

Abstract
Determining the genetic basis of cancer requires comprehensive analyses of large collections of histopathologically well-classified primary tumours. Here we report the results of a collaborative study to discover somatic mutations in 188 human lung adenocarcinomas. DNA sequencing of 623 genes with known or potential relationships to cancer revealed more than 1,000 somatic mutations across the samples. Our analysis identified 26 genes that are mutated at significantly high frequencies and thus are probably involved in carcinogenesis. The frequently mutated genes include tyrosine kinases, among them the EGFR homologue ERBB4; multiple ephrin receptor genes, notably EPHA3; vascular endothelial growth factor receptor KDR; and NTRK genes. These data provide evidence of somatic mutations in primary lung adenocarcinoma for several tumour suppressor genes involved in other cancers—including NF1, APC, RB1 and ATM—and for sequence changes in PTPRD as well as the frequently deleted gene LRP1B. The observed mutational profiles correlate with clinical features, smoking status and DNA repair defects. These results are reinforced by data integration including single nucleotide polymorphism array and gene expression array. Our findings shed further light on several important signalling pathways involved in lung adenocarcinoma, and suggest new molecular targets for treatment.
Lung cancer is the leading cause of cancer death, annually resulting in more than one million deaths worldwide. About 1.2 million new cases are diagnosed each year1 and prognoses are poor. Lung adenocarcinoma is the most common form of lung cancer and has an average 5-yr survival rate of 15%2, mainly because of late-stage detection and a paucity of late-stage treatments.
Although smoking is unquestionably the leading cause of lung cancer, approximately 10% of cases occur in patients who have never smoked3. Environmental exposures and genetic susceptibility are also thought to contribute to cancer risk4-7.Adenocarcinomas in patients who have never smoked frequently contain mutations within the tyrosine kinase domain of the epidermal growth factor receptor(EGFR)gene; those patients soften respond to tyrosine kinase inhibitor drugs such as gefitinib and erlotinib8-10, but usually develop drug resistance11,12. Conversely, KRAS mutations are more common in individuals with a history of cigarette use and are associated with resistance to EGFR-tyrosine-kinaseinhibitors13,14.
Previous gene resequencing efforts have identified several key mutations associated with human cancers15-18. The Tumour Sequencing Project (TSP) is a pilot project to characterize cancer genomes, and has allowed the discovery of somatic mutations in the coding exons of 623 candidate cancer genes in 188 lung adenocarcinomas. Here we identify significantly mutated genes not previously associated with lung adenocarcinoma, describe relationships between different genetic alterations, and report correlations between genetic alterations and clinical features. Moreover, our integration of single nucleotide polymorphism (SNP) array, gene expression array and mutation data provides a broader view of genomic alterations in lung adenocarcinomas. These findings further our understanding of lung cancer and provide clues to new therapeutic targets.
Overview of samples, genes and mutations discovered
We selected 188 primary lung adenocarcinomas, each containing a minimum of 70% tumour cells as determined by study pathologists. We screened for somatic mutations in 623 candidate genes comprising known oncogenes and tumour suppressor genes, protein kinase families, and genes in regions of copy number alteration, focusing on coding exons and splice sites (Supplementary Table 1). A total of 247 megabases of tumour DNA sequence was analysed to identify putative mutations, and non-synonymous mutations were validated by orthogonal methods or confirmed by independent polymerase chain reaction (PCR) amplification and sequencing (Supplementary Methods and Supplementary Fig. 1).
We have identified 1,013 non-synonymous somatic mutations in 163 of the 188 tumours, including 915 point mutations, 12 dinucleotide mutations (mutations affecting two consecutive bases on the same allele), 29 insertions and 57 deletions, with insertions/deletions (indels) ranging from 1 to 23 nucleotides. The point mutations include 802 missense, 75 nonsense, 1 read-through and 37 splice-site mutations (Supplementary Table 2).
A set of 12 genes was found with significantly higher frequencies of nonsense, splice-site and frameshift mutations (P, < 0.1), suggesting that they were candidate tumour suppressor genes (Supplementary Table 3a). Recurrent somatic mutations were observed at 28 sites across seven genes; these included five previously unknown sites in five genes (Supplementary Table 3b). In silico predictions suggest that 580 of the missense mutations have potential functional relevance. A comparison of the mutations to the COSMIC19 and OMIM20 databases identified 823 somatic mutations and 818 mutation sites that were not present in these databases, respectively (Supplementary Methods and Supplementary Table 2).
