

Abstract
Human apolipoprotein C-I (apoC-I) is an exchangeable apolipoprotein that binds to lipoprotein particles in vivo. In this study, we employed a LC-MS/MS assay to demonstrate that residues 38–51 of apoC-I are significantly protected from proteolysis in the presence of 1,2-dimyristoyl-3-sn-glycero-phosphocholine (DMPC). This suggests that the key lipid-binding determinants of apoC-I are located in the C-terminal region, which includes F42 and F46. To test this, we generated site-directed mutants substituting F42 and F46 for glycine or alanine. In contrast to wild-type apoC-I (WT), which binds DMPC vesicles with an apparent Kd [Kd(app)] of 0.89 μM, apoC-I(F42A) and apoC-I(F46A) possess 2-fold weaker affinities for DMPC with Kd(app) of 1.52 μM and 1.58 μM, respectively. However, apoC-I(F46G), apoC-I(F42A/F46A), apoC-I(F42G), and apoC-I(F42G/F46G) bind significantly weaker to DMPC with Kd(app) of 2.24 μM, 3.07 μM, 4.24 μM, and 10.1 μM, respectively. Sedimentation velocity studies subsequently show that the protein/DMPC complexes formed by these apoC-I mutants sediment at 6.5S, 6.7S, 6.5S, and 8.0S, respectively. This is compared with 5.0S for WT apoC-I, suggesting the shape of the particles was different. Transmission electron microscopy confirmed this assertion, demonstrating that WT forms discoidal complexes with a length-to-width ratio of 2.57, compared with 1.92, 2.01, 2.16, and 1.75 for apoC-I(F42G), apoC-I(F46G), apoC-I(F42A/F46A), and apoC-I(F42G/F46G), respectively. Our study demonstrates that the C-terminal amphipathic α-helix of human apoC-I contains the major lipid-binding determinants, including important aromatic residues F42 and F46, which we show play a critical role in stabilizing the structure of apoC-I, mediating phospholipid interactions, and promoting discoidal particle morphology.
Keywords: analytical ultracentrifugation, apo, C1, CI, electron microscopy, lipid metabolism, lipidomics, mass spectrometry, phospholipid, protein-lipid interaction
Human apolipoprotein C-I (apoC-I) is a 6.6 kDa (57 residue) apolipoprotein that is synthesized primarily in the liver (1). ApoC-I circulates in plasma at a concentration of approximately 0.06 mg/ml bound to the surface of chylomicron, VLDL, intermediate density lipoprotein (IDL), and HDL particles (2, 3). As a component of VLDL, apoC-I is reported to regulate the receptor-binding function of apolipoprotein E (4). As a constituent of the smaller HDL, apoC-I functions as an activator of lecithin:cholesterol acyltransferase (LCAT) (5, 6) and an inhibitor of cholesterol ester transfer protein (CETP) (7), which are both important regulators of reverse cholesterol transport. Like other members of the C apolipoprotein family, apoC-I possesses a high level of structural adaptability. In its lipid-free form, this small protein contains approximately 30% α-helix as shown by circular dichroism (CD) spectroscopy (8, 9). Self-association of apoC-I in the absence of lipid increases the α-helical content in a concentration-dependent manner from 30% at 0.01 mg/ml to 80% at 2.8 mg/ml (8). Structural studies in the presence of sodium dodecyl sulfate (10) and short chain phospholipids (11) demonstrate that apoC-I adopts a helix-turn-helix motif, with an N-terminal amphipathic helix spanning residues 7–29 and a C-terminal amphipathic helix spanning residues 38–52 (Fig. 1). These studies also indicate that the N-terminal helix is significantly more flexible, suggesting that it binds less tightly to a lipid surface than its C-terminal counterpart (10).
Fig. 1.

NMR structure of human apoC-I measured in the presence of SDS micelles. The backbone is depicted as ribbons for α-helical regions and wireframe for turn/unordered structure. The aromatic residues in the C-terminal amphipathic helix of apoC-I targeted for mutagenesis in this study (F42 and F46) are depicted as sticks. The coordinates were obtained from the PDB file 1IOJ (10).
More recently, Gursky and coworkers generated a series of proline mutants spanning the N- and C-terminal helices of apoC-I. They examined changes in secondary structure of the protein upon binding to 1,2-dimyristoyl-3-sn-glycero-phosphocholine (DMPC) vesicles and the gross morphologies of the complexes themselves (12). The mutants showed a marked decrease in α-helicity when compared with the wild-type protein, with the largest change noted for the proline mutation of arginine at position 23 (R23P), where the total α-helicity in the presence of DMPC decreased from 65% for wild-type apoC-I to 40% for the R23P mutant. This was also accompanied by a change in morphology of the R23P-lipid complex (12). Additional studies by Gursky (6) employing near-UV circular dichroism spectroscopy also demonstrated that mutation of W41 to alanine or proline results in significant loss of α-helical structure. This is not surprising because earlier studies have suggested that aromatic residues are important for anchoring apolipoproteins to lipid surfaces (13). Gursky (6) has subsequently proposed that the C-terminal aromatic cluster of W41, F42, and F46 are important residues for mediating helix stability and also the interactions of apoC-I with phospholipid (6). However, unlike W41, whose role in helix stability and lipid binding is well characterized (6, 10), the contributions of F42 and F46 in mediating and/or stabilizing the structure and phospholipid interactions of apoC-I are not well understood.
In this study we set out to test the hypothesis that F42 and F46 of human apoC-I play important roles in mediating phospholipid binding and the morphology of the resulting protein-lipid complexes. We first present results of protease resistance assays measured by LC-MS/MS that demonstrate the C-terminal region of human apoC-I, including F42 and F46, contains the major lipid-binding determinants. We subsequently employ circular dichroism spectroscopy, analytical ultracentrifugation, and transmission electron microscopy studies, comparing wild-type and mutant forms of apoC-I, to demonstrate the importance of F42 and F46 in mediating apoC-I/phospholipid interactions and morphology.
