

Abstract
We describe the synthesis and biological evaluation of a series of tubulin polymerization inhibitors that contain the 1,2,4-triazole ring to retain the bioactive configuration afforded by the cis double bond in combretastatin A-4 (CA-4). Several of the subject compounds exhibited potent tubulin polymerization inhibitory activity as well as cytotoxicity against a variety of cancer cells including multi-drug-resistant (MDR) cancer cell lines. Attachment of the N-methyl-5-indolyl moiety to the 1,2,4-triazole core, as exemplified by compound 7, conferred optimal properties among this series. Computer docking and molecular simulations of 7 inside the colchicine binding site of tubulin enabled identification of residues most likely to interact strongly with these inhibitors and explain their potent anti-tubulin activity and cytotoxicity. It is hoped that results presented here will stimulate further examination of these substituted 1,2,4-triazoles as potential anti-cancer therapeutic agents.
Introduction
Given their essential role in the growth and function of cells, microtubules are among the most important molecular targets for cancer chemotherapeutic agents.1 The formation of microtubules is a dynamic process that involves the polymerization and depolymerization of α and β tubulin heterodimers. Molecules binding to tubulins interfere with this dynamic equilibrium and thus induce cell cycle arrest, resulting in cell death.2 Three ligand binding sites have been identified in tubulins: the taxane,3 vinca alkaloid,4 and colchicine sites.5 Combretastatin A-4 (CA-4,a Chart 1), a natural product isolated from the South African bush willow tree Combretum caffrum, binds to the colchicine binding site and inhibits the polymerization of microtubules.6 CA-4 has been reported to exhibit potent cytotoxicity against a broad range of cancer cells including multi-drug-resistant (MDR) cell lines.7,8 However, CA-4 failed to show in vivo efficacy against murine colon 26 adenocarcinoma, partly due to its poor water solubility.9 A disodium phosphate derivative of CA-4 (CA-4P) has shown promising results in human cancer clinical trials,10 thus stimulating significant interest in a variety of CA-4 analogues.
Chart 1.

Extensive studies have been conducted to examine the structure–activity relationship (SAR) of CA-4 and its analogues. The trimethoxy substitutions on the A ring and the cis-olefin configuration at the bridge have been reported as prerequisites for potent cytotoxicity, while the B ring is tolerant of structural modifications (Figure 1).11 To retain the cis-olefin configuration of CA-4, alternative bridge groups have been introduced into CA-4 to replace the double bond. Examples of nonheterocyclic bridges are ethers, olefins, ketones, sulfonamides, sulfonates, amine, amide derivatives, and cyclopentanes.11,12 Heterocyclic bridges include five-membered rings (e.g., pyrazoles, thiazoles, triazoles, tetrazoles, oxazoles, imidazoles, furans, furanones, furazans, dioxolanes, and thiophenes)11,13 and indoles.14,15 Structural modifications on the B ring suggest that the 4-methoxy group is crucial for cytotoxicity, while the 3-hydroxy group is optional.16–18 Strategies have been pursued to improve the cytotoxity and physicochemical properties of these compounds. For example, Wang et al.19 reported a series of CA-4 analogues containing five-membered ring bridges and the N-methylindole as the B ring (1 and 2 in Chart 1) that possess potent cytotoxicity and improved bioavailability comparable to CA-4.
Figure 1.

Known SAR of CA-4 analogues.
In our efforts to discover novel tubulin polymerization inhibitors as potential anti-cancer chemotherapeutic agents, we selected the 1,2,4-triazole ring to retain the cis-olefin configuration of CA-4. Our underlying premise was that the triazole ring would augment bioavailability and chemical stability and, at the same time, maintain the in vitro and in vivo efficacy of CA-4. Additionally, insertion of N-methylindole as the B ring would yield potent cytotoxicity and tubulin polymerization inhibition. Here we report the synthesis and in vitro cytotoxicity of a series of novel triazole-based tubulin polymerization inhibitors (Chart 1, 3−9).
Chemistry
Compounds 3−8 (Chart 1) were synthesized starting from amides that were obtained by coupling of 3,4,5-trimethoxybenzoic chloride with different amines (Scheme 1). The amides were converted to thioamides by reaction with different thionation reagents. Use of P4S10 as a thionation reagent gave reactions that were very slow at temperatures below 60 °C and failed to yield the desired product above 60 °C. Curphey's method in combination with P4S10 and hexamethyldisiloxane was found to generate thioamides in quantitative yield with sufficient purity so that the products could be used directly for the next step.20 This reaction is highly versatile and proceeds smoothly with most groups as the B ring, aside from basic groups such as indole. In the latter case, Lawesson's reagent was used to synthesize the thioamides. With thioamides in hand, amidrazones could be synthesized smoothly by reaction with anhydrous hydrazine at 0 °C. The final cyclization step was performed with trimethyl orthoformate under acidic conditions. Reduction of the appropriate carbon-carbon double bond in 7 by sodium cyanoborohydride (NaCNBH3) produced indoline 8 in nearly quantitative yield.
