

Abstract
Bone morphogenetic proteins (BMPs) are known to induce ectopic bone. However, it is largely unknown how BMP signaling in osteoblasts directly regulates endogenous bone. This study investigated the mechanism by which BMP signaling through the type IA receptor (BMPR1A) regulates endogenous bone mass using an inducible Cre-loxP system. When BMPR1A in osteoblasts was conditionally disrupted during embryonic bone development, bone mass surprisingly was increased with upregulation of canonical Wnt signaling. Although levels of bone formation markers were modestly reduced, levels of resorption markers representing osteoclastogenesis were severely reduced, resulting in a net increase in bone mass. The reduction of osteoclastogenesis was primarily caused by Bmpr1a-deficiency in osteoblasts, at least through the RANKL-OPG pathway. Sclerostin (Sost) expression was downregulated by about 90% and SOST protein was undetectable in osteoblasts and osteocytes, whereas the Wnt signaling was upregulated. Treatment of Bmpr1a-deficient calvariae with sclerostin repressed the Wnt signaling and restored normal bone morphology. By gain of Smad-dependent BMPR1A signaling in mice, Sost expression was upregulated and osteoclastogenesis was increased. Finally, the Bmpr1a-deficient bone phenotype was rescued by enhancing BMPR1A signaling, with restoration of osteoclastogenesis. These findings demonstrate that BMPR1A signaling in osteoblasts restrain endogenous bone mass directly by upregulating osteoclastogenesis through the RANKL-OPG pathway, or indirectly by downregulating canonical Wnt signaling through sclerostin, a Wnt inhibitor and a bone mass mediator.
Keywords: BMP receptor IA, Bone mass, Canonical Wnt signaling, Osteoblast, Osteoclastogenesis, Sclerostin, Mouse
INTRODUCTION
BMPs were discovered as potent inducers of ectopic bone formation when implanted subcutaneously (Urist, 1965). Studies of human disorders chondrodysplasia (Thomas et al., 1996) and fibrodysplasia ossificans progressiva (Shore et al., 2006) indicate the importance of BMP signaling in cartilage and muscle, respectively, suggesting that BMP signaling plays an important role in controlling mineralization in musculoskeletogenesis. Bone is the primary component in skeletogenesis, and osteoblasts are the predominant cell type in bone. However, the mechanism by which BMP signaling in osteoblasts contributes the skeletogenesis has not been fully described.
The majority of bones, including long and ectopic bone, are formed through an endochondral process (Kronenberg, 2003). Condensed mesenchymal cells differentiate into chondrocytes to form a cartilage template that is later replaced by osteoblasts (Mackie et al., 2008; Maes et al., 2007). Mesenchymal cells and chondrocytes respond to BMP signaling to differentiate and maintain their features in vivo (Bandyopadhyay et al., 2006; Tsuji et al., 2006; Yoon et al., 2005). By contrast, during the alternate process of intramembranous bone formation, mesenchymal cells differentiate directly into osteoblasts without going through a cartilaginous phase. In this study, we genetically altered BMP signaling in osteoblasts using a mouse model. To avoid secondary effects from chondrocytes on osteoblasts in the endochondral process, we primarily examined intramembranous bone formation (e.g. calvaria) and demonstrated the direct effects that BMP signaling has on osteoblasts.
BMP receptor type IA (BMPR1A), which is abundantly expressed in bone, is activated by major BMP ligands BMP2 and BMP4. Conventional knockout of BMP2, BMP4 and BMPR1A in mice leads to embryonic death before bone development (Mishina et al., 1995; Winnier et al., 1995; Zhang and Bradley, 1996). We previously disrupted Bmpr1a during adult stages in an osteoblast-specific manner using Og2-Cre mice (Mishina et al., 2004). This study suggests that the response of osteoblasts to loss of BMP signaling is age dependent, as bone volume decreased in young mice but increased in old mice. Similarly, the mechanism by which BMP signaling regulates bone mass is not straightforward, as loss-of-function of BMP2 and gain-of-function of BMP4 both reduce bone mass (Okamoto et al., 2006; Tsuji et al., 2006). Bone mass is determined by the balance of bone formation and resorption, and osteoblasts regulate both processes. Thus, we focused on osteoblasts and addressed the complicated effect of BMP signaling on bone mass.
Human genetic studies have shown that loss-of-function mutations in components of Wnt signaling, such as the Wnt co-receptor low-density lipoprotein receptor-related protein 5 (LRP5), is associated with osteoporosis (Gong et al., 2001; Patel and Karsenty, 2002). Dominant missense LRP5 mutations are associated with high bone mass (HBM) diseases (Boyden et al., 2002; Little et al., 2002; Van Wesenbeeck et al., 2003), indicating that canonical/β-catenin Wnt signaling enhances bone mass (Baron et al., 2006; Glass and Karsenty, 2006; Krishnan et al., 2006). In vitro, Wnt signaling induces BMP expression (Bain et al., 2003; Winkler et al., 2005), whereas BMPs induce Wnt expression (Chen et al., 2007; Rawadi et al., 2003), suggesting that both BMP and Wnt signaling may synergistically regulate each other in osteoblast, possibly through autocrine/paracraine loop. Both BMP and Wnt signaling induce bone mass; however, the mechanism by which BMP and Wnt signaling cooperate to affect bone mass is not well understood, particularly during embryonic development when bone mass dramatically increases.
Here, we have employed a tamoxifen-inducible Cre-loxP system under the control of a 3.2 kb type I collagen promoter and have disrupted or upregulated BMP signaling through BMPR1A in osteoblasts during embryonic bone development. We unexpectedly found increased bone mass in response to loss of BMPR1A in osteoblasts and a new interaction between BMP and Wnt signaling through sclerostin.
MATERIALS AND METHODS
Mice and tamoxifen administration
Mice expressing the tamoxifen (TM)-inducible Cre fusion protein Cre-ER™ (Danielian et al., 1998; Hayashi and McMahon, 2002) under the control of a 3.2 kb mouse pro-collagen α1(I) promoter (Col1-CreER™), which is active in osteoblasts, odontoblasts and tendon fibroblasts (Rossert et al., 1995), were generated by pronuclear injection and crossed with floxed Bmpr1a mice (Mishina et al., 2002). Mice that conditionally express a constitutively active form of Bmpr1a (caBmpr1a), which has a mutation in the GS domain of BMPR1A that results in ligand-independent activation of Smad signaling after Cre recombination, were generated using a transgenic construct similar to one previously reported (Fukuda et al., 2006) (see Fig. S4A in the supplementary material). ROSA26 Cre reporter (R26R) (Soriano, 1999) and TOPGAL (DasGupta and Fuchs, 1999) mice were obtained from Dr Philippe Soriano and the Jackson Laboratory, respectively. Tamoxifen (TM; Sigma) was dissolved in a small volume of ethanol, diluted with corn oil at a concentration of 10 mg/ml. TM (75 mg/kg) was injected intraperitoneally daily into pregnant mice (100 to 200 ml/mouse) for at least 3 days starting at E13.5.