EXPERIMENTAL PROCEDURES
Expression and purification of recombinant wild-type apoC-I
Recombinant human apoC-I was expressed using the method described by Atcliffe et al. (11). Briefly, the pET-32a vector containing the cDNA of human apoC-I was transformed into the UT5600 strain of E. coli. The cells were grown in 6 × 500 ml of Luria-Bertani (LB) media containing 50 μg/ml ampicillin and 0.2% (w/v) glucose at 37°C in an incubator-shaker (120 opm) to an optical density at 600 nm of 0.6. Expression of the MBP-apoC-I fusion protein was induced by the addition of 1 mM isopropyl-1-β-thiogalactopyranoside, and the culture incubated for a further 2 h at 37°C (120 opm). The cells were harvested by centrifugation at 6,000 rpm (5,860 g, 4°C) using a Sorvall RC-5C preparative ultracentrifuge and GSA rotor. Cell lysates were prepared by treatment with 0.2 mM lysozyme and 1 mM EDTA at 4°C followed by sonication using an MSE Soniprep 150 sonicator at 10 micron amplitude for 30 × 30 s pulses with a 30 s break between each pulse. Cell lysates containing MBP-apoC-I fusion protein were incubated with amylose resin at 4°C overnight. The MBP-apoC-I fusion protein was eluted from the resin using 15 mM maltose. The concentration of the fusion protein was determined at 280 nm using an extinction coefficient of 70,410 M−1 cm−1 based on amino acid composition, which was calculated using the program SEDNTERP (14). The apoC-I moiety was then cleaved from the fusion protein with 150 U mg−1 of Factor Xa protease. ApoC-I was purified using an AKTA Basic LC system (GE Healthcare) on an HR 26/60 column packed with Superdex 75 size exclusion resin equilibrated with 3.5 M GuHCl. The purified recombinant protein was renatured via dialysis into 10 mM NH4OAc, pH 6.8 and stored at 4°C. The concentration was determined by measuring the absorbance at 280 nm using an extinction coefficient of 5,690 M−1 cm−1 calculated from amino acid composition (14). The recombinant product was assessed by SDS-PAGE (see supplementary Fig. I) and deemed to be greater than 95% pure.
Generation of apoC-I mutants
ApoC-I mutants were generated either recombinantly using the Stratagene QuikChange site-directed mutagenesis kit or using Fmoc peptide synthesis as described below. For recombinant apoC-I glycine mutants, mutagenesis was performed according to manufacturer's instructions (Stratagene). Primers were designed to generate two single site-directed mutants of apoC-I and a double site-directed mutant (see supplementary Table I). Briefly, the primers (125 ng) were added to 10 ng of apoC-I wild-type plasmid in the presence of PfuUltra HF DNA polymerase. The sample was subjected to 18 thermal cycles with each cycle as follows: 95°C for 50 s; 60°C for 50 s; and 68°C for 14 min. Following this, the sample was cooled to 4°C. 1 μl Dpn was added to the reaction mix and incubated for 1 h at 37°C. 3.0 M NaOAc and ethanol were then added to a final concentration of 0.1 (v/v) and 2.5 (v/v), respectively, and incubated overnight at –20°C. The sample was microfuged for 15 min at 13,200 rpm (16,100 g), and the resultant pellet was redissolved in 10 μl TAE. A volume of 4 μl was utilized for each transformation. The mutant plasmid was transformed into XL10-Gold Ultracompetent cells provided by Stratagene. The cells were allowed to sit for 30 min at 4°C after addition of the plasmid before a brief heat shock (42°C for 30 s). The cells were subsequently returned to 4°C for 2 min and then 0.5 ml LB media containing 0.2% (w/v) glucose was added. The cells were incubated for 1 h at 37°C with gentle shaking (120 opm). A 250 μl aliquot of the sample was plated onto LB agar containing 50 μg/ml ampicillin, and the plate was incubated overnight at 37°C. ApoC-I glycine mutants were subsequently expressed and purified as described for the wild-type protein above. Yields for the recombinant mutant proteins are presented and compared with the wild-type protein (see supplementary Table II).
Peptide synthesis
ApoC-I alanine mutants were synthesized by solid-phase Fmoc chemistry on a CEM Liberty microwave peptide synthesizer (AI Scientific, Australia) as C-terminal acids on Fmoc-PEG-PS resin (0.2 mmol/g loading, Applied Biosystems, Australia). All peptides were synthesized on a 0.1 mmol scale using a 5-fold molar excess of Fmoc protected amino acids (0.5 mmol) that were activated by coupling agent HCTU (1 equivalent) in the presence of DIEA (2 equivalents). Fmoc protecting groups were removed by treating the resin attached peptide with 20% piperidine in DMF (v/v, containing 0.1 M HOBt). The deprotection and coupling reactions were carried out at 75°C using 45 W microwave power for 3 min and 40 W microwave power for 5 min, respectively. Peptides were cleaved from the resin at the end of the synthesis by treatment with TFA/triisopropylsilane/water (95/2.5/2.5, 2 h at 20°C) precipitated with diethyl ether and lyophilized. The crude products were purified by reverse-phase HPLC (Agilent Zorbax C18 5 μm, 250 × 9.4 mm semi-prep column, 5 ml/min) using an Agilent 1100 LC system. Purity was assessed by analytical RP HPLC on an Agilent Zorbax C18 5uM 150 × 4.3mm column and all peptides were judged to be greater than 95% pure. The molecular weights of the products were confirmed on an Agilent 6520 LC/Q-TOF mass spectrometer with an electrospray ionizing source coupled to an Agilent 1100 LC system.
Preparation of DMPC vesicles
For a typical preparation of small unilamellar vesicles, 0.136 g lyophilized DMPC was mixed with 1.0 ml in 10 mM NH4OAc, pH 6.8. The suspension was sonicated using an MSE Soniprep 150 sonicator at 10 micron amplitude for 15 × 10 s pulses with a 5 s break between each pulse. The resultant solution was centrifuged at 13,200 rpm (16,100 g) for 5 min in a bench-top microfuge to remove nonsuspended aggregates of phospholipid and titanium particles shed from the sonicator probe. The supernatant containing DMPC vesicles was stored at 30°C for up to 24 h. All DMPC concentrations are expressed in terms of the monomer.
LC-MS/MS protease resistance assay
A typical assay mixture consisted of 20 μM apoC-I and 5 mM DMPC vesicles. Mixtures were preincubated for 4 h at 30°C then supplemented with 20 μg of trypsin or GluC and placed at 4°C within the auto-sampler of an Agilent Surveyer LC system attached to a Finnigan-MAT LTQ-FTMS (Bremen, Germany). At 40 min intervals, 20 μl samples were injected onto a μRPC C2/C18 column (4.6 mm × 10 cm, GE Healthcare). This was carried out a total of seven times for each apoC-I/DMPC mixture. The gradient was initiated with a 5 min isocratic step of Solvent A [0.1% (v/v) formic acid] followed by a 30 min linear gradient to 100% Solvent B [0.1% (v/v) formic acid in acetonitrile]. Solvent B was then run isocratically for 5 min after which the column was returned to Solvent A for over 5 min. In between injection of successive samples, the column was subjected to 10 rapid gradients from 0–100% Solvent B to ensure removal of bound DMPC. Prior to each FT-MS run, the instrument was calibrated with manufacturer-provided solutions. The ESI conditions employed were as described previously (15) with the instrument calibrated to the increased flow of 500 μl/min. Mass spectra were collected in a data-dependent manner as follows: a single high resolution FT-MS spectra covering 300–2000 m/z, followed by three IT-MS CID (m/z window = 1.5 Th, collision relative energy = 35%, collision time = 100 ms) spectra that targeted the three most abundant peaks measured in the preceding FT-MS spectra. However, once a mass had been selected for analysis, it was excluded from selection for 3 min. This allowed for peaks other than the three most abundant to be analyzed. The data obtained was then manually annotated based on expected tryptic or GluC peptides.