Scheme 1a.

Our attempts to synthesize 9 by the coupling of acid and amine via Scheme 1 were unsuccessful, likely due to the strong deactivating effect of the three methoxyl groups on aniline. Success was finally achieved via the alternative reaction pathway depicted in Scheme 2. The key intermediate, thioamide, was synthesized by the coupling of the indole Grignard reagent with 5-isothiocyanato-1,2,3-trimethoxybenzene. The Grignard reagent was obtained via standard methods or by metal exchange reaction between iodide and isopropyl magnesium chloride.21–23
Scheme 2a.

All amines used in the present synthesis were obtained through conversion of the respective nitro precursor by catalytic hydrogenation reaction. For compounds 7 and 8, additional N-methylation reactions were performed with methyl iodide as the alkylation reagent (Scheme 1).
Results and Discussion
Compounds 3−7 (Chart 1) were submitted for in vitro biological testing against the ∼60 cancer cell line screen provided by the National Cancer Institute–Developmental Therapeutics Program (NCI–DTP). The NCI–DTP screening procedure is described in detail elsewhere (http://dtp.nci.nih.gov/).24,25 The results, summarized in Table 1, reveal that compounds 3−7 exhibited appreciable growth inhibition (log10 GI50 < −4) against nearly all of the cancer cell lines reported by NCI–DTP. As expected, the subject compounds exhibited variable growth inhibition against different cancer cell lines of the same tumor type (Supporting Information, Table 1). For example, compound 7 exhibited very strong growth inhibition (log10 GI50 < −7.00) against lung cancer cell lines A549/ATCC, EKVX, HOP-62, HOP-92, NCI-H23, NCI-H322M, NCI-H460, and NCI-H522, whereas it showed only weak inhibition against the lung cancer cell line NCI-H226 (log10 GI50 = −4.07). The same phenomenon was observed for ovarian cancer and melanoma cell lines. Among compounds 3−7, 7 exhibited overall the most potent cytotoxicity against the NCI–DTP cell lines. The present results are consistent with those of Wang et al.,19 who found that insertion of the N-methyl-5-indolyl moiety as the B ring (Chart 1) was superior to 4-aminophenyl (4) and 3-hydroxyl-4-methoxyphenyl (5) groups.
Table 1.
Summary of the NCI–DTP 60-Cell-Line Screening for Compounds 3−7
| compd | no. of cell linesa | no. of cell lines with log10 GI50 < −4b | range of log10 GI50 |
|---|---|---|---|
| 3 | 50 | 49 | −4.43 to −6.63 |
| 4 | 53 | 52 | −4.00 to < −8.00 |
| 5 | 55 | 53 | −4.04 to −7.61 |
| 6 | 54 | 51 | −4.05 to −6.48 |
| 7 | 55 | 55 | −4.07 to < −8.00 |
Cell lines for which results were reported by NCI–DTP.
GI50 refers to growth inhibition of 50%.
To quantify the cytotoxicity profile of these compounds more precisely, MTT assays were conducted against several cancer cell lines: colon cancer HCT-116, breast cancer ZR-75−1, cervical cancer HeLa and KB-3−1, and P-glycoprotein-overexpressing multi-drug-resistant (MDR+) cancer cell line KB-V1 (Table 2). The cytotoxicity against these cell lines was 2−10-fold higher with 4-dimethylamino substitution (4) compared with 4-methoxy (3) on the B ring (Chart 1). Introduction of hydroxyl at the 3-position of the B ring (5) produced cytotoxicity comparable to that of 4. However, fluorine substitution at the 3-position (6) failed to improve the cytotoxicity profile. Compound 7, with N-methyl-5-indolyl as ring B, consistently showed very potent cytotoxicity (IC50 < 24 nM) against the cell lines. The cytotoxicity of 7 is comparable to that of colchicine but slightly weaker than that of CA-4 (Table 2).
Table 2.
Cytotoxicity and Tubulin Polymerization Inhibition Effects of Subject Compounds
| cytotoxicity IC50, nM |
||||||
|---|---|---|---|---|---|---|
| compd | HCT-116 (colon) | ZR-75−1 (breast) | HeLa (cervix) | KB-3−1 (cervix) | KB-V1a (cervix) | anti-tubulin activity IC50, μM |
| 3 | 230 | 522 | 290 | 373 | 907 | NDb |
| 4 | 91.5 | 308 | 29.6 | 119 | 323 | 4.1 |
| 5 | 88.9 | 141 | 25 | 57.3 | 544 | 6.0 |
| 6 | 365 | 367 | 173 | 399 | 1479 | ND |
| 7 | 7.39 | 23.8 | 8.73 | 9.9 | 20.8 | 3.0 |
| 8 | 3288 | ND | 4097 | ND | ND | >10 |
| 9 | 24.3 | ND | 18.0 | 15.0 | 32.1 | 5.6 |
| CA-4 | 0.35 | 0.24 | 0.30 | 0.78 | 0.64 | 5.8 |
| colchicine | 2.75 | 522 | 1.84 | 3.99 | 620 | ND |
| paclitaxel | ND | 308 | ND | 23.3 | >10 000 | ND |
Multiple-drug-resistant positive (MDR+) cancer cell line.