Significantly mutated genes in lung adenocarcinoma
The large size of our sample set enabled the identification of mutated genes that show evidence for positive selection in lung adenocarcinoma. We used three different methods (Supplementary Methods and Supplementary Tables 4 and 5) to determine the significance of the difference between the observed versus expected numbers of mutations in 188 tumours. We identified a total of 26 significantly mutated genes, among them 17 genes are designated as significant by at least two approaches (Fig. 1 and Supplementary Table 6a). Note that LRP1B, despite its large number of mutations, was found to be significant by only one method, mostly owing to its long coding sequence.
Figure 1. Significantly mutated genes in lung adenocarcinomas.
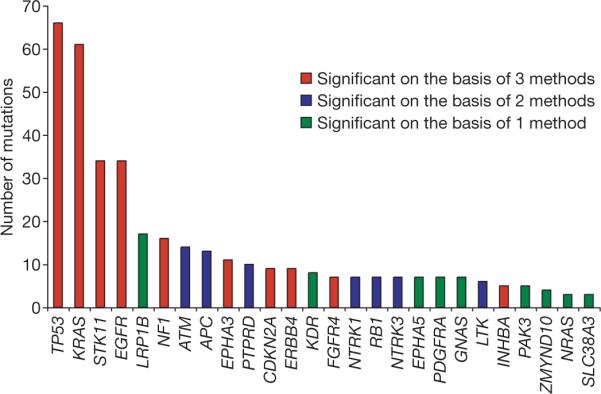
The height of the bars represents the number of somatic mutations in each indicated gene in 188 tumour and normal pairs. Standard, gene-specific and category-based tests were used for this analysis (Supplementary Information). Ten genes were found to be significantly mutated by all three statistical methods (red bars), 7 genes by at least two methods (blue bars) and 9 genes by one of the three methods (green bars), for up to 26 significantly mutated genes in total.
The study identified many genes previously known to be mutated in lung adenocarcinoma, including several tumour suppressor genes (TP53 (ref. 21), CDKN2A (ref. 22) and STK11 (ref. 23)) and oncogenes (KRAS24, EGFR8 and NRAS25). In addition, we found several new genes that were significantly mutated in this disease.
Bona fide and putative tumour suppressor genes
The most prominent case for a tumour suppressor gene is NF1, for which inactivating mutations are found in neurofibromatosis type I patients26. In this study, 16 NF1 mutations (4 nonsense, 5 splice-site and 1 frame-shift mutations) were identified in 13 patients (Supplementary Table 2). Three tumours harboured two mutations each, although it is not known whether these mutations are in cis or in trans. This suggests potential bi-allelic inactivation of NF1 in these three patients.
Another previously unknown mutated tumour suppressor gene in lung adenocarcinoma is ATM, encoding a cell-cycle checkpoint kinase that functions as a regulator of p53 (ref. 27). Genetic polymorphisms of ATM are known to affect lung cancer risk28, but only isolated instances of ATM somatic mutation have been reported in lung adenocarcinoma15. We found 14 ATM mutations in 13 tumours, including 1 nonsense, 1 splice-site and 2 frameshift mutations (Supplementary Table 2).
Another tumour suppressor gene harbouring frequent mutations is RB1, which was first identified as the susceptibility gene for retinoblastoma29. Given that DNA tumour viruses such as papillomaviruses typically target RB1 and TP53 simultaneously30, it is interesting to note that five of the seven RB1 mutations occur in tumours with TP53 mutations, and two occur in tumours with ATM mutations, suggesting that an ATM mutation may substitute functionally for a TP53 mutation.
APC mutations have been reported in lung squamous cell carcinoma and small-cell lung carcinoma31, but not in lung adenocarcinoma. We observed 13 mutations in 11 tumours confirmed by pathology evaluation to be lung tumour samples and not metastatic colorectal carcinomas. Mutations (G34E and S37F) of the CTNNB1 gene were observed in two other tumours.
Deletion and epigenetic silencing of LRP1B have been previously observed in lung cancer cell lines and oesophageal tumours32,33. Our finding of 17 mutations in LRP1B further supports the notion that LRP1B genomic alterations are significant in lung cancer pathogenesis (Fig. 1). PTPRD, previously shown to be deleted in lung adenocarcinoma34,35, is also found to be frequently mutated34. Owing to the absence of nonsense, splice-site or frameshift mutations in both of these genes in our tumour set, further evidence is required to determine whether they are tumour suppressors or another category of genes.
Possible proto-oncogenes
Although the involvement of EGFR and ERBB2 mutations in lung cancer has been reported previously, we also found mutations at a significant frequency in ERBB4 (Fig. 1). The discovery of nine mutations in ERBB4, two of which are putatively deleterious with respect to the protein tyrosine kinase domain and five of which are clustered in the receptor ligand binding domain, indicates its involvement in lung cancer (Fig. 2). We also discovered four mutations in ERBB2 and three in ERBB3.