Circular dichroism spectroscopy
Spectra were collected between wavelengths of 190 and 250 nm in a Jasco J-815 CD spectrometer at 25°C using a 1 mm pathlength quartz curvette, 0.5 nm step size, 1.0 nm bandwidth, and 2 s averaging time. Spectra of wild-type and mutant apoC-I solubilized in 10 mM NH4OAc, pH 6.8 were recorded at a protein concentration of 20 μM. CD spectra of apoC-I were also collected in the presence of DMPC vesicles ranging from a concentration of 40 μM through to 5 mM. All CD spectra were analyzed by nonlinear least-squares regression using the CONTINLL algorithm and SP43 database available in the CDPro software package downloaded from lamar.colostate.edu/∼sreeram/CDPro/ (16). For apoC-I/DMPC binding measurements, the ellipticity at 222 nm was plotted as a function of total lipid concentration and the data analyzed by nonlinear regression using SIGMAPLOT 10.0 to determine the apparent affinity (KDapp) of apoC-I (expressed per DMPC monomer) for the phospholipid surface. Nonlinear least-squares fits were weighted by the reciprocal of the variance across three separate experiments. Given the complexity of the experimental system (i.e., polydispersity with respect to vesicle preparation, and potential multivalence with respect to apoC-I binding) data were analyzed in terms of a generic hyperbolic decay rather than a discrete binding model as follows:
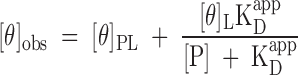 |
Eq. 1 |
where [θ]obs represents the experimentally observed mean residue ellipticity at 222 nm, and [θ]L and [θ]PL represent the mean residue ellipticity at 222 nm of free and bound apoC-I, respectively, KDapp represents the apparent dissociation constant for the protein-lipid complex, and [P] represents the total monomeric phospholipid concentration.
Analytical ultracentrifugation
Samples of between 250 and 380 μl were analyzed in a Beckman XL-I ultracentrifuge using an An50 Ti eight-hole rotor against a reference of buffer alone (270 or 400 μl). An initial wavelength and radial scan was carried out at a rotor velocity of 3,000 rpm to determine the optimal wavelength for data collection. Data were acquired at a rotor velocity of 25,000 or 50,000 rpm at 225 nm and collected continuously or at 20 min intervals using a step size of 0.003 cm without averaging. Solvent densities and viscosities were computed with the program SEDNTERP (14). The partial specific volume of the apoC-I/DMPC complexes were obtained from a previous study (11). Sedimentation velocity data were analyzed in terms of a noninteracting discrete single species (17) or a continuous-size distribution (18) using the program SEDFIT (available from www.analyticalultracentrifugation.com).
Transmission electron microscopy
Samples of DMPC (800 μM) in the presence of wild-type and mutant apoC-I proteins (20 μM) were incubated for 4 h at 30°C prior to deposition onto an EM grids. Transmission electron microscopy (TEM) sample preparation was carried out by adsorbing a 5.0 μl aliquot of the sample onto a carbon-coated Formvar film (Sigma Chemical Co.., St. Louis, MO) mounted on 300 mesh copper grids. The grids were rendered hydrophilic by glow discharge in a reduced atmosphere of air for 15 s prior to adsorption. After 30 s, samples were blotted and negatively stained with 1.5% (w/v) uranyl acetate. Specimens were investigated in a FEI Co. (Hillsborough, OR,) Tecnai TF30 fitted with a Gatan (Pleasanton, CA) US1000 2k × 2k CCD Camera.
RESULTS
LC-MS/MS protease protection assay
To map the major lipid-binding determinants of human apoC-I, an automated LC-MS/MS protease protection assay was employed using trypsin and GluC. Peptide fragments resulting from limited proteolysis were sampled, separated by reverse-phase HPLC and characterized by FT-MS every 40 min following addition of protease. The resulting total ion count (TIC) chromatograms, high resolution FT-MS spectra, and MS/MS spectra were then compared in the absence and presence of DMPC. For example, Fig. 2A shows the TIC chromatogram of apoC-I in the absence of phospholipid obtained 40 min after the addition of trypsin. The chromatogram shows four major peaks, eluting at 12.30, 12.92, 13.44, and 13.88 min, respectively (Fig. 2A). By comparison, only three peaks are observed in the TIC chromatogram in presence of DMPC eluting at 12.98, 13.49, and 13.96 min (Fig. 2B). The first peptide in Fig. 2A, which eluted at 12.30 min, is missing in the corresponding TIC chromatogram obtained in the presence of phospholipid (Fig. 2B). This peptide has an exact mass of 1003.5626 (Fig. 2C) and corresponds to amino acids 29-37 of human apoC-I as deduced by MS/MS analyses (Fig. 2D). This indicates that residues 29–37 are protected from cleavage in the presence of 5 mM DMPC for at least 40 min after the addition of trypsin.
Fig. 2.

Limited trypsinolysis of wild-type apoC-I before and after incubation with DMPC vesicles. A: Total ion current (TIC) chromatogram of wild-type apoC-I in the absence of lipid sampled 40 min after addition of trypsin. B: TIC chromatogram of wild-type apoC-I in the presence of 5 mM DMPC sampled 40 min after addition of trypsin. The crossed arrow denotes the absence of the peak eluting at 12.3 min in panel A. C: FT-MS spectrum of the tryptic peptide eluting at 12.30 min in panel A. D: IT-MS CID spectrum of the peptide with m/z 1003.5826 shown in panel C. The asterisk indicates the parent ion and the y- and b- fragment ions are for the peptide sequence IKQSELSAK, which corresponds to residues 29–37 of human apoC-I.
Similar comparisons were made immediately following, and at 80, 120, 160, 200, and 240 min after addition of trypsin and GluC. A summary of the LC-MS/MS results obtained are presented in Fig. 3. The amino acid sequence of mature human apoC-I is presented at the top of Fig. 3 with the trypsin and GluC cleavage sites indicated with asterisks in Fig. 3A and Fig. 3C, respectively. Also shown in Fig. 3A and Fig. 3C are the sequence coverages for trypsin and GluC in the absence of phospholipid corresponding to 89% and 100%, respectively. The lower coverage for trypsin is due to the challenge of the mass spectrometer to detect dipeptides under the conditions used. For reference, these correspond to residues 22–23, 49–50, and 51–52 (Fig. 3A). Plotted in Fig. 3B as black and gray bars are the peptide fragments observed immediately upon and at 40, 80, 120, 160, 200, and 240 min after addition of trypsin. Black bars indicate peptides that have been detected in the FT-MS and verified by data-dependent MS/MS sequencing. White spaces signify that peptides were not detected at the indicated time point. Gray bars denote the presence of a peptide with mass matching the appropriate sequence in the FT-MS but not confirmed by MS/MS. This indicates that the ion signal was less than 2% of the total ion abundance and, therefore, was not selected by the software for data-dependent MS/MS. Instead, these peaks were found by manual searches of the known elution regions of each peptide.