ND, not determined.
The tubulin assembly assay suggested that these new derivatives function as tubulin polymerization inhibitors (Table 2). The inhibition potency on tubulin polymerization of 4, 5, 7, and 9 is similar to that of CA-4. The cytotoxicity is lower for these compounds than for CA-4, suggesting that the latter compound may exert its cytotoxic effects through additional targets besides tubulin. Compound 7 yielded the most potent anti-tubulin polymerization activity and cytotoxicity against the cancer cell lines, in concordance with the NCI–DTP screening results.
Compound 8, in which one double bond in the indole ring of 7 is reduced (Chart 1), lacked activity against the two cell lines, suggesting that the aromaticity and/or planarity of the B ring is critical for the potent activity. Quantum mechanical calculations confirmed that the absence of this double bond in 8 renders the B ring nonplanar (see Experimental Section). Compound 9 (Chart 1), in which the substitution pattern with respect to the triazole ring is switched relative to 7, demonstrated potent anti-tubulin polymerization activity (5.6 μM) and cytotoxicity (∼20 nM) against the cell lines. Compounds 7 and 9 also exhibited potent cytotoxicity (21 and 32 nM, respectively) against MDR+ cancer cell line KB-V1, indicating that they are not substrates of the MDR efflux pump. In contrast, the inhibitory activity of both colchicine and paclitaxel diminished substantially against KB-V1, suggesting that they are MDR substrates. Association with MDR leads to diminished intracellular drug concentrations, thereby reducing or even abolishing cytotoxicity.26,27
The high-resolution crystal structure (resolution = 3.58 Å) of the tubulin–colchicine–stathmin-like domain complex revealed the location of the colchicine binding site.28 We then proceeded to examine the interactions of the subject compounds with tubulin. Adopting the conformation found in its X-ray crystal structure (Figure 2), 7 was computer docked inside the colchicine binding site of tubulin. Molecular dynamics (MD) simulations on the tubulin–7 complex indicated that 7 interacts extensively with β tubulin but with only two residues (Ala α180 and Val α181) in α tubulin via hydrophobic interactions (Figure 3, upper panel), which is consistent with the fact that the colchicine binding site is largely buried within β tubulin.28
Figure 2.

X-ray crystal structure of compound 7. Atoms are depicted as 50% thermal probability ellipsoids.
Figure 3.
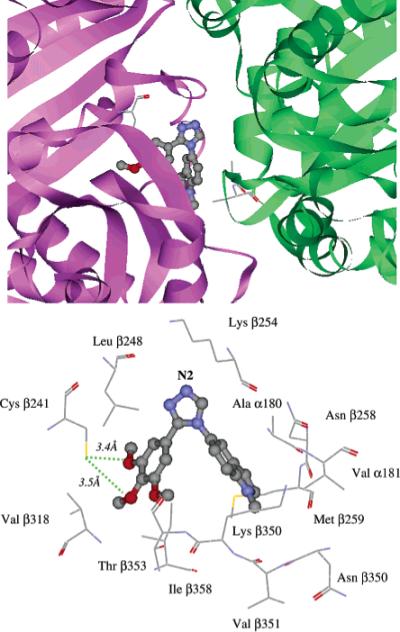
Binding conformation of compound 7 docked inside colchicine binding site of tubulin: perspective view (upper panel) and close-up view (lower panel) of bound ligand inside tubulin binding site. Compound 7 is rendered in ball-and-stick mode and colored by atom type. In the upper panel, tubulin is displayed as a flat ribbon with α-tubulin colored green and β-tubulin colored magenta. Residues Ala α180, Val α181, and Cys β239 are displayed in stick mode. In the lower panel, the residues interacting with the ligand are rendered in stick mode and labeled. Hydrogen bonds are depicted as green dotted lines. The triazole N2 atom is labeled accordingly.
More detailed analysis of the inhibitor–tubulin complex (Figure 3, lower panel) revealed several key interactions that appear to play a role in the binding of 7. Two methoxy O atoms on the A ring are located within hydrogen-bond distances (3.4 and 3.5 Å) of the thiol group of Cys β241. The trimethoxyphenyl moiety occupies a pocket bounded by Leu β248, Val β318, Thr β353, and Ile β358, while the indolic ring of 7 is embedded in a hydrophobic pocket consisting of the side chains of residues of Ala α180, Val α181, Lys β254, Asn β258, Met β259, Val β351, and Lys β352. The protonated side-chain N atom of residue Lys β254 remained in close proximity (≤5 Å) to the triazole N2 atom (Figure 3, lower panel) for 180 ps of the entire 200 ps MD run. Similarly, Nguyen et al.29 proposed that residues within the loop region of nearby Lys β254 formed hydrogen bonds with known colchicine site inhibitors (e.g., podophyllotoxin and methylchalcone). Also consistent with the results of both Nguyen et al.29 and Ravelli et al.,28 our MD simulations indicated that the side-chain N atom of Lys β254 forms hydrogen bonds with the phosphate O atoms of R tubulin-bound GTP. Taken together, these observations suggest that the triazole ring in the subject compounds interacts with the Lys β254 side chain via attractive contacts with the triazole's N2 atom (Figure 3, lower panel).