Histological analysis and skeletal preparation
Whole-mount β-gal staining was performed as previously described (Mishina et al., 2004). For histological analysis, fetuses were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned frontally for calvariae and sagittally for long bones at 6 μm. Sections were stained with Hematoxylin and Eosin or Eosin alone for β-gal stained samples. For von Kossa staining to detect mineral deposition, sections were covered with filtered 5% silver nitrate (Sigma), exposed to ultraviolet light for 45 minutes and placed in 5% sodium thiosulfate (Sigma) for a few seconds. For BrdU (bromodeoxyuridine) incorporation, 100 μM of BrdU (Roche) was injected into pregnant females intraperitoneally 2 hours before collecting calvariae. TRAP (tartrate resistant acid phosphatase) staining was performed using the leukocyte acid phosphatase kit (Sigma). Immunostaining was performed using primary antibodies against BMPR1A (Orbigen) (Yoon et al., 2005) and phospho-Smad1, -Smad5, -Smad8 (Cell Signaling) and sclerostin (R&D). Alexa Fluor (488, 594, Molecular Probes) and ABC kit (Santa Cruz Biotechnology) were used for detection. Frozen frontal sections at 10 μm were prepared for phospho-Smad1, -Smad5 and -Smad8 antibodies. For skeletal preparations, mice were dissected and fixed in 100% ethanol, and then stained with Alcian Blue and Alizarin Red. To count total cell number in sections, 1 μM of DAPI was treated for 10 minutes.
Quantitative real time RT-PCR (QRT-PCR)
RNA was isolated from calvariae using the Micro-FastTrack 2.0 Kit (Invitrogen). cDNA was synthesized using the SuperScript Preamplification System (GIBCO). PCR reactions, data quantification and analysis were performed (Applied Biosystems). Values were normalized to Gapdh using the 2−ΔΔCt method (Livak and Schmittgen, 2001).
Calvaria and osteoblast culture
For ex vivo bone culture, newborn calvariae were dissected at the sagittal suture and cultured in modified BGJ (Invitrogen) supplemented with ascorbic acid (Sigma, 50 mg/ml) and 5% fetal bovine serum for the first 24 hours in culture. Hemicalvariae were treated with 4-hydroxyl TM (Sigma, 100 ng/ml) and sclerostin (R&D, 50 ng/ml) in the absence of serum for 5 days. For in vitro culture, osteoblasts were isolated from conditional knockout (cKO) and wild-type newborn calvariae, and cultured in the same media with addition of 4-hydroxyl TM (Sigma, 100 ng/ml) every other day.
RESULTS
Tissue specificity and efficiency of Cre recombinase in Col1-CreER™ mice
Tamoxifen (TM)-inducible Col1-CreER™ mice were mated to ROSA26 reporter mice (R26R) to monitor Cre activity assessed by staining for β-galactosidase (β-gal). When TM was injected at embryonic day (E) 13.5 or earlier (E10.5), β-gal positive cells were first visible at E14.5 both in intramembranous and endochondral bones (data not shown). Daily injections for 3 days from E13.5 established strong β-gal activity at E18.5 (Fig. 1A). Neither Cre negative littermates nor Cre-positive embryos that did not receive TM showed β-gal activity (Fig. 1A, data not shown). Cre is active in immature periosteum wrapping growth plates, in osteogenic centers and in bone collars, but not in chondrocytes (Fig. 1B). By β-gal staining of whole calvariae, the positive areas started at the temporal border of parietal bones and expanded gradually as intramembranous bone formation progressed from E15.5 to E18.5 (Fig. 1C), where Cre was active in osteoblasts by histology. These results demonstrate the specificity and efficiency of Cre recombinase in Col1-CreER™ mice during embryonic bone development.
Fig. 1. Tamoxifen (TM)-dependent and osteoblast-specific Cre activity using Cre-ER™ mice.

(A) Cre-ER™ transgenic mice mated with ROSA26 Cre reporter mice (R26R). TM was injected daily into pregnant females for 3 days from E13.5 to E15.5. Cre-negative and -positive littermates were stained for β-gal at E18.5. (B) Cre activity in long bones. Forelimbs were stained for β-gal at E18.5. C, chondrocytes; black arrow, periosteum; red arrow, osteogenic center. Scale bars: 50 μm. (C) Cre recombination in calvariae at E15.5, E16.5 and E18.5 in whole head (lateral and overhead views) and histology of E18.5 calvariae. Broken line, areas of parietal bones (P). Scale bar: 20 μm. (D) BMPR1A as evaluated by immunohistochemistry using cKO calvariae at E18.5. BMPR1A, green; DAPI (nuclei), blue. Scale bars: 10 μm. (E) Immunoblotting for phosphorylated Smad 1/5/8 using E18.5 calvariae. The membrane was treated with polyclonal rabbit anti-phospho-Smad1/5/8 (1:1000) and monoclonal mouse anti-beta-actin (1:2000), and visualized by ECL.
Developmental abnormalities in BMPR1A cKO bones
Col1-CreER™ mice were bred into mice homozygous for floxed Bmpr1a. Bmpr1a cKO fetuses (cKO, Cre+; Bmpr1a fx/fx) and wild-type controls (WT, Cre-; Bmpr1a fx/fx) were collected after daily TM injection into pregnant female from E13.5. Gross morphology of E18.5 cKO was normal (data not shown). Production of BMPR1A was suppressed in osteoblasts and osteocytes in the cKO (Fig. 1D), demonstrating efficient and specific loss-of-function of BMPR1A in cKO osteoblasts. Phosphorylated Smad 1/5/8 was modestly reduced in cKO (Fig. 1E), presumably owing to remaining signals from other type I receptors, such as BMPR1B and ACVR1.