Fig. 3.

Summary of limited proteolysis of wild-type apoC-I before and after incubation with DMPC vesicles. Amino acid sequence and position number of mature apoC-I appear at top of figure. A: Asterisks denote the putative trypsin cleavage sites, and black bars indicate the degree of sequence coverage in the absence of lipid. B: Peptides observed by mass spectrometry following incubation of apoC-I in the presence of lipid with trypsin. Data were acquired immediately and at 40, 80, 120, 160, 200, and 240 min after addition of trypsin. Black areas represent peptide sequences of human apoC-I detected by FT-MS and subsequently verified by data-dependent MS/MS. Gray areas represent peptide sequences detected by FT-MS but not of sufficient intensity to be confirmed by subsequent data-dependent MS/MS analyses. White areas represent undetected peptide sequences. C and D: Same as panels A and B, except that Glu C was used instead of trypsin.
CD spectroscopy of apoC-I wild-type and mutants in the absence of lipid
To determine the role F42 and F46 play in mediating interactions of human apoC-I with phospholipid, four mutants containing a single point mutation involving either alanine or glycine substitutions were prepared, namely, apoC-I(F42A), apoC-I(F42G), apoC-I(F46A) and apoC-I(F46G), as well as two mutants containing both point mutations, apoC-I(F42A/F46A) and apoC-I(F42G/F46G). The mutants were either overexpressed in E. coli and subsequently purified in a similar manner to the wild-type protein or prepared by Fmoc peptide synthesis. Mutant proteins were assayed for purity by electrospray ionization mass spectrometry (see supplementary Table I) and/or SDS-PAGE (see supplementary Fig. I).
Circular dichroism spectroscopy was used to compare the degree of secondary structure of the wild-type and mutant apoC-I constructs first in the absence of phospholipid. CD spectra were analyzed using the CDPro program employing the CONTINLL algorithm and SP43 database, which afforded the best fits to the CD spectra generated for the apoC-I wild-type and mutant constructs. In the absence of lipid, wild-type apoC-I comprises 44% α-helix; whereas several of the apoC-I mutants possess significantly lower α-helical structure, ranging from 20% for the apoC-I(F46G) to as high as 39% for apoC-I(F46A) (Table 1).
TABLE 1.
Physical properties of wild-type and mutant apoC-I derivatives before and after incubation with DMPC vesicles
| % α-helix |
% β-strand |
% Turn |
% Unordered |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ApoC-I Construct | Predicted MW (Da) | Measured MW (Da) | - Lipid | + Lipid | - Lipid | + Lipid | - Lipid | + Lipid | - Lipid | + Lipid |
| Wild-type | 6630.6 | 6630.7 | 44 | 84 | 9 | 0 | 18 | 1 | 29 | 16 |
| ApoC-I(F42A) | 6554.5 | 6554.5 | 36 | 85 | 13 | 0 | 20 | 0 | 31 | 15 |
| ApoC-I(F46A) | 6554.5 | 6554.5 | 39 | 89 | 10 | 0 | 20 | 0 | 32 | 11 |
| ApoC-I(F42A/F46A) | 6478.4 | 6478.4 | 29 | 66 | 18 | 2 | 21 | 8 | 33 | 24 |
| ApoC-I(F42G) | 6540.5 | 6540.5 | 25 | 68 | 18 | 2 | 23 | 8 | 34 | 22 |
| ApoC-I(F46G) | 6540.5 | 6540.5 | 20 | 56 | 25 | 6 | 23 | 14 | 32 | 24 |
| ApoC-I(F42G/F46G) | 6450.5 | 6450.5 | 29 | 73 | 17 | 1 | 21 | 4 | 33 | 22 |
ApoC-I, apolipoprotein C-I. The predicted molecular weight (MW) was calculated from the amino acid sequence, while the measured molecular weight was determined by electrospray ionization mass spectrometry (as described in Materials and Methods). Secondary structure content was obtained by analysis of CD spectra using the CONTINLL algorithm and SP43 database within the CDPro program (16). CD spectroscopy was performed with 20 μM apoC-I in the absence and presence of 5 mM DMPC. All percentages derived from CDPro analyses are rounded to the nearest whole number. rmsd 9 0.01 for all CDPro nonlinear least squares best fits.
Phospholipid binding of apoC-I constructs measured by CD spectroscopy
To examine the effect of each alanine or glycine mutation in a more biologically relevant setting, we repeated the CD measurements in the presence of DMPC vesicles. To ensure that apoC-I was completely bound, we titrated the proteins with increasing concentrations of DMPC vesicles and recorded the CD spectrum at each point in the titration. The apoC-I proteins were considered to be completely bound when the addition of DMPC vesicles was no longer accompanied by a change in the CD spectrum. This was achieved at a DMPC concentration in the range from 2.0 to 10.0 mM (Fig. 4, supplementary Figs. II and III). CD spectra of the double glycine mutant, apoC-I(F42G/F46G), in the presence of increasing DMPC concentrations are shown in Fig. 4A, while the comparable data for wild-type apoC-I, apoC-I(F42G), apoC-I(F46G), apoC-I(F42A), apoC-I(F46A), and apoC-I(F42A/F46A) are shown in supplementary Fig. II. One of the most striking features is the progressive decrease in signal at 222 nm, which indicates that the apoC-I constructs become proportionately more α-helical upon binding to the vesicle surface. We note that little, if any, change in ellipticity results above 5 mM DMPC. Using this, we calculate that bound wild-type apoC-I comprises 84% α-helix at 5 mM DMPC (Table 1), which is consistent with previously published values (12, 19). By contrast, apoC-I(F42A), apoC-I(F46A), apoC-I(F42G), apoC-I(F46G), apoC-I(F42A/F46A), and apoC-I(F42G/F46G) are composed of between 56% and 89% α-helix in the presence of saturating DMPC vesicles (Table 1). Although the proportion of α-helix is slightly lower than the wild-type protein for the single glycine mutants, the uniformity of ellipticities of the mutant and wild-type proteins at the endpoint of the titrations suggests that all derivatives share a similar structure in the DMPC-bound state.
Fig. 4.

Circular dichroism analyses of apoC-I in the absence and presence of DMPC vesicles. A: The mean residue ellipticity [θ] is plotted as a function of wavelength for 20 μM apoC-I(F42G/F46G) in the presence of 0 mM DMPC (solid line); 0.04 mM DMPC (solid line + diamond); 0.08 mM DMPC (dashed line); 0.1 mM DMPC (dashed line + square); 0.2 mM DMPC (dash-dot line); 0.5 mM DMPC (dash-dot line + triangle); 1.0 mM DMPC (dotted line); 2.0 mM DMPC (dash-dot-dot line); 5.0 mM DMPC (dash-dot-dot line + circle); and 10 mM DMPC (solid line + XXX). Spectra were acquired after samples had been incubated for 40 min at 30°C. B: The mean residue ellipticity at 222 nm is plotted as a function of increasing DMPC concentration for wild-type apoC-I (closed circles); apoC-I(F42G) (closed triangles); apoC-I(F46G) (open triangles); and apoC-I(F42G/F46G) (open circles). The error bars indicate the standard deviation of triplicate measurements. The solid lines represent the nonlinear least squares best fits to Equation 1. The resulting parameters from the nonlinear least squares analyses are presented in Table 2.