Figure 2. Diagrams of mutations found in the members of several receptor families in lung adenocarcinomas.

a-e, Mutations in members of the EGF (a), EPH (b), FGF (c), NTRK (d) and VEGF (e) receptor families are shown. Protein domains are determined by using HMMPFAM. The PFAM domains include `L' (receptor ligand binding domain), Fn3 (fibronectin type III domain), Ig (immunoglobulin domain), LRR (leucine rich repeat domain), LRRNT (leucine rich repeat amino-terminal domain), PTK (protein tyrosine kinase domain) and SAM (sterile α-motif). The locations of mutations are indicated by diamonds, circles and triangles, with filled shapes representing new mutations and open shapes denoting known mutations. The size of the shapes is positively proportional to the degree of conservation at the mutated residue. Representative scheme for each family is constructed based on the ClustalW2 alignment. Recurrent mutations are outlined in black.
The most significantly mutated gene in the ephrin family is EPHA3 (Fig. 1). Although isolated mutations in this gene have been reported15,17, this is to our knowledge the first demonstration of statistical significance of EPHA3 mutations in lung adenocarcinoma. The 11 mutations in EPHA3 are distributed along the length of the gene, with 8 mutations in the extracellular domain and 3 in the kinase domain, but no hotspot positions in which mutations cluster. One observed mutation in EPHA3, K761N, is located in the kinase domain at a highly conserved position analogous to FGFR2(K641)—part of a newly described “molecular brake”36. In total, we identified 37 mutations in 10 of the 13 ephrin receptors sequenced, finding high mutation rates in several family members (Figs 1 and 2).
Previous mutational screening of the tyrosine kinase domain of NTRKs identified 9 mutations in 29 large-cell neuroendocrine carcinomas, but found no mutations in 443 non-small-cell lung cancers37. In contrast we discovered 20 mutations in NTRKs (Fig. 1) of which 7 mutations occur within their tyrosine kinase domains, suggesting that the role of NTRKs is not restricted to large-cell neuroendocrine carcinomas. A significant number of mutations have also been identified in VEGFR and FGFR family members. In particular, four and three kinase domain mutations were found in KDR and FGFR4 (ref. 38), respectively (Fig. 2 and Supplementary Table 2).
Notably, several known oncogenes and tumour suppressor genes fell below the borderline of significance in our study. These genes include the proto-oncogenes AKT1 (in which we found two mutations, including one (E17K) described as a transforming mutation in other cancers39), CTNNB1, ERBB2 (ref. 40) and BRAF41, as well as the PTEN tumour suppressor gene42. These results offer enriched data for investigating mutated functional domains (Supplementary Methods and Supplementary Table 6b) and for analysing interactions among mutations and pathways.
Concurrent and mutually exclusive mutations
We searched for correlations among mutations in 29 genes with at least 6 mutations each. The strongest positive correlations were for mutations in PIK3C3 and PTPRD, NTRK2 and PDGFRA, FGFR4 and NTRK2, and FGFR4 and PDGFRA (P ≤ 0.01; Supplementary Table 7a, c). The well-known example of negative correlation of mutations in EGFR and KRAS14 was confirmed in this study (P < 1 × 10-07), with no sample having mutations in both genes (Fig. 3). We also found negative correlation between mutations in EGFR and STK11 (P = 7 × 10-06), consistent with a previous report43. Notably, samples with mutations in several receptor tyrosine kinase genes do not harbour any mutations in EGFR (Fig. 3). We also detected a strong negative correlation between mutations in ATM and TP53 (P = 9.5 × 10-05; Fig. 3), suggesting that mutations in ATM and TP53 may be independently sufficient for the loss of cell-cycle checkpoint control.
Figure 3. Concurrent and mutual exclusion of mutations observed across genes in lung adenocarcinomas.
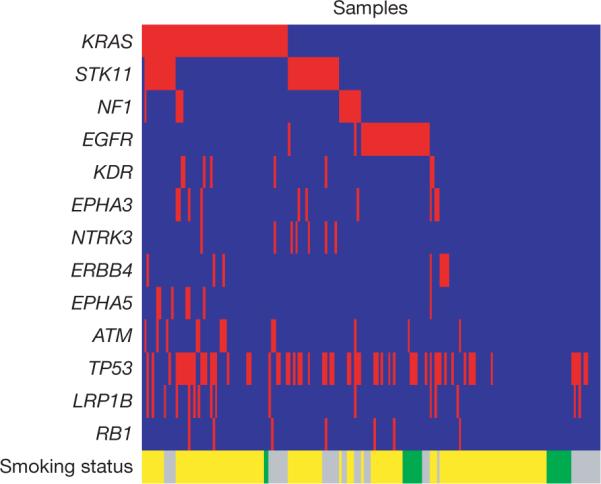
Tumours with and without mutations in the indicated genes are labelled in red and blue in the corresponding columns, respectively. Tumours from smokers (former and current) and from individuals who have never smoked are labelled in yellow and green, respectively. Tumours without smoking status are labelled in grey.