Mineralization was increased in cKO parietal bones when assessed by Alizarin Red staining, although skeletal shape in the forelimb and hindlimb were unchanged (Fig. 2A). The ratio of bone volume to total tissue volume (BV/TV) was 30% higher in cKO calvariae and femora at E18.5 when assessed by micro computed tomography (μCT) (Fig. 2B). Hematoxylin and Eosin staining of E18.5 calvariae demonstrated that cKO parietal bones were markedly thicker than wild type (Fig. 2C). The cKO bony area positive for Eosin was loose, discontinuous and disorganized, whereas in wild type it was compact and lamellar in structure. Von Kossa staining for Ca2+ in mineralized tissue showed increases in mineralized area of cKO calvariae where bone continuity was disrupted (Fig. 2D). In cKO femora where bone mass was increased (Fig. 2B), woven bone was increased in the primary spongiosa (Fig. 2E). By BrdU incorporation for detection of proliferating cells in vivo, positive cells per total cells in bone were unchanged in cKO calvaria (Fig. 2F). These facts suggest that bone mass of cKO was increased both in calvariae and femora, which follow intramembranous and endochondral ossification, respectively.
Fig. 2. Increased bone mass in Bmpr1a cKO mice.

(A) Skeletal preparation of the skull and long bones at E18.5 using Alizarin Red/Alcian Blue staining. Broken line, areas of parietal bones (P). (B) Bone volume over total tissue volume (BV/TV) obtained from μCT analysis on parietal bones of the skull (left) and femur (right) at E18.5. Values are expressed as mean±s.d. (wild type, n=5; cKO, n=4, Student’s t-test; *P<0.01). (C) Hematoxylin and Eosin staining of E18.5 calvariae. Boxed areas in frontal sections were magnified. Scale bars: 25 μm. (D) Von Kossa staining for Ca2+ using E18.5 calvariae. Scale bars: 25 μm. (E) Hematoxylin and Eosin staining of E18.5 humerus. Boxed areas in sagittal sections are magnified. Scale bars: 100 μm. (F) BrdU incorporation using E18.5 calvariae. BrdU-positive cells (arrows) per total cells in bone were unchanged. Scale bars: 25 μm.
Effects of BMPR1A signaling on bone formation and resorption markers
Bone mass is determined by the balance between bone formation and resorption. We examined changes in bone formation markers using calvariae and QRT-PCR. Expression of Runx2 and osterix (Sp7), genes that are required for osteoblast differentiation, and bone sialoprotein (Ibsp), which is induced rapidly prior to calcification, were reduced in E16.5 cKO (Fig. 3A). However, expression of these genes, as well as of alkaline phosphatase (Akp2; Alpl – Mouse Genome Informatics) and osteocalcin (Bglap2) was unchanged at E18.5. In addition, alkaline phosphatase activity was also unchanged at E18.5 (data not shown). These results suggest that bone formation is modestly reduced in E16.5 cKO calvariae, which could impact the phenotype of E18.5.
Fig. 3. Alteration of bone formation and resorption in Bmpr1a cKO mice.

(A) QRT-PCR for bone formation markers (Runx2, Sp7, Ibsp, Akp2 and Bglap2 using E16.5 and E18.5 calvariae). Values are expressed relative to wild type at E16.5. (B) QRT-PCR for bone resorption markers expressed by osteoclasts (Mmp9, Ctsk and Trap), and Rankl and Opg expressed by osteoblasts using E18.5 and E19.5 calvariae. Values are expressed relative to wild type at E18.5. (C) Relative ratio of Rankl to Opg expression levels calculated from Fig. 3B. Values are expressed relative to wild type at E18.5. (D) Evaluation of osteoclast activity by TRAP staining using E18.5 calvariae. The positive cells localized randomly in cKO calvariae compared with wild type (left two panels, red arrows). The percent of TRAP-positive cells per total cells in bone area detected by DAPI was significantly reduced in cKO calvariae (wild type, 13.9%; cKO, 6.4%, right panel). Scale bars: 50 μm. (E) QRT-PCR for BMP type I receptors (Bmpr1b, Acvr1), type II receptors (Bmpr2, Acvr2a and Acvr2b) and potential ligands for these receptors (Bmp2, Bmp4, Bmp6 and Bmp7) using E18.5 calvariae. Values in A–E represent mean±s.d. from a minimum of three independent experiments using wild-type and cKO bones. Student’s t-test; *P<0.05.
Among bone resorption markers expressed by osteoclasts, expressions of Mmp9 and cathepsin K (Ctsk) were decreased 25% and 35% in E19.5 cKO calvariae, respectively, and that of tartrate resistant acid phosphatase (Trap) was reduced 30% and 35% at E18.5 and E19.5, respectively (Fig. 3B). Osteoclastogenesis is regulated by RANKL (receptor activator of NFκB ligand), an osteoclast differentiation factor, and OPG (osteoprotegerin), a decoy receptor for RANKL, both expressed by osteoblasts (Simonet et al., 1997). Rankl expression was reduced 30% at E19.5, and Opg was increased 50% and 80% at E18.5 and E19.5, respectively (Fig. 3B), resulting in the reduction of the ratio, Rankl to Opg, by 30% and 60% at E18.5 and E19.5, respectively (Fig. 3C). As RANKL stimulates osteoclastogenesis, whereas OPG inhibits it, these changes were consistent with the reduced osteoclastogenesis in cKO calvariae (Fig. 3B). Consistent with these data, osteoclast activity evaluated by TRAP staining was significantly reduced in cKO calvariae at E18.5 (Fig. 3D). In addition, expressions of other BMP receptors type I (Bmpr1b, Acvr1), type II (Bmpr2, Acvr2a, Acvr2b) and their ligands (Bmp2, Bmp4, Bmp6 and Bmp7), were not increased in cKO calvariae at E18.5 when assessed using QRT-PCR (Fig. 3E), suggesting that a compensation for the loss of BMPR1A by other BMP receptors or ligands is unlikely. These results suggest that osteoclastogenesis in cKO calvariae was decreased by loss of BMPR1A.
Effects of BMPR1A signaling on OPG and RANKL in vitro
Bmpr1a-deficient osteoblasts were reproduced by infecting primary osteoblasts from Bmpr1a fx/fx calvariae with recombinant adenovirus expressing Cre protein. Expressions of Bmpr1a and Rankl were reduced 70% and 40%, respectively, whereas Opg increased 2.5-fold in cKO osteoblasts (CRE) compared with control (Mock) (see Fig. S1A in the supplementary material), resulting in reduced ratio of Rankl to Opg by 80% (see Fig. S1A in the supplementary material). These indicate reduced osteoclastogenesis in Bmpr1a-deficient osteoblasts at least through the RANKL-OPG pathway, consistent with in vivo data (Fig. 3).