Conclusion
We have synthesized and biologically evaluated a series of novel tubulin polymerization inhibitors that contain a core 1,2,4-triazole ring to retain the cis configuration required for bioactivity. Attachment of the N-methyl-5-indolyl moiety to the triazole ring (e.g., 7 and 9) conferred optimal bioactivity. These compounds exhibited potent tubulin polymerization inhibitory activity and cytotoxicity against a variety of cancer cells including MDR cancer cell lines. Molecular docking and dynamics simulation were performed to study the inhibitor–protein interactions. Analysis of the inhibitor binding conformation in the colchicine binding site revealed specific residues that may play an important role in tubulin polymerization inhibitory activity and cytotoxicity. We are presently utilizing insights gleaned from these molecular modeling studies to design new tubulin inhibitors with strong affinity, potent tubulin inhibitory activity, and high cytotoxicity.
Experimental Section
1H NMR spectra were recorded at 400 MHz on a Varian Gemini-400 spectrometer and in deuterated chloroform (CDCl3) or dimethyl sulfoxide (DMSO-d6) solution at room temperature. Tetramethylsilane (TMS, 0.00 ppm) was used as the internal standard, and the spectra were reported in parts per million (ppm). Analytical thin-layer chromatography (TLC) was performed on precoated plastic-backed plates purchased from Aldrich (silica gel 60 F254; 0.25 mm thickness). Flash column chromatography was conducted with silica gel 60 (230−400 mesh) (Natland Co.). Standard (MS) and tandem (MS–MS) mass spectrometry were conducted on a Finnigan LCQ Duo mass spectrometer (Thermo Quest Co.). Melting points were taken on a Mel-temp melting point apparatus in open capillary tubes without calibration.
All reactions were carried out with anhydrous solvents in oven-dried and argon-charged glassware. All anhydrous solvents except as mentioned were freshly distilled and stored in 4 Å molecular sieves. All solvents used in workup were used as received from the commercial supplier without further purification. All reagents were purchased from Aldrich Chemical Co., St. Louis, MO.
General Procedure for the Synthesis of 3−7
3,4,5-Trimethoxybenzoyl chloride was added slowly to 1.0 equiv of amine solution, which was dissolved in CHCl3 and mixed with 1.0 equiv of triethylamine at 0−5 °C. After the reaction was complete, the CHCl3 solution was extracted three times by water and concentrated under vacuum. The resulting amide, which precipitated out immediately when the concentrated syrup was cooled to rt, possessed sufficient purity for direct use in the next step.
The amide was dissolved in anhydrous CHCl3, and 0.16 equiv of P4S10 and 1.67 equiv of hexamethyldisiloxane were introduced [for 7, 1.05 equiv of Lawesson's reagent was added at rt]. The solution was heated to 62 °C under inert gas. The CHCl3 solution slowly became yellow in color. After the reaction was complete, the product was purified by extraction by water and then concentrated. Gas chromatography–mass spectrometry (GC–MS) indicated thioamides synthesized by this method were sufficiently pure to be used directly for the next step. For 7, the thioamide was purified by column chromatography with EtOAc and hexane as eluent.
The yellow thioamide was suspended in anhydrous alcohol under inert gas atmosphere. The reactants were cooled to 0 °C, and 5.0 equiv of anhydrous hydrazine was added slowly with vigorous stirring. The mixture was stirred at rt overnight. Alcohol and excess hydrazine were evaporated under vacuum, and the syrup was dissolved in CHCl3 and extracted by water three times. The organic layer was dried and concentrated under vacuum to yield amidrazone as a white or off-white solid.
The amidrazone was dissolved in alcohol and 5.0 equiv of trimethyl orthoformate was added at rt. Several drops of sulfuric acid were added as catalyst, and the solution was stirred vigorously for 2 h. After neutralization, the alcohol was evaporated, and the triazole was purified by column chromatography.21
4-(4-Methoxyphenyl)-3-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (3)
Overall yield 86%; mp 163−165 °C; 1H NMR (400 MHz, CDCl3) δ 3.64 (s, 6H), 3.83 (s, 3H), 3.84 (s, 3H), 6.70 (s, 2H), 6.98 (d, J = 2.0 Hz, 2H), 7.19 (d, J = 3.8 Hz, 2H), 8.25 (s, 1H); MS (ESI) m/z = 342.1 (M+ + 1); Anal. (C18H19O4N3) C, H, O, N.