Distributions of mutations in individual cancer genomes
We studied the spectrum of mutations observed across tumours, in relation to the overall mutation rate and to clinical phenotypes. We found that mutations in TP53, PRKDC, SMG1 and a set of other genes (Supplementary Table 8) are positively correlated with higher mutation rates. Of particular interest, four of the six most highly mutated tumours have mutations in PRKDC, which encodes a protein involved in the repair of double-stranded DNA breaks44 (Fig. 4a). The average of 24.3 mutations in tumours having PRKDC mutations is significantly higher than the average of 4.7 mutations in tumours without PRKDC mutations (P = 3.52 × 10-59).
Figure 4. Mutation distributions in individual lung adenocarcinoma genomes.

a, Tumours with mutations in PRKDC showed higher than average mutation rates, and conversely tumours with mutations in EGFR had lower than average mutation rates. b, Smokers have on average threefold higher mutation rates compared to individuals who have never smoked.
We also determined that a set of genes including EGFR (P = 0.05) and PTEN (P = 0.03) tended to be mutated in tumours with lower-than-average mutation rates. Mutations in EGFR and PTEN may have strong tumour-growth-promoting capability and thereby reduce the selection pressure for acquiring further mutations.
Integration with copy number and gene expression data
Subsets of the TSP tumour collection were analysed using SNP array (n = 383), re-sequencing (n = 188) and gene expression array (n = 75). All tumours used for sequencing and expression studies have been analysed using SNP array. Significant correlation (false discovery rate < 0.05) between copy number and expression level in 75 tumours was observed, similar to the trend seen in a previous study45 (Supplementary Information and Supplementary Table 9).
Comparison of mutation data with copy number analysis34 shows that several significantly mutated genes are present in peaks of copy number gain (EGFR and KRAS) or loss (CDKN2A, PTPRD and RB1). Other amplified genes are subject to recurrent mutations (for example ERBB2, MDM2 and TERT) although the mutation frequency does not reach statistical significance. In parallel, several significantly mutated genes show rare amplifications or deletions. The NRAS oncogene is subject to rare amplification in lung adenocarcinoma (Supplementary Fig. 4). The amplification of EPHA3 and KDR (Supplementary Figs 4 and 5) seen in two tumours each, indicates that these genes are probably proto-oncogenes. Conversely, we found NF1 to be homozygously deleted in one tumour (Supplementary Fig. 4).
Furthermore, we found that mutations in PTEN, APC and TP53 were correlated with copy number loss (Supplementary Table 10a), suggesting that these three genes might each undergo homozygous loss of function. Conversely, mutations in EGFR, HCK, KRAS and EPHB1 were associated with copy number gain (Supplementary Table 10a), consistent with a proto-oncogene function. Notably, three of the six tumours with the highest EGFR amplification also have mutations in EGFR, and five of the six tumours with the highest KRAS amplification also harbour KRAS mutations (Supplementary Table 11). In many cases, the mutant allele is preferentially amplified (Supplementary Fig. 6) but larger sample sets are required to determine the statistical significance.
We investigated the correlation among mutations, copy number and gene expression in 41 lung adenocarcinomas with all three types of data. Mutations in TP53 (Fig. 5a) and APC (Fig. 5b) are correlated with lower copy number and lower messenger RNA expression levels. Correlations with lower gene expression are also seen for STK11 and ATM mutations (Supplementary Table 10b). Mutations in these tumour suppressor genes could cause instability of their cognate mRNAs. Conversely, mutations in EGFR (Fig. 5c) and KRAS (Fig. 5d) are associated with higher mRNA expression levels as well as higher copy number, as are EPHB1 mutations (Supplementary Table 10b).
Figure 5. DNA copy number, gene expression, and mutation distributions in lung adenocarcinomas.

a-d, Copy number, gene expression and mutation status at TP53 (a), APC (b), EGFR (c) and KRAS (d) loci in 41 lung adenocarcinomas. Normalized gene expression and log2 DNA copy number ratio in each sample were determined as described in Supplementary Information.
Integrated analysis of significantly mutated pathways
Further insight into the role of genomic alterations underlying lung adenocarcinoma was gained by examining the distribution of mutations across Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Fig. 6, Supplementary Methods and Supplementary Tables 11-13).