Closer examination of the CD spectra (Fig. 4A, supplementary Fig. II) showed that there are marked differences in the concentration of DMPC vesicles required to elicit the decrease in the CD signal. This is best exemplified by plotting the ellipticity at 222 nm as a function of DMPC concentration (Fig. 4B, supplementary Fig. III). In each case, stepwise addition of DMPC vesicles leads to an apparently hyperbolic decrease in signal. The data were fit to a phenomenological model (Equation 1) to extract the apparent dissociation constant (KDapp, expressed per phospholipid monomer or KDapp* expressed per phospholipid particle) (Table 2). Unsurprisingly, the wild-type system had the lowest KDapp* of 0.89 ± 0.18 μM. By contrast, each of the mutant systems was characterized by higher KDapp* values (Table 2). The single alanine mutants, apoC-I(F42A) and apoC-I(F46A) possess approximately 2-fold weaker affinity for DMPC vesicles, whereas the other mutants showed significantly weaker affinity, ranging from 2.5-fold weaker for apoC-I(F46G) to greater than 11-fold weaker for the double glycine mutant, apoC-I(F42G/F46G) (Table 2). These results suggest that while the mutant apoC-I derivatives have the capacity to adopt comparable secondary structure to wild-type apoC-I once saturated with lipid, they nonetheless possess a significantly lower affinity for the phospholipid surface, particularly when both phenylalanine residues at positions 42 and 46 are mutated.
TABLE 2.
Summary of CD titrations of apoC-I with DMPC vesicles
| ApoC-I Construct | [θ]initial × 103 | [θ]50 × 103 | KDapp (mM) | KDapp* (μM)a |
|---|---|---|---|---|
| Wild-type (recombinant) | −12.47 | −17.10 | 0.30 ± 0.06 | 0.89 ± 0.18 |
| ApoC-I(F42A) | −10.9 | −22.2 | 0.51 ± 0.12 | 1.52 ± 0.36 |
| ApoC-I(F46A) | −11.9 | −23.3 | 0.53 ± 0.15 | 1.58 ± 0.45 |
| ApoC-I(F42A/F46A) | −9.08 | −18.2 | 1.03 ± 0.28 | 3.07 ± 0.84 |
| ApoC-I(F42G) | −7.71 | −16.5 | 1.42 ± 0.20 | 4.24 ± 0.60 |
| ApoC-I(F46G) | −5.58 | −11.8 | 0.75 ± 0.07 | 2.24 ± 0.21 |
| ApoC-I(F42G/F46G) | −6.05 | −13.3 | 3.40 ± 0.41 | 10.1 ± 1.22 |
apoC-I, apolipoprotein C-I; CD, circular dichroism; DMPC, 1,2-dimyristoyl-3-sn-glycero-phosphocholine. CD spectra of 20 μM apoC-I samples were collected before and during titration with DMPC vesicles (Fig. 4A, Supplemental Fig. 2). The observed mean residue ellipticity at 222 nm, [θ]obs was plotted as a function of total DMPC concentration (Fig. 4B, supplementary Fig. III) and fit to Equation 1 to yield best-fit estimates of the apparent dissociation constant per phospholipid monomer (KDapp) and the characteristic ellipticity of the protein when 50% bound ([θ]50). The uncertainty in KDapp (and KDapp*) represents the standard error of the weighted fits of triplicate data sets. R2 9 0.98 was obtained for all nonlinear regression analysis best fits to Equation 1.
Calculated assuming 335 DMPC molecules per particle (24).
Sedimentation studies of apoC-I constructs in the presence of DMPC
To explore the hydrodynamic properties of the apoC-I/DMPC complexes, we conducted a series of sedimentation velocity measurements in the analytical ultracentrifuge. Experiments were initially carried out on the individual protein and lipid components and the resultant data analyzed in terms of continuous size [c(s)] distributions (18). The c(s) distribution for wild-type apoC-I, apoC-I(F46G), apoC-I(F42G/F46G), and DMPC vesicles are presented in Fig. 5A. The protein species all exhibited reasonably narrow distributions characterized by a modal sedimentation coefficient of approximately 1.0 S. The sedimentation velocity profile of the DMPC vesicles appeared broader and, hence, more heterogeneous (Fig. 5A, inset), with major DMPC species noted at approximately 10 S and 20 S.
Fig. 5.

Sedimentation velocity analyses of apoC-I before and after incubation with DMPC vesicles. A: Continuous size [c(s)] distribution of wild-type apoC-I (solid line); apoC-I(F46G) (dotted line); and apoC-I(F42G/F46G) (dashed line) plotted as a function of sedimentation coefficient (Svedberg). (i)–(iii): Residuals (observed data minus predicted data) as a function of radial position corresponding to the wild-type apoC-I, apoC-I(F46G), and apoC-I(F42G/F46G) data sets, respectively. Inset: c(s) distribution of 800 μM DMPC vesicles. Residuals plotted as a function of radial position are presented as a sub-inset. B and (iv)–(vi): Same as panels A and (i)–(iii), except data are for apoC-I/DMPC mixtures. In all cases, continuous size distribution analyses were performed using maximum entropy regularization with a resolution of 100 species and P value of 0.95. The frictional ratio (f/f0) values obtained were less than 1.2. The range of sedimentation coefficients used for protein only and protein/lipid samples was 0.05–12 S. The range of sedimentation coefficients used for the lipid only sample was 0.1–150 S. The resulting root mean square deviation (rmsd) and Runs test Z values obtained for each c(s) analysis are presented in supplementary Table III.
Analogous data for the apoC-I/phopholipid mixtures are presented in Fig. 5B. In the presence of saturating concentrations of DMPC, the modal sedimentation coefficients of the wild-type apoC-I, apoC-I(F46G), and apoC-I(F42G/F46G) species are shifted to values of approximately 5.0 S, 6.5 S, and 8.0 S, respectively, indicating the formation of protein-lipid complexes. A similar increase in modal sedimentation coefficient to that observed for apoC-I(F46G) results for the apoC-I(F42G) and apoC-I(F42A/F46A) mutants in the presence of DMPC (see supplementary Fig. IV). By contrast, the sedimentation velocity profiles of the single alanine mutants apoC-I(F42A) and apoC-I(F46A) are almost identical to the wild-type protein (supplementary Fig. IV). Given that the apoC-I constructs are all of a similar size in the absence of phospholipid and that the same preparation of phospholipid vesicles were used for all measurements, we anticipate the binding capacity of the vesicles to be independent of the particular apoC-I species employed (i.e., complexes should have the same molar mass irrespective of protein). Since the modal sedimentation coefficients of the various apoC-I/DMPC complexes are indeed different, with the exception of the single alanine mutants which were almost identical to the wild-type protein (supplementary Fig. IV), we hypothesize that the differences in sedimentation of the particles result from changes in shape. We sought a method to further characterize the shape of the apoC-I/DMPC complexes.