Monocytic cells, precursors of osteoclasts, were isolated from adult cKO and wild-type spleens, and were induced by RANKL and M-CSF. There was no difference both in osteoclast number and Bmpr1a expression levels between wild-type and cKO cells (see Fig. S1B,C in the supplementary material), suggesting that cKO osteoclasts are intact and able to fully accomplish osteoclastogenesis by responding to these cytokines. Next, a mixture of osteoblasts and osteoclasts was isolated from adult cKO and wild-type bone marrow cells independently, and induced by 1α, 25-dihydroxyvitamin D3. The cKO mixture was defective in osteoclast number and Bmpr1a expression levels (see Fig. S1B,C in the supplementary material). There was no Cre activity in osteoclasts in bone marrow (data not shown). These results suggest that osteoclasts in cKO bone marrow are intact but cannot accomplish osteoclastogenesis, partly because of a defect in RANKL-OPG signaling in Bmpr1a-deficient osteoblasts. Taken together, these data suggest that the reduced osteoclastogenesis in cKO calvaria (Fig. 3) is primarily caused by defects in osteoblasts.
Upregulation of canonical Wnt signaling in cKO bones
Both BMP and Wnt signaling (Baron et al., 2006; Glass and Karsenty, 2006; Hartmann, 2006; Krishnan et al., 2006) regulate bone mass. However, there is no described interaction between the two in either mouse models or human mutation. We first investigated canonical Wnt signaling in cKO bones using TOPGAL Wnt reporter mice. The Wnt signaling assessed by β-gal staining was increased in cKO calvariae at E17.5 (Fig. 4A). Relative number of β-gal-positive cells was 8.5 times higher in cKO calvariae compared with wild type, which was significant (Fig. 4C). Although it is known that endogenous levels of canonical Wnt signaling assessed using TOPGAL mice are low in osteoblasts during embryonic bone development (Hens et al., 2005), the intensity of β-gal staining was dramatically increased in cKO long bones compared with wild type (Fig. 4D,E). These suggest that canonical Wnt signaling in cKO mice was stimulated during both endochondral and intramembranous ossification. Thus, Bmpr1a deficiency in osteoblasts upregulates Wnt signaling at least through the canonical pathway.
Fig. 4. Upregulation of canonical Wnt signaling in Bmpr1a cKO mice.

(A) Canonical Wnt signaling assessed by TOPGAL mice using E17.5 calvariae (upper panel, lateral view; lower panel, top view). Two independent parietal bones from two littermates in each group after dissection are shown in the right-hand panel. Red arrow indicates enhanced Wnt signaling. Broken line, areas of parietal bones (P). (B) Histological analysis assessed by TOPGAL mice using E17.5 calvariae. Scale bars: 50 μm. (C) Relative cell number of DAPI and β-gal positive cells. Cells were counted in 50 fields of E17.5 calvariae from cKO (n=4) and wild type (n=4). Total cell number was obtained by counting DAPI-positive nuclei in bone. Cell number of wild type is set as 1.0. There were 2.3 times more total cells in cKO calvariae in a given section than in wild type owing to the thicker tissues, but the number of β-gal-positive cells was 19.7 times greater, resulting in an 8.5-fold increase in the proportion of β-gal positive cells in cKO mice. (D) Canonical Wnt signaling assessed by TOPGAL mice using humerus, ulna and radius at E17.5. Red arrow indicates enhanced Wnt signaling. (E) Histological analysis assessed by TOPGAL mice using radius at E17.5. Asterisks, growth plates. Scale bars: 100 μm.
Downregulation of sclerostin in cKO bones
Alternation of Wnt-related genes were further examined. Sclerostin (Sost) expression was consistently reduced by 95% in cKO calvariae from E16.5 to E18.5 when assessed with QRT-PCR (Fig. 5A) and was the most severely downregulated at E18.5 on microarray data (−5.68-fold, P=3.27E−24) (see Fig. S2 in the supplementary material). Expression levels of Wnt target genes Axin2 and Ctgf were significantly increased in cKO calvariae at E18.5 when assessed with QRT-PCR (Fig. 5B), but those of Wnt ligands (Wnt3a, Wnt5a, Wnt7a, Wnt7b and Wnt9a), other Wnt inhibitors [dickkopf 1 (Dkk1), Dkk2 and secreted frizzled-related proteins] and co-receptor Lrp5 were unchanged both when assessed with microarray and QRT-PCR at E18.5 (Fig. 5A, data not shown). Immunohistochemistry using E17.5 cKO calvariae confirmed that Bmpr1a deficiency in osteoblasts and osteocytes correlated with increased levels of β-gal staining and reduced production of sclerostin at cellular levels (Fig. 5C,D). The reduction of sclerostin was reproduced in vitro using primary osteoblasts from newborn cKO mice (Fig. 5E). Similarly, Sost expression was 90% reduced by loss of BMPR1A when assessed using adenoviral Cre infection in vitro (see Fig. S1A in the supplementary material). These results suggest that sclerostin is a downstream effector of BMPR1A on Wnt signaling, and that Bmpr1a deficiency in osteoblasts increased canonical Wnt signaling at least by the suppression of sclerostin.
Fig. 5. Inhibition of canonical Wnt signaling by sclerostin in Bmpr1a cKO mice.

(A) QRT-PCR analysis for Sost, Dkk1, Dkk2 and Lrp5 using calvariae at E16.5, E17.5 and E18.5. Values are expressed relative to wild type at E16.5. Student’s t-test; *P<0.05. (B) QRT-PCR analysis for Wnt target gene Axin2 and Ctgf using E18.5 calvariae. Expression levels of Axin2 and Ctgf were significantly increased in cKO calvariae. Student’s t-test; *P<0.05. (C) Immunohistochemical staining of sclerostin (brown) counterstained with Hematoxylin (blue) using E17.5 calvariae. Broken line, osteoblasts. Scale bars: 50 μm, left panel; 10 μm, right panel. (D) Detection of BMPR1A (green), canonical Wnt signaling (blue) and sclerostin (red) using E17.5 calvariae. Canonical Wnt signaling was assessed by β-gal staining using TOPGAL mice. Nuclei were stained with DAPI (blue). Broken line, osteoblasts. Scale bars: 20 μm. (E) Immunohistochemical staining for sclerostin (red) in primary osteoblasts from Bmpr1a cKO and wild-type control. Nuclei were stained with DAPI (blue). Scale bars: 20 μm.