Figure 6. Significantly mutated pathways in lung adenocarcinomas.
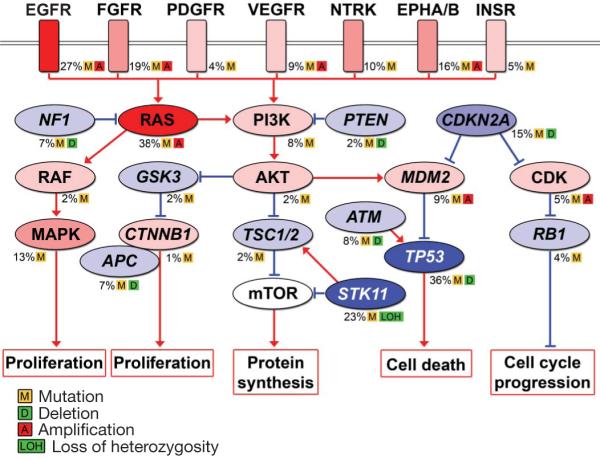
Genetic alterations in lung adenocarcinoma frequently occur in genes of the MAPK signalling, p53 signalling, Wnt signalling, cell cycle and mTOR pathways. Oncoproteins are indicated in pink to red and tumour suppressor proteins are shown in light to dark blue. The darkness of the colours is positively correlated to the percentage of tumours with genetic alterations. Frequency of genetic alterations for each of these pathway members in 188 tumours is indicated. Genes (EGFR, FGFR1, FGFR4, KDR, EPHA3, KRAS, NRAS, MDM2 and CDK6) lying in regions of focal amplification were analysed for the percentage of samples with copy number amplification. Samples with greater than 2.5 and fewer than 1.5 DNA copies were considered as amplified and deleted, respectively. Selected components of each pathway are shown in the figure.
In the MAPK pathway we found 289 mutations in 56 genes, including members of EGF, FGF and NTRK receptor families, and KRAS and NF1 (Fig. 6). Notably, 132 of the 188 tumours sequenced have at least one mutation in the MAPK pathway, underscoring its pivotal role in lung cancer.
We identified mutations in multiple components of the Wnt pathway, including APC, CTNNB1, SMAD2, SMAD4 and GSK3B. Of the 188 lung adenocarcinomas 29 showed mutations in this pathway (not including mutations in TP53, which is included in the Wnt pathway in KEGG), which is to our knowledge the first demonstration of Wnt alteration in lung adenocarcinoma. At least one mutation in the p53 pathway was seen in 85 tumours. In addition to the 66 TP53 mutations, frequent mutations were found in ATM and amplifications were identified in MDM2 (Fig. 6).
We have found an array of mutations in PTEN, PI3K genes and AKT genes—all members of the insulin/PI3K/AKT signalling arm of this pathway (Fig. 6). In addition, 13 tumours were found to carry 16 NF1 mutations, the deficiency of which has been implicated in RAS-and PI3K-dependent hyperactivation of the mTOR pathway46. More than 30 mutations were also discovered in STK11, a member of the AMP-dependent protein kinase signalling pathway. By sequencing 70 polymorphic STK11 SNP sites, we identified 17 tumours with loss of heterozygosity (LOH) (as defined by at least three consecutive heterozygous loci that reduced to homozygosity in the tumour; Supplementary Table 14). Two tumours having clear regions of LOH at STK11 also harboured one nonsense mutation and one deletion, suggesting possible homozygous loss of function. Six tumours have mutations in the tuberous sclerosis complex 1 and 2 (TSC1 and TSC2). In summary, mTOR pathway components are mutated in 17 genes and in more than 30% of tumours sequenced, not including tumours with KRAS mutations. Our finding suggests that dysregulation of mTOR is important for lung carcinogenesis and hence is a potential therapeutic target. The effectiveness of rapamycin and its analogues in the treatment of lung adenocarcinoma should be further tested.
There are nine mutations in CDKN2A and one each in CDKN2B and CDKN2C, as well as seven mutations in RB1. Furthermore, as described there are frequent focal amplifications of CDK4 and CDK6 as well as CCND1 and CCNE1, and frequent deletions of RB1, CDKN2A and CDKN2B (ref. 34; Fig. 6).
Mutations correlated with clinical features
We investigated the distribution of mutations across different clinical subgroups, including smoking status, tumour grade, tumour stage and histological subtype (Fig. 4, Supplementary Fig. 7 and Supplementary Table 15).