Transmission electron microscopy (TEM) of apoC-I/DMPC complexes
To facilitate interpretation of our sedimentation velocity data (Fig. 5, supplementary Fig. IV), we examined the apoC-I/phospholipid complexes via TEM. In the absence of apoC-I (Fig. 6A), DMPC forms a heterogeneous mixture of small unilamellar vesicles (SUV) of ∼24 nm in diameter and large unilamellar vesicles (LUV) of ∼60 nm in diameter. Addition of wild-type apoC-I followed by prolonged incubation at 30°C results in a significant change in morphology of the complexes from the previously noted spheres to discoidal particles (Fig. 6B). The most common species observed over the grid was characterized by a long axis of ∼19.7 nm and a short axis of ∼7.7 nm (Table 3), in accordance with previously published data of wild-type apoC-I/phospholipid complexes (5, 20).
Fig. 6.

Transmission electron microscopy of protein/DMPC complexes formed by apoC-I wild-type and glycine mutant constructs. A: Negatively-stained electron micrographs of 800 μM DMPC vesicles. The bar displayed in the bottom left corner represents a distance of 20 nm. B, C, D: Same as panel A, except that samples also contained 20 μM wild-type apoC-I, 20 μM apoC-I(F46G), and 20 μM apoC-I(F42G/F46G), respectively.
TABLE 3.
TEM analysis of protein/DMPC complexes formed by wild-type and mutant apoC-I derivatives
| Sample | Length (L) (nm) | Width (W) (nm) | Ratio (L/W) |
|---|---|---|---|
| Lipid Alone | 33.2 ± 8.8 | 31.2 ± 9.2 | 1.07 |
| ApoC-I wild-type | 19.7 ± 4.0 | 7.7 ± 3.3 | 2.57 |
| ApoC-I(F42A) | 18.7 ± 3.1 | 6.2 ± 1.4 | 3.02 |
| ApoC-I(F46A) | 18.2 ± 3.0 | 7.1 ± 1.6 | 2.56 |
| ApoC-I(F42A/F46A) | 26.4 ± 4.8 | 12.2 ± 3.7 | 2.16 |
| ApoC-I(F42G) | 36.8 ± 8.6 | 19.2 ± 8.3 | 1.92 |
| ApoC-I(F46G) | 28.7 ± 5.1 | 14.3 ± 4.9 | 2.01 |
| ApoC-I(F42G/F46G) | 27.7 ± 5.6 | 15.8 ± 6.0 | 1.75 |
apoC-I, apolipoprotein C-I; DMPC, 1,2-dimyristoyl-3-sn-glycero-phosphocholine; TEM, transmission electron microscopy. The distances across the longest and shortest axis of 100 particles (Fig. 6 and supplementary Fig. V) were measured to determine the average length and average width values ± standard deviation.
By comparison, TEM analysis of DMPC in the presence of the three glycine mutants and the double alanine mutant reveals discoidal particles noticeably different in shape than those formed in the presence of the wild-type protein and the single alanine mutants (Fig. 6, supplementary Fig. V). For apoC-I(F42G), the protein-lipid complexes measured by TEM have a long axis of ∼36.8 nm and a short axis of ∼19.2 nm [Table 3, supplementary Fig V(a)]. By contrast, the corresponding measurements for the apoC-I(F42G/F46G) protein-lipid system are ∼27.7 nm and ∼15.8 nm (Fig. 6D, Table 3), with similar measurements also obtained for the apoC-I(F46G) and apoC-I(F42A/F46A) (Table 3). Based on these results, we can confidently assign discrete species to the sedimentation velocity profiles shown in Fig. 5 and supplementary Fig. IV. We propose that that the spherical DMPC SUVs observed in the absence of protein (Fig. 6A) correspond to the ∼10 S species; the LUVs observed in the absence of protein correspond to the ∼20 S species; the discoidal particles common in the presence of the wild-type or single alanine mutants correspond to the 5.0 S species; and the more oblate spheroids represented by the complexes of DMPC vesicles with the single glycine or double alanine mutant correspond to the 6.5-6.7 S species. The double glycine mutant with a length-to-width ratio of 1.75 represents the 8.0 S species.
DISCUSSION
The initial aim of this study was to identify amino acid residues important in mediating the interaction of human apolipoprotein C-I with phospholipid surfaces. Accordingly, we utilized a LC-MS/MS protease shaving assay employing recombinant human apoC-I and the proteases trypsin and GluC in the absence and presence of DMPC vesicles. Our results demonstrate that the C-terminal region of apoC-I spanning residues 38–51 is protected from proteolytic activity in the presence of phospholipid for significantly longer periods of time when compared with all other regions of the protein (Fig. 3). This suggests that the C-terminal amphipathic helix (Fig. 1) spanning residues 38–52 (10) contains the major lipid-binding determinants for apoC-I. By contrast, the N-terminal region of apoC-I, which includes an amphipathic α-helix spanning residues 7–29 (Fig. 1) (10), is considerably more susceptible to proteolysis in the presence of phospholipid (Fig. 3), suggesting this region possesses lower lipid binding affinity than its C-terminal counterpart. This is consistent with a previous NMR-based study employing SDS micelles, which indicated that the N-terminal helix of apoC-I has lower lipid-binding potential than the C-terminal helix (10). Therefore, we propose that the C-terminal amphipathic helix of apoC-I plays a key role in anchoring the protein to the lipoprotein surface in vivo, while the N-terminal amphipathic helix dissociates more readily from the lipoprotein surface. This has significant implications for the function of apoC-I in lipoprotein metabolism as follows. First, we propose that while the C-terminal region anchors apoC-I to the lipoprotein surface, the N-terminal region of the protein is free to interact with other lipoprotein-bound proteins whose activity is regulated by apoC-I, such as CETP and LCAT (5–7). This is supported by previous studies (21, 22) suggesting that the major amino acid determinants responsible for regulating the activity of CETP (21) and LCAT (22) are located within the N-terminal region of the protein. Second, we propose that the N-terminal region of apoC-I initiates the dissociation of the protein from the lipoprotein particle, thus enhancing the exchangeability of apoC-I; whereas the C-terminal region promotes binding to the lipoprotein surface. This is also consistent with previous structural studies that demonstrate the N-terminal helix of apoC-I is more flexible than the C-terminal region (10).