N,N-Dimethyl-4(3-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazol-4-yl)benzenamine (4)
Overall yield 81%; mp 145−147 °C; 1H NMR (400 MHz, CDCl3) δ 2.99 (s, 6H), 3.65 (s, 6H), 3.83 (s, 3H), 6.71 (d, J = 3.8 Hz, 2H), 6.76 (s, 2H), 7.09 (d, J = 6.0 Hz, 2H), 8.22 (s, 1H); MS (ESI) m/z = 355.2 (M+ + 1); Anal. (C19H20O3N4) C, H, O, N.
2-Methoxy-5-[3-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazol-4-yl]phenol (5)
Overall yield 83%; mp 197−198 °C; 1H NMR (400 MHz, CDCl3) δ 3.66 (s, 6H), 3.83 (s, 3H), 3.94 (s, 3H), 6.18 (s, 1H), 6.74 (m, 3H), 6.89 (m, 2H), 8.24 (s, 1H); MS (ESI) m/z = 358.3 (M+ + 1); Anal. (C18H19O5N3) C, H, O, N.
4-(3-Fluoro-4-methoxyphenyl)-3-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazole (6)
Overall yield 88%; mp 151−152 °C; 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 6H), 3.89 (s, 3H), 3.93 (s, 3H), 3.69 (s, 2H), 7.05 (m, 3H), 8.25(s, 1H); MS (ESI) m/ z = 360.3 (M+ + 1); Anal. (C18H18O4N3F) C, H, O, N, F.
1-Methyl-5-[3-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazol-4-yl]-1H-indole (7)
Overall yield 69%; mp 157−159 °C; 1H NMR (400 MHz, CDCl3) δ 3.52 (s, 6H), 3.80 (s, 3H), 3.84 (s, 3H), 6.51 (s, 1H), 6.73 (s, 2H), 7.04 (d, 1H), 7.17 (s, 1H), 7.37 (d, J = 1.6 Hz, 1H), 7.559 (s, 1H), 8.30 (s, 1H); MS (ESI) m/z = 365.3 (M+ + 1); Anal. (C20H20O3N4) C, H, O, N.
1-Methyl-5-[3-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazol-4-yl]-indoline (8)
Compound 7 (25 mg) was dissolved in 2 mL of HOAc at rt. NaCNBH3 (1.5 equiv) was added in one portion while stirring, and the solution was stirred overnight. TLC indicated that the reaction was complete. The acidic solution was neutralized by adding NaOH solution dropwise and extracted by CHCl3. After concentration, the residue was purified by column chromatography with CHCl3/MeOH (20:1) as eluent to obtain 20 mg of 7 as a white solid: mp 158−160 °C; 1H NMR (400 MHz, CDCl3) δ 2.83 (s, 3H), 3.00 (t, J = 8.0 Hz, 2H), 3.49 (t, J = 9.0 Hz, 2H), 3.67 (s, 6H), 3.87 (s, 3H), 6.46 (d, J = 8.0 Hz, 1H), 6.75 (s, 1H), 6.92 (s, 1H), 7.01 (d, J = 7.0 Hz, 1H), 8.65 (s, 1H); MS (ESI) m/z = 367.3 (M+ + 1); Anal. (C20H22O3N4) C, H, O, N.
1-Methyl-5-[4-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazol-3-yl]-1H-indole (9)
5-Iodo-1-methyl-1H-indole (1.0 g, 3.89 mmol) was dissolved in THF (10 mL) and cooled to −10 °C. To the solution was added isopropylmagnesium chloride (2.5 mL, 2.0 M in THF), and the reaction mixture was kept at −10 °C for 30 min. 5-Isothiocyanato-1,2,3-trimethoxybenzene (1.0 equiv) was added dropwise, and the resulting brown solution was stirred at rt overnight. Water was added to quench the reaction, and the mixture was extracted by CHCl3. After drying and concentration, the product was purified by column chromatography (EtOAc/hexane 1:4) to give thioamide 1.0 g as a yellow solid, yield 75%.
The thioamide (0.5 g, 1.37 mmol) was suspended in anhydrous alcohol under inert gas atmosphere. The reactants were cooled to 0 °C and 5.0 equiv of anhydrous hydrazine was added slowly with vigorous stirring, after which the thioamide was stirred at rt overnight. The alcohol and excess hydrazine were evaporated under vacuum, and the syrup was dissolved in CHCl3 and extracted by water three times. The organic layer was dried and concentrated under vacuum to yield 0.4 g of amidrazone as a white solid.
The amidrazone was dissolved in alcohol and 5.0 equiv of trimethyl orthoformate was added at rt. Several drops of sulfuric acid were added as catalyst, and the solution was stirred vigorously for 2 h. After neutralization, the alcohol was evaporated and the triazole was purified by column chromatography (EtOAc/hexane 4:1) to obtain 366 mg of triazole with a yield of 71% from thioamide: mp 157−159 °C; 1H NMR (400 MHz, CDCl3) δ 3.20 (s, 6H), 3.26 (s, 3H), 3.36 (s, 3H), 5.92−5.94 (m, 3H), 6.55 (d, J = 3.0 Hz, 1H), 6.72−6.75 (m, 3H), 6.83 (d, J = 1.5 Hz, 1H), 7.77 (s, 1H); MS (ESI) m/z = 365.1 (M+ + 1); Anal. (C20H20O3N4) C, H, O, N.