The average number of mutations in smokers is significantly higher than in individuals who have never smoked (P=0.021, t-test), and notably none of the tumours from those who have never smoked had more than five mutations in the resequenced genes, whereas smokers had as many as 49 mutations (Fig. 4b). Consistent with previous findings47, we observed that EGFR mutations correlate with the status of patients who have never smoked (P=0.0046, Fisher's exact test), whereas KRAS mutations correlate with smoker status (P=0.021). We also have observed correlation between mutations in STK11 and smokers (P=0.044), consistent with a previous report43.
As expected, tumours with higher grade had accumulated more mutations than tumours of lower grade (P=0.001; Supplementary Fig. 7a). Some genes showed a clear increase in the frequency of somatic mutation with tumour grade, suggesting that these genes may have a role in transformation or progression. A clear example is TP53, with somatic mutations in 13%, 24% and 52% of tumours of grade 1, 2 and 3, respectively (correlation P=7.8×10-06), consistent with a previous report48. Other genes in which the mutation frequency positively correlated with tumour grade were LRP1B (P=0.013), INHBA (P=0.013) and PRKDC (P=0.018). Conversely, other genes showed no significant correlation with tumour grade, which could indicate that mutations in this group of genes are critical early in tumorigenesis. A clear example is KRAS, with somatic mutations in 38%, 32% and 32% of tumours of grades 1, 2 and 3, respectively.
Our analysis shows that tumours of higher stage had accumulated more mutations than tumours of lower stage (P=0.006; Supplementary Fig. 7b), although this rate varies widely among individual tumours. We found significant correlations between tumour stage and mutations in NTRK2 (P=0.003), EPHA7 (P=0.003), PRKCG (P=0.0087) and FLT4 (P=0.0093).
There are several subclasses of lung adenocarcinoma, including acinar, papillary, BAC (bronchioloalveolar carcinoma) and solid, on the basis of World Health Organization standards49,50. Our most notable finding was that mutations in LRP1B, TP53 and INHBA show various levels of negative correlation with acinar, papillary and BAC subtypes, but significant positive correlation with solid subtype (LRP1B, P=2.29×10-05; TP53, P=0.002; INHBA, P=0.0023) in 152 tumours with subtype information. On the other hand mutations in EGFR showed moderate negative correlation with the solid subtype (P=0.13) and significant positive correlation with the papillary sub-type (P=0.041), consistent with a previous report50.
Furthermore, our analysis shows that the 25 patients in which no mutations were found have diverse clinical features and some show a comparable extent of copy number alterations compared to samples having mutations (Supplementary Table 16). Of note, 16 of 25 tumours without discovered mutations in the 623 genes are from the group with higher stromal contamination rate (Supplementary Table 17), suggesting that stromal contamination might reduce the sensitivity in discovering mutations.
Discussion
Our study represents to our knowledge the largest effort so far to characterize genomic alterations in lung adenocarcinoma. Before this study, there were five genes known to be mutated at high frequency in lung adenocarcinoma—TP53, KRAS, STK11, EGFR and CDKN2A— as well as several known genes with lower mutation frequencies— PTEN, NRAS, ERBB2, BRAF and PIK3CA. After sequencing 623 genes in 188 tumours, we have identified further significantly mutated genes, more than doubling the list. The newly identified genes include tumour suppressor genes (NF1, RB1, ATM and APC) along with tyrosine kinase genes (ephrin receptor genes, ERBB4, KDR, FGFR4 and NTRK genes) that may function as proto-oncogenes. We have demonstrated that many of these genes are also targeted by copy number alterations and/or gene expression changes. Additionally, there is a significant excess of mutations and copy number alterations in genes from the MAPK, p53, Wnt, cell cycle and mTOR signalling pathways, suggesting links to the disease. Our results also demonstrate that lung adenocarcinomas are heterogeneous, with diverse combinations of mutations yet commonality in the main pathways affected by these mutations. The mutation rate varies across tumour samples and is probably influenced by DNA mismatch repair defects and clinical features. The newly discovered genes and pathways may expand the range of potential therapeutic options for treatment of lung adenocarcinoma. For example, inhibitors of the MEK kinase could be tested in tumours with NF1 mutations, whereas inhibitors of KDR, such as sorafenib and sunitinib, might be tested in tumours with KDR mutations.
Although the analysis of the 188 TSP tumours is the largest tumourtype-specific screen for mutations to date, it does not have complete power to detect some genes known to be associated with lung cancer. Thus, larger sample sizes will be desirable. Moreover, these approaches should be extended to other types of lung cancer, metastatic lung cancer, and other cancers to determine the underlying genetic basis of those diseases and to highlight potential approaches for diagnosis and therapy. These studies can also be extended by comprehensive resequencing of the entire transcriptome, the entire collection of exons or the entire genome in large collections of cancers. Such studies should be feasible with next-generation sequencing technologies and at present are being prototyped within this programme.