Wnt inhibitor sclerostin regulates bone mass
The effect of sclerostin on Wnt signaling and bone mass was further examined using Bmpr1a-deficient calvariae treated with 4-hydroxyl TM for 5 days ex vivo. Cre-dependent DNA recombination and upregulated Wnt signaling were confirmed by β-gal staining using R26R and cKO:TOPGAL newborn calvariae, respectively (Fig. 6A, parts a,b). Expression levels of Bmpr1a and Sost were significantly reduced by 80% and by 90%, respectively, but Dkk1, Dkk2 and Lrp5 were unchanged (Fig. 6A, parts c,d). After treatment with 4-hydroxyl TM for 5 days, expression levels of bone resorption markers Mmp9, Ctsk and Trap were significantly reduced when assessed by QRT-PCR (Fig. 6A, part d), consistent with in vivo results (Fig. 4A, Fig. 5A). Next, when sclerostin was added in the culture, both canonical Wnt signaling and bone thickness were reduced in cKO calvariae (cKO+Scl, Fig. 6B), and the ratio of β-gal positive cells was significantly reduced (cKO, 65.3%; cKO+Scl, 17.2%). The expression levels of osteoclast markers Rankl and Opg, and the ratio of Rankl to Opg were partially restored in the cKO compared with wild type (Fig. 6C). Moreover, TRAP staining pattern in the cKO resembled that in the wild type in response to sclerostin treatment (Fig. 6D, compare with Fig. 3D). These results suggest that exogenous sclerostin in Bmpr1a-deficienct calvariae partially restore morphological changes, Wnt signaling and osteoclastogenesis. In addition, Noggin treatment ex vivo reduced Sost expression by 85% and upregulated the Wnt signaling (see Fig. S3A,B in the supplementary material). Noggin antagonizes both BMP2 and BMP4, which are potent ligands of BMPR1A. Thus, we conclude that loss of BMP signaling reduces Sost expression, and sclerostin inhibits canonical Wnt signaling as a bone mass regulator during embryonic bone development.
Fig. 6. Suppressed expression of sclerostin by loss of BMP signaling ex vivo.

(A) Newborn mouse calvariae were cultured for 5 days treated with 4OH-TM (100 ng/ml). (a) Confirmation of Cre activity in the calvariae from CreER:R26R mice assessed by β-gal staining. (b) Upregulation of canonical Wnt signaling in the calvariae from CreER:Bmpr1afx/fx:TOPGAL mice assessed by β-gal staining. Broken lines, areas of parietal bones (P). (c) Expression of Wnt inhibitors Sost, Dkk1, Dkk2 and Lrp5 assessed by QRT-PCR. (d) Expressions of bone resorption markers Mmp9, Ctsk, TRAP and Bmpr1a. Values are expressed relative to those of wild type (Cre-, TM-). (B) Sclerostin treatment of wild-type (WT+Scl) and cKO (cKO+Scl) calvariae ex vivo. The ratio of β-gal to DAPI-positive cells was evaluated from 50 fields in frontal sections (n=3, in each condition). Broken line, parietal bones. Scale bars: 50 μm. (C) Expression of bone resorption markers Rankl and Opg, and relative ratio of Rankl to Opg using wild-type and cKO calvariae in sclerostin treated versus untreated groups. Values are expressed relative to those of wild type without sclerostin treatment. (D) TRAP staining of cKO calvariae treated with sclerostin and non-treated ex vivo. Values in A–C represent mean±s.d. from three independent experiments. Student’s t-test; * P<0.05.
Smad-dependent BMPR1A signaling upregulates sclerostin expression in vivo
To reveal the effects of Smad-dependent BMPR1A signaling on sclerostin expression and bone morphology in vivo, we generated inducible transgenic mice expressing caBmpr1a (constitutively activated Bmpr1a) in osteoblasts (see Fig. S4A in the supplementary material). After daily TM injection from E13.5 to E17.5, gross morphology of caBmpr1a fetuses was relatively normal at E18.5 (data not shown). Histology showed moderately reduced thickness in caBmpr1a calvariae (Cre+, caBmpr1a+) at E18.5, where levels of phosphorylated Smads were enhanced compared with controls (Cre-, caBmpr1a+) (Fig. 7A). Sost expression when assessed by QRT-PCR increased approximately sevenfold in the transgenic calvariae at E18.5, indicating that Smad signaling positively controls Sost expression. Expressions of bone resorption markers were all increased over fourfold, and Rankl levels increased four times while Opg levels decreased 40% (Fig. 7B), resulting in a 6.5-fold increase in the ratio of Rankl to Opg (Fig. 7C). Expression of bone formation markers was also increased in the caBmpr1a (see Fig. S4B in the supplementary material). These changes observed in the caBmpr1a were the opposite of those seen in Bmpr1a cKO calvariae (Fig. 3). We next generated rescue mice expressing caBmpr1a on a Bmpr1a cKO background (Cre+, caBmpr1a+, Bmpr1afx/fx) and compared them with littermate Bmpr1a cKO mice (cKO: Cre+, caBmpr1a−, Bmpr1afx/fx). In E18.5 calvariae from rescued mice, expression of Sost, Rankl and osteoclast markers increased over four times (Fig. 7D), and the ratio of Rankl to Opg increased about 2.5-fold (Fig. 7E) when compared with the cKO. There was also a reduction in morphological changes observed in littermate Bmpr1a cKO mice (Fig. 7F). These results strongly suggest that Sost expression is regulated by BMPR1A signaling at least through the Smad pathway.
Fig. 7. Enhanced BMPR1A signaling upregulates both sclerostin and osteoclastogenesis.

(A) Constitutively active Bmpr1a (caBmpr1a) mouse fetuses at E18.5 induced daily by TM injection from E13.5. Hematoxylin and Eosin staining showed moderately reduced thickness in the caBmpr1a calvariae (Cre+, caBmpr1a+) where levels of phosphorylated Smad1/5/8 (brown) were enhanced compared with littermate controls (Cre−, caBmpr1a+). Scale bars: 50 μm. (B) Expressions of Sost and the bone resorption markers Rankl and Opg assessed by QRT-PCR using caBmpr1a and control calvariae at E18.5. Values are expressed relative to control. (C) Relative ratio of Rankl to Opg calculated based on the expression levels in Fig. 7B. (D) Expressions of Sost, bone resorption markers, Rankl and Opg by QRT-PCR using rescued (black bar: Cre+, caBmpr1a+, Bmpr1a fx/fx) and littermate cKO (gray bar: Cre+, caBmpr1a−, Bmpr1a fx/fx) calvariae at E18.5. Values of rescued mice are expressed relative to littermate cKO mice. (E) Relative ratio of Rankl to Opg calculated based on expression levels in Fig. 7D. (F) Hematoxylin and Eosin staining of rescued and cKO calvariae at E18.5. Scale bars: 100 μm. Values in B–E are mean±s.d. from three independent experiments. Student’s t-test; *P<0.05.