In Vitro Cytotoxicity Assay
Cytotoxic effects were examined in a human breast carcinoma cell line (ZR-75−1), a human colon carcinoma cell line (HCT-116), a human cervix epithelial adenocarcinoma cell line (HeLa), and a human cervix carcinoma cell line (KB-3−1) and its MDR+ subclone (KB-V1). Cells in logarithmic phase were diluted to a density of 20 000−30 000 cells/mL in culture medium based on growth characteristics. For each well of a 96-well microplate, 100 μL of cell dilution was seeded, allowed to attach overnight, and then exposed to varying concentrations (10−5−10−12 M) of compounds for 72 h (37 °C, 5% CO2 atmosphere). The number of living cells was estimated by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (Sigma, St. Louis MO). Absorbance at 595 nm was recorded on a TECAN GENios multifunction microplate reader (TECAN U.S. Inc., Research Triangle Park, NC). Compounds were tested in triplicate in at least three independent assays. The IC50 values were determined by a nonlinear regression analysis with Prism 3.03 (Graphpad Software Inc.). Average values were reported.
In Vitro Tubulin Polymerization Assay
Tubulin polymerization assays were conducted with the CytoDYNAMIX Screen 03 assay system (Cytoskeleton Inc.) following the manufacturer's instructions. Tubulin (>99% pure, 3 mg/mL) in 100 μL of G-PEM buffer was placed in a prewarmed 96-well plate in the presence of tested compounds at varying concentrations (ranging from 0.03 to 30 μM). The OD340nm was recorded every 60 s on a TECAN GENios multifunction microplate reader (TECAN U.S. Inc., Research Triangle Park, NC) at 37 °C for 1 h. Apparent IC50 values for the compounds were determined by nonlinear regression analysis with Prism 3.03 (Graphpad Software Inc.) by use of area under curve (AUC) from the polymerization profiles.
Molecular Modeling
All calculations were conducted on SGI Octane R12000 and SunFire 6900 machines. The crystal structure of the tubulin–colchicine–stathmin-like domain complex (PDB ID = 1SA0) was retrieved from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (http://www.rcsb.org/pdb).30 The crystal structure of compound 7 was assigned partial atomic charges and force constants by use of the MMFF94 force field in Sybyl v7.1.31 Compound 7 was aligned to the structure of colchicine in the binding site of the tubulin crystal structure by atom-fitting the trimethoxyphenyl ring and the centroids of the B ring (Figure 1), followed by manually docking 7 in the colchicine binding site. Energy minimization (EM) and molecular dynamics (MD) with AMBER832 were performed to refine the ligand–protein interactions.
The Hawkins–Cramer–Truhlar33,34 pairwise generalized Born model was used for the EM and MD calculations of the tubulin system. The standard force-field parameter set parm99 in AMBER832 was selected with dielectric constant ε = 1, and cutoff distance = 12.0 Å was applied for both electrostatic and van der Waals interactions. The concentration of mobile counterions in solution was set to 0.2 M35 and surface area was computed by use of the linear combination of pairwise overlaps (LCPO) model.36 The system was energy-minimized in two steps: (i) 1000 iterations of steepest descent and (ii) 4000 iterations of conjugate gradient optimization. MD simulations were then run on the resulting system with the same force-field parameters as EM. The SHAKE algorithm37 was implemented for bonds involving hydrogen atoms. The system was coupled to a Berendsen bath at 300 K by using a coupling constant τT = 1.0 ps.38 A 2.0 fs time step was applied during MD simulations. Restrained MD was first run for 10 ps to equilibrate the tubulin system by freezing the protein and ligand. Unconstrained MD was then applied to the entire system for 200 ps; the time-averaged structure derived from the final 150 ps was energy-minimized by 500 iterations of steepest descent and 500 iterations of conjugate gradient optimization. The hydrogen bonds and hydrophobic interactions between the ligand and protein were identified by LIGPLOT 4.439 and displayed in DSviewerPro5.0.40
Compound 8 was constructed in Sybyl7.1, based on the crystal structure of 7, and energy-minimized with the MMFF force field and charges. Ab initio molecular orbital calculations were performed on 7 and 8 at the HF/6−31G** level of theory, accessed through the Spartan ’02 program (Wavefunction, Inc., Irvine CA). By comparison of the resulting structures, it was found that the B ring was essentially planar (0°) in 7, consistent with the extended aromatic character of the indolic moiety, but was distinctly nonplanar (14°) in 8, reflecting a loss of aromatic character.
Acknowledgment
Access to the computational facilities at the UMDNJ Informatics Institute, supported in part by the National Library of Medicine (Grant G08 LM6230−07), is gratefully acknowledged. We thank Dr. Nigam Rath, Department of Chemistry & Biochemistry, University of Missouri–St. Louis, St. Louis, MO, for providing the X-ray crystal structure of 7.