Interestingly, the C-terminal helix of apoC-I contains three aromatic residues, namely W41, F42, and F46, which form a hydrophobic cluster (6, 10). Previous studies by Gursky (6) demonstrated that mutation of tryptophan 41 to proline or alanine significantly attenuates the helical structure of apoC-I. However, prior to this study it was unclear what contribution F42 and F46 make to the structural and functional properties of apoC-I. We decided to mutate F42 and F46. We generated four single mutants with glycine or alanine at position 42 and 46, and we generated two double mutants with glycine or alanine at both positions. Not surprisingly, CD spectroscopy analyses in lipid-free solution demonstrate that apoC-I(F42G), apoC-I(F46G), apoC-I(F42A/F46A), and apoC-I(F42G/F46G) show approximately 2-fold reduction in α-helical content compared with the wild-type protein (Table 1). This is similar to the results observed for the apoC-I(W41P) and apoC-I(W41A) mutants previously characterized by Gursky (6). By comparison, the mutants apoC-I(F42A) and apoC-I(F46A) show only slight reductions in α-helical content, which indicates that single phenylalanine-to-alanine substitutions can be tolerated at positions 42 and 46 without excessively perturbing the overall helical nature of the protein. In addition, phospholipid vesicle binding assays measured by CD spectroscopy show that the single alanine and glycine mutants as well as the double alanine mutant possess 2- to 3.5-fold reduced binding affinity for DMPC vesicles compared with wild-type apoC-I (Table 2). By contrast, the double glycine mutant possesses over 11-fold weaker affinity for DMPC vesicles compared with the wild-type protein, and 2.3- to 4.5-fold lower affinity than the single glycine mutants (Table 2). These results suggest that the effect of the glycine point mutations at positions 42 and 46 may be additive with respect to phospholipid binding potential. If this is the case, we would expect that the sum of the differences in ΔG° of the single glycine mutants compared with the wild-type protein (i.e., ΔΔG° values) would approximate the ΔΔG° value for the double glycine mutant. Table 4 shows that this is indeed the case. Moreover, the ΔΔG° for apoC-I(F42G/F46G) of –6.0 kJ/mol is similar to the sum of ΔΔG° values for the single glycine mutants, which is calculated to be –6.2 kJ/mol (Table 4). Overall, these results suggest that F42 and F46 play a significant role in mediating helix stability and lipid-binding function of human apoC-I. Our studies are supported by earlier studies that show aromatic residues are important in promoting binding to phospholipid surfaces (13, 23). The study by Sanderson and Whelan (23) indicates that tryptophan and tyrosine residues bind to the phosphate and choline head groups of phospholipids, whereas phenylalanine residues interact with the acyl chains. It is plausible that F42 and F46 of human apoC-I interact with the acyl chains within the phospholipid monolayer of lipoproteins in vivo.
TABLE 4.
ΔΔG°analyses for the binding of wild-type and glycine mutants of apoC-I to DMPC vesicles
| ApoC-I Construct | KDapp (mM)a | ΔG° (kJ/mol)b | ΔΔG° (kJ/mol)c |
|---|---|---|---|
| Wild-type | 0.30 ± 0.06 | −20.1 ± 0.5 | N/A |
| ApoC-I(F42G) | 1.42 ± 0.20 | −16.2 ± 0.3 | −3.9 ± 0.6 |
| ApoC-I(F46G) | 0.75 ± 0.07 | −17.8 ± 0.2 | −2.3 ± 0.5 |
| ApoC-I(F42G/F46G) | 3.40 ± 0.41 | −14.1 ± 0.3 | −6.0 ± 0.6 |
apoC-I, apolipoprotein C-I; DMPC, 1,2-dimyristoyl-3-sn-glycero-phosphocholine. Theoretical ΔΔG° if the two single mutations were additive is –6.2 ± 0.8 kJ/mol.
Uncertainties represent the standard error of the parameter estimate.
Uncertainties are propagated from the original error in a.
Uncertainties are propagated from the error in b.
One of the most exciting results in this study was the finding that the shape of the protein/phospholipid complexes formed by wild-type and glycine and double alanine mutants of apoC-I were markedly different (Figs. 5 and 6). Table 3 reports the average length-to-width ratio of the discoidal particles formed by these apoC-I constructs. The wild-type/DMPC complex adopts a significantly asymmetric shape both in solution and on the TEM grid, with a length-to-width ratio of 2.57 and a modal sedimentation coefficient of 5.0 S (Figs. 5 and 6, Table 3). Similar results are also obtained for the apoC-I(F42A) and apoC-I(F46A) mutants (Table 3), which again indicates that single alanine mutations at either position 42 and 46 can be tolerated in the C-terminal amphipathic helix without altering the morphology of the resulting complex. By comparison, the single glycine mutants and the double alanine mutant form complexes with DMPC that have a length-to-width ratio of 1.92-2.16 and a modal sedimentation coefficient of 6.5-6.7 S; whereas the double glycine mutant is appreciably more symmetrical with a length-to-width ratio of 1.75 and modal sedimentation coefficient of 8.0 S. By contrast, earlier work by Mehta et al. (12) shows that protein/DMPC complexes formed by apoC-I constructs with mutations in the N-terminal region, namely at positions 23, 31, and 34, adopt similar shapes to those formed by the wild-type protein. The data presented in this study (Figs. 5 and 6) together with the results described by Mehta et al. (12) indicate a major role for F42 and F46 of apoC-I in promoting the formation of asymmetric discoidal particle morphology, whereas R23, Q31, and L34 within the N-terminal region of apoC-I have no effect on remodeling particle shape.
In summary, this study demonstrates that the C-terminal amphipathic α-helix of human apoC-I contains the major lipid-binding determinants, including important aromatic residues F42 and F46, which we show play a critical role in stabilizing the structure of apoC-I, mediating phospholipid interactions, and promoting discoidal particle morphology.
Supplementary Material
Acknowledgments
The authors thank the Australian National Heart Foundation, the Australian National Health and Medical Research Council, Australian Research Council (ARC), the Victorian Institute for Chemical Sciences, and the ARC Centre of Excellence for Free Radical Chemistry and Biotechnology for their support.
Abbreviations
apoC-I, apolipoprotein C-I
CD, circular dichroism
CETP, cholesterol ester transfer protein
CID, collision induced dissociation
DMPC, 1,2-dimyristoyl-3-sn-glycero-phosphocholine
EM, electron microscopy
ESI, electrospray ionization
FT, Fourier transform
IDL, intermediate density lipoprotein
IT, ion trap
LB, Luria-Bertani
LC, liquid chromatography
LUV, large unilamellar vesicle
MBP, maltose binding protein
MS, mass spectrometry
MS/MS, tandem mass spectrometry
rmsd, root mean square deviation
SUV, small unilamellar vesicle
TAE, Tris-acetate-EDTA buffer
This work was supported by a scholarship from the Australian National Heart Foundation (P.F.J.); C.J. Martin Fellowship 145843 from the Australian National Health and Medical Research Council (M.F.B.); a post-doctoral fellowship and financial support from the Australian Research Council (ARC) (M.A.P.); and financial support from the ARC Centre of Excellence for Free Radical Chemistry and Biotechnology (R.A.J.O.). An ARC LIEF grant and funding from the Victorian Institute for Chemical Sciences are acknowledged for the purchase of the LTQ-FTMS (R.A.J.O.).