DISCUSSION
This study showed that loss of BMPR1A increased bone mass and leads to two possibilities: (1) that BMPR1A signaling directly regulates osteoclastogenesis through RANKL-OPG pathway; or (2) indirectly regulates them via secondary mediator sclerostin as a Wnt inhibitor (Fig. 8). Both indicate that BMP signaling in osteoblasts restrains bone mass.
Fig. 8. A model of the relationship between BMPR1A and canonical Wnt signaling in mouse bone.
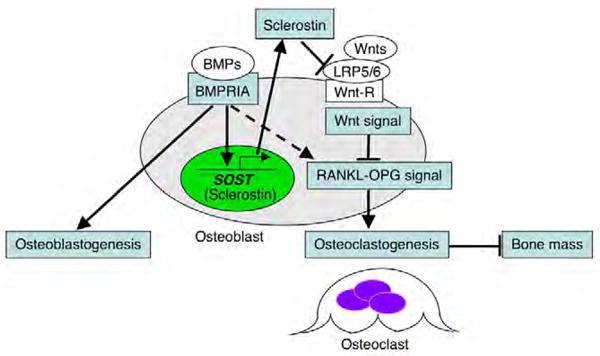
BMPR1A signaling upregulates sclerostin expression, leading to an inhibition of canonical Wnt signaling and a decrease in bone mass by upregulating osteoclastogenesis through the RANKL-OPG pathway. Sclerostin, the SOST gene product, acts as a downstream effector of BMPR1A signaling, an inhibitor of canonical Wnt signaling and a bone mass-determining factor. Broken line indicates another possibility: that BMP signaling directly upregulates osteoclastogenesis through the RANKL-OPG pathway.
BMP signaling, bone formation and bone mass
By using a 3.2 kb mouse pro-collagen α1(I) promoter (Rossert et al., 1995), we demonstrate that reduction in BMP signaling through BMPR1A in osteoblasts increased bone mass during embryonic stages. This is consistent with our previous report that showed increased bone volume (BV/TV) at 10 months by loss of BMP signaling in osteoblasts, using Og2-Cre mice in which osteoblasts initiate Cre expression postnatally. However, it is not consistent with 3-month-old mice, which showed decreased bone mass (Mishina et al., 2004). The bone phenotype observed in Bmpr1a-deficient mice with Og2-Cre was milder than that with Col1-CreER™. These discrepancies may be due to differences in the timing of recombination between promoters. As characteristics of osteoblasts change as they mature (Aubin, 1998), the effects of disrupting BMPR1A signaling may be influenced by the stages of osteoblast maturation. In Col1-CreER™ mice, Cre activity was detected in immature periosteum that wraps growth plates, in osteogenic centers and in bone collars (Fig. 1B), indicating that Cre activation occurs just after commitment of mesenchymal cells towards osteoblastic cells. Therefore, Col1-CreER™ mice can induce recombination earlier in osteoblastogenesis, including in preosteoblasts, compared with Og2-Cre mice, which could explain why the Bmpr1a-deficient mice using Og2-Cre mice did not change expression levels of early osteogenic markers Runx2 and Bsp, and showed mild bone phenotype (Mishina et al., 2004).
Expression levels of bone formation markers (Runx2, Sp7, Ibsp, Akp2 and Bglap2) were increased more than twofold in caBmpr1a mice (see Fig. S4B in the supplementary material), indicating that BMP signaling through Smad1/5/8 induces osteoblastogenesis, as is well known in vitro (Chen et al., 2004). Some of these markers (Runx2, Sp7 and Ibsp) were reduced in cKO bones as expected (Fig. 3A), consistent with the modest decrease in Smad phosphorylation levels (Fig. 1E) and our previous report that loss of BMP signaling decreases bone formation rate over bone surface (BFR/BS) during adulthood (Mishina et al., 2004). It is also suggested that normal bones respond to endogenous BMPs by phosphorylating Smads at a very low level, which is difficult to detect by immunostaining (Fig. 7A). It is possible that proliferation of osteoblasts is increased in cKO bones, which could influence increased bone mass; however, this is less likely because the number of BrdU-positive cells per total cells in bone was unchanged (Fig. 2E). Histomorphometric analysis of bone is an established method to assess bone formation but is technically unfeasible in fetuses. Thus, we applied histomorphometry to adult cKO mice, where we confirmed osteoblasts failed to support osteoclastogenesis (see Fig. S1B in the supplementary material). Bone volume (BV/TV) was significantly increased, but formation rate (BFR/BS) and osteoclast number per bone area (N.Oc/T.Ar) were significantly decreased and osteoblast surface per bone surface (Ob.S/BS) were unchanged in the adult cKO (Kamiya et al., 2008). Similar to embryonic cKO data (Fig. 3), expression levels of osteoclast markers and the ratio of Rankl to Opg were significantly reduced when assessed by QRT-PCR in the adult cKO (Kamiya et al., 2008). These results suggest that osteoblast number is unchanged and bone formation is modestly reduced, while resorption markedly decreases in cKO bones, resulting in a net increase in bone mass. In addition, BMP signaling can control bone formation and resorption by inducing both osteoblastogenesis and osteoclastogenesis in vivo.
RANKL-OPG pathway involved in BMP signaling
This study found that osteoblasts respond to BMP signaling to support differentiation of osteoclasts at least through the RANKL-OPG pathway. The link between BMP signaling and the RANKL-OPG pathway has been reported in vitro, showing that BMP2 enhances osteoclastogenesis by upregulating Rankl (Itoh et al., 2001; Usui et al., 2008). However, the link is not detected in those mice overexpressing Noggin or Bmp4 in osteoblasts using a 2.3 kb type I collagen promoter (Okamoto et al., 2006) or by knocking out BMPR1A using an osteocalcin promoter (Og2-Cre) (Mishina et al., 2004), whereas osteoclastogenesis is upregulated by BMP signaling. As the 2.3 kb type I collagen and osteocalcin promoter are first activated in mature osteoblasts (Ducy and Karsenty, 1995; Kalajzic et al., 2002), immature osteoblasts, where the 3.2 kb type I collagen promoter is active, may be crucial for regulating osteoclastogenesis through the RANKL-OPG pathway. Furthermore, it is possible that BMP signaling directly controls osteoclastogenesis through the RANKL-OPG signaling pathway probably by downregulating Opg expression in addition to upregulating Rankl, because BMP response elements were identified in the Opg promoter by Evolutionary Conserved Regions (ECR) browser search (data not shown). Further analysis is necessary to clarify the direct regulation of RANKL-OPG pathway by BMP signaling.