METHODS SUMMARY
Source DNAs were extracted from primary lung adenocarcinoma tumours and adjacent normal tissue (or peripheral blood lymphocytes). Collection and use of all tissue samples were approved by the human subjects Institutional Review Boards of participating institutions. These samples were snap-frozen, anonymized and contributed along with matched normal samples by the Dana-Farber Cancer Institute, MD Anderson Cancer Center, Memorial Sloan-Kettering Cancer Center, University of Michigan, and Washington University in St Louis. Affymetrix 250K StyI Array data were used to estimate the level of stromal contamination and thereby to select 188 tumours and matched normals for the resequencing study. Whole-genome amplification was performed using Qiagen REPLI-g Service before sequencing. All coding exons and splice-site sequences of 623 target genes were PCR amplified and sequenced on both strands for all of the tumours. Additional data were generated until more than 90% of targeted exonic and splice-site bases were covered by at least one sequence read. Traces were automatically processed to identify SNPs and indels. Sequence data were obtained for the matched normals from a variety of platforms to determine the somatic status of new variants and unvalidated dbSNPs. Further data were generated using orthogonal technologies to validate the candidate somatic mutations. Synonymous somatic mutations identified in 250 genes were used to estimate the background mutation rate, which was used in statistical calculations to identify significantly mutated genes. Statistical approaches were used to identify significantly mutated pathways. Expression profiles were determined for 75 TSP tumours using the Affymetrix U133Plus2 GeneChip. Further analyses were performed to determine correlation between mutation and copy number variation, mutation and gene expression, copy number variation and gene expression, as well as mutation and clinical attributes.
Supplementary Material
Acknowledgements
We thank A. Lash, M. F. Zakowski, M. G. Kris and V. Rusch for intellectual contributions, and many members of the Baylor Human Genome Sequencing Center, the Broad Institute of Harvard and MIT, and the Genome Center at Washington University for support. This work was funded by grants from the National Human Genome Research Institute to E.S.L., R.A.G. and R.K.W.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Juergens RA, Brahmer JR. Adjuvant treatment in non-small cell lung cancer: Where are we now? J. Natl Compr. Canc. Netw. 2006;4:595–600. doi: 10.6004/jnccn.2006.0049. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Hecht SS. Tobacco smoke carcinogens and lung cancer. J. Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 4.Hung RJ, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 5.Sellers TA, et al. Evidence for mendelian inheritance in the pathogenesis of lung cancer. J. Natl Cancer Inst. 1990;82:1272–1279. doi: 10.1093/jnci/82.15.1272. [DOI] [PubMed] [Google Scholar]
- 6.Sellers TA, Weaver TW, Phillips B, Altmann M, Rich SS. Environmental factors can confound identification of a major gene effect: results from a segregation analysis of a simulated population of lung cancer families. Genet. Epidemiol. 1998;15:251–262. doi: 10.1002/(SICI)1098-2272(1998)15:3<251::AID-GEPI4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 7.Thorgeirsson TE, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pao W, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl Acad. Sci. USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 10.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 12.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Calvez F, et al. TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res. 2005;65:5076–5083. doi: 10.1158/0008-5472.CAN-05-0551. [DOI] [PubMed] [Google Scholar]
- 14.Pao W, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies H, et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005;65:7591–7595. doi: 10.1158/0008-5472.CAN-05-1855. [DOI] [PubMed] [Google Scholar]
- 16.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 18.Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 19.Forbes S, et al. Cosmic 2005. Br. J. Cancer. 2006;94:318–322. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKusick VA. Mendelian inheritance in man and its online version, OMIM. Am. J. Hum. Genet. 2007;80:588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi T, et al. p53: a frequent target for genetic abnormalities in lung cancer. Science. 1989;246:491–494. doi: 10.1126/science.2554494. [DOI] [PubMed] [Google Scholar]
- 22.Packenham JP, et al. Homozygous deletions at chromosome 9p21 and mutation analysis of p16 and p15 in microdissected primary non-small cell lung cancers. Clin. Cancer Res. 1995;1:687–690. [PubMed] [Google Scholar]
- 23.Sanchez-Cespedes M, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659–3662. [PubMed] [Google Scholar]