Published, JLR Papers in Press, November 4, 2008.
Footnotes
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of two tables and five figures.
References
- 1.Lauer S. J., D. Walker, N. A. Elshourbagy, C. A. Reardon, B. Levywilson, and J. M. Taylor. 1988. Two copies of the human apolipoprotein C–I gene are linked closely to the apolipoprotein E-gene. J. Biol. Chem. 263 7277–7286. [PubMed] [Google Scholar]
- 2.Schaefer E. J., S. Eisenberg, and R. I. Levy. 1978. Lipoprotein apoprotein metabolism. J. Lipid Res. 19 667–687. [PubMed] [Google Scholar]
- 3.Windler E., and R. J. Havel. 1985. Inhibitory effects of C apolipoproteins from rats and humans on the uptake of triacylglyceride-rich lipoproteins and their remnants by the perfused rat liver. J. Lipid Res. 26 556–565. [PubMed] [Google Scholar]
- 4.Weisgraber K. H., R. W. Mahley, R. C. Kowal, J. Herz, J. L. Goldstein, and M. S. Brown. 1990. Apolipoprotein C–I modulates the interaction of apolipoprotein E with β-migrating very low density lipoproteins (β-VLDL) and inhibits binding of β-VLDL to low density lipoprotein receptor-related protein. J. Biol. Chem. 265 22453–22459. [PubMed] [Google Scholar]
- 5.Jonas A., S. A. Sweeny, and P. N. Herbert. 1984. Discoidal complexes of A and C apolipoproteins with lipids and their reactions with lecithin: cholesterol acyltransferase. J. Biol. Chem. 259 6369–6375. [PubMed] [Google Scholar]
- 6.Gursky O. 2001. Solution conformation of human apolipoprotein C-1 inferred from proline mutagenesis: far- and near-UV CD study. Biochemistry. 40 12178–12185. [DOI] [PubMed] [Google Scholar]
- 7.Gautier T., D. Masson, J. P. de Barros, A. Athias, P. Gambert, D. Aunis, M. H. Metz-Boutigue, and L. Lagrost. 2000. Human apolipoprotein C–I accounts for the ability of plasma high density lipoproteins to inhibit the cholesterol ester transfer protein activity. J. Biol. Chem. 275 37504–37509. [DOI] [PubMed] [Google Scholar]
- 8.Osborne J. C., T. J. Bronzert, and H. B. Brewer. 1977. Self-association of apo-C-I from human high-density lipoprotein complex. J. Biol. Chem. 252 5756–5760. [PubMed] [Google Scholar]
- 9.Gursky O., and D. Atkinson. 1998. Thermodynamic analysis of human plasma apolipoprotein C-1: high-temperature unfolding and low-temperature oligomer dissociation. Biochemistry. 37 1283–1291. [DOI] [PubMed] [Google Scholar]
- 10.Rozek A., J. T. Sparrow, K. H. Weisgraber, and R. J. Cushley. 1999. Conformation of human apolipoprotein C–I in a lipid-mimetic environment determined by CD and NMR spectroscopy. Biochemistry. 38 14475–14484. [DOI] [PubMed] [Google Scholar]
- 11.Atcliffe B. W., C. A. MacRaild, P. R. Gooley, and G. J. Howlett. 2001. The interaction of human apolipoprotein C–I with sub-micellar phospholipid. Eur. J. Biochem. 268 2838–2846. [DOI] [PubMed] [Google Scholar]
- 12.Mehta R., D. L. Gantz, and O. Gursky. 2003. Effects of mutations in apolipoprotein C-1 on the reconstitution and kinetic stability of discoidal lipoproteins. Biochemistry. 42 4751–4758. [DOI] [PubMed] [Google Scholar]
- 13.Wang G., G. K. Pierens, W. D. Treleaven, J. T. Sparrow, and R. J. Cushley. 1996. Conformations of human apolipoprotein E(263–286) and E(267–289) in aqueous solutions of sodium dodecyl sulfate by CD and 1H NMR. Biochemistry. 35 10358–10366. [DOI] [PubMed] [Google Scholar]
- 14.Laue, T. M., B. D. Shah, T. M. Ridgeway, and S. L. Pelletier. 1992. Computer-aided interpretation of analytical sedimentation data for proteins. The Royal Society of Chemistry, Cambridge.
- 15.James P. F., M. A. Perugini, and R. A. J. O'Hair. 2006. Sources of artefacts in the electrospray ionization mass spectra of saturated diacylglycerophosphocholines: from condensed phase hydrolysis reactions through to gas phase intercluster reactions. J. Am. Soc. Mass Spectrom. 17 384–394. [DOI] [PubMed] [Google Scholar]
- 16.Sreerama N., and R. W. Woody. 2000. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287 252–260. [DOI] [PubMed] [Google Scholar]
- 17.Schuck P. 1998. Sedimentation analysis of non-interacting and self-associating solutes using numerical solutions to the Lamm equation. Biophys. J. 75 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjwal S., S. Jayaraman, and O. Gursky. 2007. Role of secondary structure in protein-phospholipid surface interactions: reconstitution and denaturation of apolipoprotein C–I:DMPC complexes. Biochemistry. 46 4184–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gursky O., Ranjana, and D. L. Gantz. 2002. Complex of human apolipoprotein C–I with phospholipid: thermodynamic or kinetic stability. Biochemistry. 41 7373–7384. [DOI] [PubMed] [Google Scholar]
- 21.Kushwaha R. S., H. C. McGill, Jr., and F. H. Hausheer. 2004. Effect of synthetic truncated apolipoprotein C-I peptide on plasma lipoprotein cholesterol in nonhuman primates. J. Biomed. Biotechnol. 4 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soutar A. K., G. F. Sigler, L. C. Smith, A. M. Gotto, Jr., and J. T. Sparrow. 1978. Lecithin:cholesterol acyltransferase activation and lipid binding by synthetic fragments of apolipoprotein C–I. Scand. J. Clin. Lab. Invest. Suppl. 150 53–58. [PubMed] [Google Scholar]
- 23.Sanderson J. M., and E. J. Whelan. 2004. Characterisation of the interactions of aromatic amino acids with diacetyl phosphatidylcholine. Phys. Chem. Chem. Phys. 6 1012–1017. [Google Scholar]
- 24.Denisov I. G., M. A. McLean, A. W. Shaw, Y. V. Grinkova, and S. G. Sligar. 2005. Thermotropic phase transition in soluble nanoscale lipid bilayers. J. Phys. Chem. B. 109 15580–15588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong L. M., and K. H. Weisgraber. 1996. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J. Biol. Chem. 271 19053–19057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.