It is also possible that secondary mediators such as Wnts regulate expression of RANKL and OPG. Two recent studies have suggested that Wnt signaling in osteoblasts through the canonical β-catenin pathway not only boosts bone formation by fostering osteoblast activity but can also inhibit bone resorption by affecting osteoclasts (Goldring and Goldring, 2007). One study provided evidence that the Wnt pathway positively regulates osteoblast expression of osteoprotegerin (OPG) by overexpressing stabilized β-catenin in osteoblasts in mice, resulting in decreased osteoclast differentiation and increased bone volume (Glass et al., 2005). Another study showed that osteoblasts lacking the β-catenin gene exhibited impaired maturation and mineralization with elevated expression of Rankl and diminished Opg, suggesting that the Wnt pathway can suppress osteoclast-mediated bone resorption (Holmen et al., 2005). In our study, expression levels of Opg and Rankl as well as osteoclast markers (Mmp9, Ctsk and Trap) were partially restored by exogenous sclerostin ex vivo with a concomitant reduction in Wnt signaling (Fig. 6). Similarly, Wnt inhibitors Dkk1 and Dkk2 can induce osteoclastogenesis by changing the RANKL-OPG pathway in vitro (Fujita and Janz, 2007). These facts suggest that Wnt inhibitors in the canonical/β-catenin pathway enhance osteoclastogenesis. In addition, Wnt/β-catenin-responsive LEF1-binding sites were identified both in the Opg promoter and the Rankl promoter by an ECR browser search (data not shown). These facts strongly suggest that the changes in Bmpr1a-deficient bones are due to decreased bone resorption and osteoclastogenesis through the RANKL-OPG pathway, and that sclerostin is presumably involved in the pathway as a Wnt signaling inhibitor (Fig. 8).
Sclerostin, a downstream target of BMPR1A
Sclerostin, which is encoded by Sost, has homology to the DAN family of BMP antagonists, which can control both BMP and Wnt signaling in Xenopus (Bell et al., 2003; Itasaki et al., 2003; Piccolo et al., 1999). However, although sclerostin directly binds BMPs (Kusu et al., 2003; Winkler et al., 2003), its antagonistic effects on BMP activity in mammals are still controversial. Resent in vitro studies showed that sclerostin is not a BMP antagonist (van Bezooijen et al., 2004), as it binds only weakly to BMPs and does not inhibit direct BMP-induced responses (ten Dijke et al., 2008). Together with the evidence from human mutations in SOST or its co-receptor LRP5, in vitro studies revealed that sclerostin is a Wnt inhibitor that binds LRP5 (Li et al., 2005; Semenov et al., 2005; van Bezooijen et al., 2007). Loss-of-function and hypomorphic mutations in SOST cause sclerosteosis (Balemans et al., 2001; Brunkow et al., 2001) and Van Buchem disease (Balemans et al., 2002; Staehling-Hampton et al., 2002), respectively, and bone mass is increased in these disorders, similar to Sost KO mouse (Li et al., 2008). In human and mouse, these Sost mutants share the high bone mass (HBM) phenotype with humans who have gain-of-function mutations in LRP5 (Boyden et al., 2002; Little et al., 2002; Van Wesenbeeck et al., 2003) and by overexpression of Lrp5 in mice (Babij et al., 2003). By contrast, loss-of-function of LRP5 leads to osteoporosis pseudoglioma syndrome in humans with low bone mass (Gong et al., 2001; Patel and Karsenty, 2002), which is similar to the bone phenotype of mice that overexpress Sost (Winkler et al., 2003). Consistent with these facts, Bmpr1a cKO mice show a HBM phenotype with lack of sclerostin expression. In addition, sclerostin differs from the BMP antagonist Noggin with respect to effects on Wnt signaling, as sclerostin inhibits Wnt (Fig. 6) whereas Noggin upregulates it (see Fig. S3A in the supplementary material). Thus, our study strongly suggests that sclerostin physiologically acts as an inhibitor of canonical Wnt signaling in vivo during embryonic bone development, and loss of sclerostin largely directs the Bmpr1a cKO bone phenotype.
The phenotype of Sost KO mice (Li et al., 2008) is different from that observed in Bmpr1a cKO with respect to the morphology of the bone, as Bmpr1a cKO mice showed disorganized bone structure (Fig. 2), which is not described in Sost KO mice (Li et al., 2008) or SOST human disorders. This fact implies that BMPR1A is not only an upstream effector of sclerostin but also has other roles beyond regulating Sost in the osteoblasts. In the Sost KO there is no increase in osteoclast number or erosion surface, which is consistent with mice overexpressing LRP5 with HBM phenotype (Babij et al., 2003), suggesting that bone formation and resorption are uncoupled in these mutants. However, as we discussed earlier, loss of BMP signaling reduced both bone formation and resorption together. In addition, the diameter of collagen fibrils in Bmpr1a cKO bones were heterogeneous, aggregations of collagen fibers were disorganized and MMP expression levels were reduced (data not shown), suggesting that BMP signaling additionally regulates proper bone structure and turnover. In support of our speculation, reduction in osteoclastogenesis was not fully restored by sclerostin treatment ex vivo (Fig. 6C). These facts suggest that BMP signaling in osteoblasts directly regulates bone resorption independently of Wnt signaling through sclerostin, which in turn maintains proper bone structure and bone mass.
Sost expression was dramatically downregulated by removal of Bmpr1a in vivo, whereas it was upregulated by enhancing Smad-dependent BMPR1A signaling using caBmpr1a. It is reported that both BMP2 and BMP4, potent ligands of BMPR1A, induce Sost expression in mouse and human osteoblasts (Ohyama et al., 2004; Sutherland et al., 2004). Although sclerostin is produced primarily by osteocytes in adult bones (Poole et al., 2005; van Bezooijen et al., 2004), it is also detected in osteoblasts before birth (Fig. 5), suggesting the positive regulatory role of BMPR1A signaling in Sost expression both in osteoblasts and osteocytes during embryonic bone development. Interestingly, the promoter region of the Sost gene contains putative BMP response elements that were detected by an ECR browser search (data not shown), suggesting possible regulation of Sost by BMP signaling at the transcriptional level.
Interaction of BMP and Wnt signaling in bone
Accumulating evidence suggests that both BMP and Wnt signaling may regulate each other in a context and age-dependent manner (Barrow et al., 2003; Guo et al., 2004; He et al., 2004; Huelsken et al., 2001). Only a few cascades such as Pten/Akt (Zhang et al., 2006) and Smad1/Dvl1 (Liu et al., 2006) are reported in intracellular crosstalk between the BMP and Wnt pathways. However, as in bone, the interaction between BMP and Wnt signaling is not well described generally in vivo, partly because alteration of BMP signaling has not been studied in Wnt signaling mutants and vice versa. Some in vitro studies have demonstrated that both BMP and Wnt pathways synergistically regulate each other possibly through autocrine/paracraine loop, as BMPs induce Wnts in C2C12 cells and primary osteoblasts (Chen et al., 2007; Rawadi et al., 2003), and Wnt signaling, on the contrary, enhances BMPs expression in C3H10T1/2 cells (Bain et al., 2003; Winkler et al., 2005). These studies indicate that BMP signaling may upregulate Wnt signaling.
By contrast, our in vivo study suggests that BMP signaling downregulates Wnt signaling, as loss of BMPR1A signaling upregulates Wnt signaling by inhibiting Sost expression. These discrepancies are partly due to the differences of cell type examined, as both osteoblasts and osteoclasts always affect each other as coupling factors in vivo, and coupling effects on the two signaling pathways are difficult to address using osteoblasts alone in vitro. Furthermore, it is also difficult for in vitro studies to address age-dependent changes in both signaling pathways and their interactions. Similar to our results, other studies have shown antagonistic interaction of BMP and Wnt signaling in lung (Dean et al., 2005), intestine (He et al., 2004), hair (Zhang et al., 2006) and joints (Guo et al., 2004). Moreover, Noggin treatment ex vivo upregulated canonical Wnt signaling and downregulated Sost expression (see Fig. S3A,B in the supplementary material), presumably by antagonizing BMP2 and BMP4, ligands for BMPR1A. Thus, our study strongly suggests that BMPR1A signaling in osteoblasts regulates negatively canonical Wnt signaling through downstream effector sclerostin during embryonic bone development.
In conclusion, we demonstrate a new interaction between the BMP and Wnt signaling pathways in osteoblasts through sclerostin, a Wnt inhibitor and a bone mass regulator. BMP signaling via BMPR1A directs osteoblasts to reduce bone mass in part by upregulating sclerostin expression and supporting osteoclastogenesis through the RANKL-OPG pathway.
Supplementary Material
Acknowledgments
We gratefully thank Tomokazu Fukuda and Greg Scott for generation of caBmpr1a mouse line and Donald Lucas for critical reading of this manuscript. This work was supported by NIH grants P01 DK56246 (H.K.), R01 AR051587 (J.F.), R21 AR052824 (M.Y.), ES071003-11 and a conditional gift from RIKEN Brain Science Institute (Y.M.) and Lilly Fellowship Foundation (N.K.).
Footnotes
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/135/22/3801/DC1
References
- Aubin JE. Advances in the osteoblast lineage. Biochem Cell Biol. 1998;76:899–910. [PubMed] [Google Scholar]
- Babij P, Zhao W, Small C, Kharode Y, Yaworsky PJ, Bouxsein ML, Reddy PS, Bodine PV, Robinson JA, Bhat B, et al. High bone mass in mice expressing a mutant LRP5 gene. J Bone Miner Res. 2003;18:960–974. doi: 10.1359/jbmr.2003.18.6.960. [DOI] [PubMed] [Google Scholar]
- Bain G, Muller T, Wang X, Papkoff J. Activated beta-catenin induces osteoblast differentiation of C3H10T1/2 cells and participates in BMP2 mediated signal transduction. Biochem Biophys Res Commun. 2003;301:84–91. doi: 10.1016/s0006-291x(02)02951-0. [DOI] [PubMed] [Google Scholar]
- Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10:537–543. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216. doi: 10.1371/journal.pgen.0020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron R, Rawadi G, Roman-Roman S. Wnt signaling: a key regulator of bone mass. Curr Top Dev Biol. 2006;76:103–127. doi: 10.1016/S0070-2153(06)76004-5. [DOI] [PubMed] [Google Scholar]
- Barrow JR, Thomas KR, Boussadia-Zahui O, Moore R, Kemler R, Capecchi MR, McMahon AP. Ectodermal Wnt3/beta-catenin signaling is required for the establishment and maintenance of the apical ectodermal ridge. Genes Dev. 2003;17:394–409. doi: 10.1101/gad.1044903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E, Munoz-Sanjuan I, Altmann CR, Vonica A, Brivanlou AH. Cell fate specification and competence by Coco, a maternal BMP, TGFbeta and Wnt inhibitor. Development. 2003;130:1381–1389. doi: 10.1242/dev.00344. [DOI] [PubMed] [Google Scholar]
- Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. New Engl J Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–589. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22:233–241. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- Chen Y, Whetstone HC, Youn A, Nadesan P, Chow EC, Lin AC, Alman BA. Beta-catenin signaling pathway is crucial for bone morphogenetic protein 2 to induce new bone formation. J Biol Chem. 2007;282:526–533. doi: 10.1074/jbc.M602700200. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Dean CH, Miller LA, Smith AN, Dufort D, Lang RA, Niswander LA. Canonical Wnt signaling negatively regulates branching morphogenesis of the lung and lacrimal gland. Dev Biol. 2005;286:270–286. doi: 10.1016/j.ydbio.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol. 1995;15:1858–1869. doi: 10.1128/mcb.15.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K, Janz S. Attenuation of WNT signaling by DKK-1 and -2 regulates BMP2-induced osteoblast differentiation and expression of OPG, RANKL and M-CSF. Mol Cancer. 2007;6:71. doi: 10.1186/1476-4598-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Scott G, Komatsu Y, Araya R, Kawano M, Ray MK, Yamada M, Mishina Y. Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis. 2006;44:159–167. doi: 10.1002/dvg.20201. [DOI] [PubMed] [Google Scholar]
- Glass DA, 2nd, Karsenty G. Molecular bases of the regulation of bone remodeling by the canonical Wnt signaling pathway. Curr Top Dev Biol. 2006;73:43–84. doi: 10.1016/S0070-2153(05)73002-7. [DOI] [PubMed] [Google Scholar]
- