li, and E. J. Schaefer. 1996. Differences in LDL subspecies involve alterations in lipid composition and conformational changes in apolipoprotein B. J. Lipid Res. 37: 1924–1935. [PubMed] [Google Scholar]
- 31.Ibdah J. A., S. Lund-Katz, and M. C. Phillips. 1989. Molecular packing of high-density and low-density lipoprotein surface lipids and apolipoprotein A-I binding. Biochemistry. 28: 1126–1133. [DOI] [PubMed] [Google Scholar]
- 32.Nolte, R. T. 1994. Structural Analysis of the Human Apolipoproteins: An Integrated Approach Utilizing Physical and Computational Methods. PhD Thesis. Boston University School of Medicine, Boston.
- 33.Segrest J. P., M. K. Jones, V. K. Mishra, G. M. Anantharamaiah, and D. W. Garber. 1994. apoB-100 has a pentapartite structure composed of three amphipathic alpha-helical domains alternating with two amphipathic beta-strand domains. Detection by the computer program LOCATE. Arterioscler. Thromb. 14: 1674–1685. [DOI] [PubMed] [Google Scholar]
- 34.Anderson T. A., D. G. Levitt, and L. J. Banaszak. 1998. The structural basis of lipid interactions in lipovitellin, a soluble lipoprotein. Structure. 6: 895–909. [DOI] [PubMed] [Google Scholar]
- 35.Mann C. J., T. A. Anderson, J. Read, S. A. Chester, G. B. Harrison, S. Kochl, P. J. Ritchie, P. Bradbury, F. S. Hussain, J. Amey, et al. 1999. The structure of vitellogenin provides a molecular model for the assembly and secretion of atherogenic lipoproteins. J. Mol. Biol. 285: 391–408. [DOI] [PubMed] [Google Scholar]
- 36.Segrest J. P., M. K. Jones, and N. Dashti. 1999. N-terminal domain of apolipoprotein B has structural homology to lipovitellin and microsomal triglyceride transfer protein: a “lipid pocket” model for self-assembly of apoB-containing lipoprotein particles. J. Lipid Res. 40: 1401–1416. [PubMed] [Google Scholar]
- 37.Read J., T. A. Anderson, P. J. Ritchie, B. Vanloo, J. Amey, D. Levitt, M. Rosseneu, J. Scott, and C. C. Shoulders. 2000. A mechanism of membrane neutral lipid acquisition by the microsomal triglyceride transfer protein. J. Biol. Chem. 275: 30372–30377. [DOI] [PubMed] [Google Scholar]
- 38.Stillemark P., J. Borén, M. Andersson, T. Larsson, S. Rustaeus, K. A. Karlsson, and S. O. Olofsson. 2000. The assembly and secretion of apolipoprotein B-48-containing very low density lipoproteins in McA-RH7777 cells. J. Biol. Chem. 275: 10506–10513. [DOI] [PubMed] [Google Scholar]
- 39.Herscovitz H., M. Hadzopoulou-Cladaras, M. T. Walsh, C. Cladaras, V. I. Zannis, and D. M. Small. 1991. Expression, secretion, and lipid-binding characterization of the N-terminal 17% of apolipoprotein B. Proc. Natl. Acad. Sci. USA. 88: 7313–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herscovitz H., A. Derksen, M. T. Walsh, C. J. McKnight, D. L. Gantz, M. Hadzopoulou-Cladaras, V. Zannis, C. Curry, and D. M. Small. 2001. The N-terminal 17% of apoB binds tightly and irreversibly to emulsions modeling nascent very low density lipoproteins. J. Lipid Res. 42: 51–59. [PubMed] [Google Scholar]
- 41.Weinberg R. B., V. R. Cook, J. A. DeLozier, and G. S. Shelness. 2000. Dynamic interfacial properties of human apolipoproteins A-IV and B-17 at the air/water and oil/water interface. J. Lipid Res. 41: 1419–1427. [PubMed] [Google Scholar]
- 42.Ledford A. S., V. A. Cook, G. S. Shelness, and R. B. Weinberg. 2009. Structural and dynamic interfacial properties of the lipoprotein initiating domain of apolipoprotein B. J. Lipid Res. 50: 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang Z. G., M. Carraway, and C. J. McKnight. 2005. Limited proteolysis and biophysical characterization of the lipovitellin homology region in apolipoprotein B. Biochemistry. 44: 1163–1173. [DOI] [PubMed] [Google Scholar]
- 44.Jiang Z. G., D. Gantz, E. Bullitt, and C. J. McKnight. 2006. Defining lipid-interacting domains in the N-terminal region of apolipoprotein B. Biochemistry. 45: 11799–11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang Z. G., M. N. Simon, J. S. Wall, and C. J. McKnight. 2007. Structural analysis of reconstituted lipoproteins containing the N-terminal domain of apolipoprotein B. Biophys. J. 92: 4097–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitsche M. A., L. Wang, Z. G. Jiang, C. J. McKnight, and D. M. Small. 2009. Interfacial properties of a complex multi-domain 490 amino acid peptide derived from apolipoprotein B (residues 292–782). Langmuir. 25: 2322–2330. [DOI] [PubMed] [Google Scholar]
- 47.Yang C-Y., Z-W. Gu, S-A. Weng, T. W. Kim, S-H. Chen, H. J. Pownall, P. M. Sharp, S-W. Liu, W-H. Li, A. M. Gotto, Jr., et al. 1989. Structure of apolipoprotein B-100 of human low density lipoproteins. Arteriosclerosis. 9: 96–108. [DOI] [PubMed] [Google Scholar]
- 48.Maget-Dana R., D. Lelièvre, and A. Brack. 1999. Surface active properties of amphiphilic sequential isopeptides: comparison between α-helical and β-sheet conformations. Biopolymers. 49: 415–423. [DOI] [PubMed] [Google Scholar]
- 49.Castano S., B. Desbat, and J. Dufourcq. 2000. Ideally amphipathic β-sheeted peptides at interfaces: structure, orientation, affinities for lipids and hemolytic activity of (KL)mK peptides. Biochim. Biophys. Acta. 1463: 65–80. [DOI] [PubMed] [Google Scholar]
- 50.Rapaport H., G. Möller, C. M. Knobler, R. T. Jensen, K. Kjaer, L. Leiserowitz, and D. A. Tirrell. 2002. Assembly of triple-stranded β-sheet peptides at interfaces. J. Am. Chem. Soc. 124: 9342–9343. [DOI] [PubMed] [Google Scholar]
- 51.Isenberg H., K. Kjaer, and H. Rapaport. 2006. Elasticity of crystalline β-sheet monolayers. J. Am. Chem. Soc. 128: 12468–12472. [DOI] [PubMed] [Google Scholar]