Footnotes
Abbreviations: CA-4, combretastatin A-4; CA-4P, combretastatin A 4-phosphate; MDR, multi-drug-resistant; SAR, structure–activity relationship; NCI–DTP, National Cancer Institute–Developmental Therapeutics Program; EM, energy minimization; MD, molecular dynamics.
Supporting Information Available: Results from the NCI–DTP 60 cell-line assays on compounds 3–7 and tabulated X-ray crystal structure data for compound 7. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ducki S, Mackenzie G, Lawrence NJ, Snyder JP. Quantitative structure–activity relationship (5D-QSAR) study of combretastatin-like analogues as inhibitors of tubulin assembly. J. Med. Chem. 2005;48:457–465. doi: 10.1021/jm049444m. [DOI] [PubMed] [Google Scholar]
- 2.Jordan A, Hadfield JA, Lawrence NJ, McGown AT. Tubulin as a target for anticancer drugs: agents that interact with the mitotic spindle. Med. Res. Rev. 1998;18:259–296. doi: 10.1002/(sici)1098-1128(199807)18:4<259::aid-med3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 3.Andreu JM, Barasoain I. The interaction of baccatin III with the taxol binding site of microtubules determined by a homogeneous assay with fluorescent taxoid. Biochemistry. 2001;40:11975–11984. doi: 10.1021/bi010869+. [DOI] [PubMed] [Google Scholar]
- 4.Rai SS, Wolff J. Localization of the vinblastine-binding site on beta-tubulin. J. Biol. Chem. 1996;271:14707–14711. doi: 10.1074/jbc.271.25.14707. [DOI] [PubMed] [Google Scholar]
- 5.ter Haar E, Rosenkranz HS, Hamel E, Day BW. Computational and molecular modeling evaluation of the structural basis for tubulin polymerization inhibition by colchicine site agents. Bioorg. Med. Chem. 1996;4:1659–1671. doi: 10.1016/0968-0896(96)00158-7. [DOI] [PubMed] [Google Scholar]
- 6.Lin CM, Singh SB, Chu PS, Dempcy RO, Schmidt JM, Pettit GR, Hamel E. Interactions of tubulin with potent natural and synthetic analogs of the antimitotic agent combretastatin: a structure–activity study. Mol. Pharmacol. 1988;34:200–208. [PubMed] [Google Scholar]
- 7.Pettit GR, Rhodes MR, Herald DL, Hamel E, Schmidt JM, Pettit RK. Antineoplastic agents. 445. Synthesis and evaluation of structural modifications of (Z)- and (E)-combretastatin A-41. J. Med. Chem. 2005;48:4087–4099. doi: 10.1021/jm0205797. [DOI] [PubMed] [Google Scholar]
- 8.McGown AT, Fox BW. Differential cytotoxicity of combretastatins A1 and A4 in two daunorubicin-resistant P388 cell lines. Cancer Chemother. Pharmacol. 1990;26:79–81. doi: 10.1007/BF02940301. [DOI] [PubMed] [Google Scholar]
- 9.Ohsumi K, Nakagawa R, Fukuda Y, Hatanaka T, Morinaga Y, Nihei Y, Ohishi K, Suga Y, Akiyama Y, Tsuji T. Novel combretastatin analogues effective against murine solid tumors: design and structure–activity relationships. J. Med. Chem. 1998;41:3022–3032. doi: 10.1021/jm980101w. [DOI] [PubMed] [Google Scholar]
- 10.Thorpe PE, Chaplin DJ, Blakey DC. The first international conference on vascular targeting: meeting overview. Cancer Res. 2003;63:1144–1147. [PubMed] [Google Scholar]
- 11.Nam NH. Combretastatin A-4 analogues as antimitotic antitumor agents. Curr. Med. Chem. 2003;10:1697–1722. doi: 10.2174/0929867033457151. [DOI] [PubMed] [Google Scholar]
- 12.Maya AB, Perez-Melero C, Mateo C, Alonso D, Fernandez JL, Gajate C, Mollinedo F, Pelaez R, Caballero E, Medarde M. Further naphthylcombretastatins. An investigation on the role of the naphthalene moiety. J. Med. Chem. 2005;48:556–568. doi: 10.1021/jm0310737. [DOI] [PubMed] [Google Scholar]
- 13.Flynn BL, Flynn GP, Hamel E, Jung MK. The synthesis and tubulin binding activity of thiophene-based analogues of combretastatin A-4. Bioorg. Med. Chem. Lett. 2001;11:2341–2343. doi: 10.1016/s0960-894x(01)00436-x. [DOI] [PubMed] [Google Scholar]
- 14.Flynn BL, Hamel E, Jung MK. One-pot synthesis of benzo-[b]furan and indole inhibitors of tubulin polymerization. J. Med. Chem. 2002;45:2670–2673. doi: 10.1021/jm020077t. [DOI] [PubMed] [Google Scholar]
- 15.Liou JP, Chang YL, Kuo FM, Chang CW, Tseng HY, Wang CC, Yang YN, Chang JY, Lee SJ, Hsieh HP. Concise synthesis and structure–activity relationships of combretastatin A-4 analogues, 1-aroylindoles and 3-aroylindoles, as novel classes of potent antitubulin agents. J. Med. Chem. 2004;47:4247–4257. doi: 10.1021/jm049802l. [DOI] [PubMed] [Google Scholar]