- 24.Rodenhuis S, et al. Incidence and possible clinical significance of K-ras oncogene activation in adenocarcinoma of the human lung. Cancer Res. 1988;48:5738–5741. [PubMed] [Google Scholar]
- 25.Sasaki H, et al. Nras and Kras mutation in Japanese lung cancer patients: Genotyping analysis using LightCycler. Oncol. Rep. 2007;18:623–628. [PubMed] [Google Scholar]
- 26.Cawthon RM, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62:193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- 27.Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 2000;14:278–288. [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JH, et al. Genetic polymorphisms of ataxia telangiectasia mutated affect lung cancer risk. Hum. Mol. Genet. 2006;15:1181–1186. doi: 10.1093/hmg/ddl033. [DOI] [PubMed] [Google Scholar]
- 29.Friend SH, et al. Deletions of a DNA sequence in retinoblastomas and mesenchymal tumors: organization of the sequence and its encoded protein. Proc. Natl Acad. Sci. USA. 1987;84:9059–9063. doi: 10.1073/pnas.84.24.9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howley PM, Scheffner M, Huibregtse J, Munger K. Oncoproteins encoded by the cancer-associated human papillomaviruses target the products of the retinoblastoma and p53 tumor suppressor genes. Cold Spring Harb. Symp. Quant. Biol. 1991;56:149–155. doi: 10.1101/sqb.1991.056.01.019. [DOI] [PubMed] [Google Scholar]
- 31.Ohgaki H, et al. APC mutations are infrequent but present in human lung cancer. Cancer Lett. 2004;207:197–203. doi: 10.1016/j.canlet.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 32.Liu CX, et al. LRP-DIT, a putative endocytic receptor gene, is frequently inactivated in non-small cell lung cancer cell lines. Cancer Res. 2000;60:1961–1967. [PubMed] [Google Scholar]
- 33.Sonoda I, et al. Frequent silencing of low density lipoprotein receptor-related protein 1B (LRP1B) expression by genetic and epigenetic mechanisms in esophageal squamous cell carcinoma. Cancer Res. 2004;64:3741–3747. doi: 10.1158/0008-5472.CAN-04-0172. [DOI] [PubMed] [Google Scholar]
- 34.Weir BA, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao X, et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005;65:5561–5570. doi: 10.1158/0008-5472.CAN-04-4603. [DOI] [PubMed] [Google Scholar]
- 36.Chen H, et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol. Cell. 2007;27:717–730. doi: 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marchetti A, et al. Frequent mutations in the neurotrophic tyrosine receptor kinase gene family in large cell neuroendocrine carcinoma of the lung. Hum. Mutat. 2008;29:609–616. doi: 10.1002/humu.20707. [DOI] [PubMed] [Google Scholar]
- 38.Marks JL, et al. Mutational analysis of EGFR and related signaling pathway genes in lung adenocarcinomas identifies a novel somatic kinase domain mutation in FGFR4. PLoS One. 2007;2:e426. doi: 10.1371/journal.pone.0000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carpten JD, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 40.Stephens P, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–526. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 41.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 42.Forgacs E, et al. Mutation analysis of the PTEN/MMAC1 gene in lung cancer. Oncogene. 1998;17:1557–1565. doi: 10.1038/sj.onc.1202070. [DOI] [PubMed] [Google Scholar]
- 43.Koivunen JP, et al. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Br. J. Cancer. 2008;99:245–252. doi: 10.1038/sj.bjc.6604469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA doublestrand breaks. DNA Repair (Amst.) 2004;3:909–918. doi: 10.1016/j.dnarep.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 45.Pollack JR, et al. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl Acad. Sci. USA. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sandsmark DK, et al. Nucleophosmin mediates mammalian target of rapamycindependent actin cytoskeleton dynamics and proliferation in neurofibromindeficient astrocytes. Cancer Res. 2007;67:4790–4799. doi: 10.1158/0008-5472.CAN-06-4470. [DOI] [PubMed] [Google Scholar]
- 47.Subramanian J, Govindan R. Lung cancer in never smokers: a review. J. Clin. Oncol. 2007;25:561–570. doi: 10.1200/JCO.2006.06.8015. [DOI] [PubMed] [Google Scholar]
- 48.Ahrendt SA, et al. p53 mutations and survival in stage I non-small-cell lung cancer: results of a prospective study. J. Natl Cancer Inst. 2003;95:961–970. doi: 10.1093/jnci/95.13.961. [DOI] [PubMed] [Google Scholar]
- 49.Beasley MB, Brambilla E, Travis WD. The 2004 World Health Organization classification of lung tumors. Semin. Roentgenol. 2005;40:90–97. doi: 10.1053/j.ro.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Motoi N, et al. Lung adenocarcinoma: modification of the 2004 WHO mixed subtype to include the major histologic subtype suggests correlations between papillary and micropapillary adenocarcinoma subtypes, EGFR mutations and gene expression analysis. Am. J. Surg. Pathol. 2008;32:810–827. doi: 10.1097/PAS.0b013e31815cb162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.