- 16.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. Medicinal chemistry of combretastatin A4: present and future directions. J. Med. Chem. 2006;49:3033–3044. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 17.Cushman M, Nagarathnam D, Gopal D, Chakraborti AK, Lin CM, Hamel E. Synthesis and evaluation of stilbene and dihydro-stilbene derivatives as potential anticancer agents that inhibit tubulin polymerization. J. Med. Chem. 1991;34:2579–2588. doi: 10.1021/jm00112a036. [DOI] [PubMed] [Google Scholar]
- 18.Cushman M, Nagarathnam D, Gopal D, He HM, Lin CM, Hamel E. Synthesis and evaluation of analogues of (Z)-1-(4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)ethene as potential cytotoxic and antimitotic agents. J. Med. Chem. 1992;35:2293–2306. doi: 10.1021/jm00090a021. [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Woods KW, Li Q, Barr KJ, McCroskey RW, Hannick SM, Gherke L, Credo RB, Hui YH, Marsh K, Warner R, Lee JY, Zielinski-Mozng N, Frost D, Rosenberg SH, Sham HL. Potent, orally active heterocycle-based combretastatin A-4 analogues: synthesis, structure–activity relationship, pharmacokinetics, and in vivo antitumor activity evaluation. J. Med. Chem. 2002;45:1697–1711. doi: 10.1021/jm010523x. [DOI] [PubMed] [Google Scholar]
- 20.Curphey TJ. Thionation with the reagent combination of phosphorus pentasulfide and hexamethyldisiloxane. J. Org. Chem. 2002;67:6461–6473. doi: 10.1021/jo0256742. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q, Keenan SM, Peng Y, Nair AC, Yu SJ, Howells RD, Welsh WJ. Discovery of novel triazole-based opioid receptor antagonists. J. Med. Chem. 2006;49:4044–4047. doi: 10.1021/jm0601250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Q, Peng Y, Welsh WJ. Efficient new approach for the synthesis of N,N-dialkylamino-1,2,4-triazoles. Heterocycles. 2006 in press. [Google Scholar]
- 23.Dohle W, Lindsay DM, Knochel P. Copper-mediated cross-coupling of functionalized arylmagnesium reagents with functionalized alkyl and benzylic halides. Org. Lett. 2001;3:2871–2873. doi: 10.1021/ol0163272. [DOI] [PubMed] [Google Scholar]
- 24.Boyd MR, Paull KD. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995;34:91–109. [Google Scholar]
- 25.Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- 26.Cole SP, Deeley RG. Multidrug resistance mediated by the ATP-binding cassette transporter protein MRP. Bioessays. 1998;20:931–940. doi: 10.1002/(SICI)1521-1878(199811)20:11<931::AID-BIES8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 27.Dumontet C, Sikic BI. Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. J. Clin. Oncol. 1999;17:1061–1070. doi: 10.1200/JCO.1999.17.3.1061. [DOI] [PubMed] [Google Scholar]
- 28.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen TL, McGrath C, Hermone AR, Burnett JC, Zaharevitz DW, Day BW, Wipf P, Hamel E, Gussio R. A Common Pharmacophore for a Diverse Set of Colchicine Site Inhibitors Using a Structure-Based Approach. J. Med. Chem. 2005;48:6107–6116. doi: 10.1021/jm050502t. [DOI] [PubMed] [Google Scholar]
- 30.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sybyl 7.1. Tripos, Inc.; St. Louis, MO: [Google Scholar]
- 32.Case DA, Pearlman DA, Caldwell JW, III TEC, Wang J, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer RJ, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER 8. University of California; San Francisco, CA: [Google Scholar]
- 33.Hawkins GD, Cramer CJ, Truhlar DG. Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 1995;246:122–129. [Google Scholar]
- 34.Hawkins GD, Cramer CJ, Truhlar DG. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996;100:19824–19839. [Google Scholar]
- 35.Srinivasan J, Trevathan MW, Beroza P, Case DA. Application of a pairwise generalized Born model to proteins and nucleic acids: inclusion of salt effects. Theor. Chem. Acc. 1999;101:426–434. [Google Scholar]
- 36.Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999;20:217–230. [Google Scholar]
- 37.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of Cartesian equation of motion of a system with constraints: molecular dynamics of N-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 38.Berendsen HJC, Postm JPM, Van Gunsteren WF, Di Nola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 39.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 40.DSviewerPro 5.0. Accelrys Inc.; San Diego, CA: [Google Scholar]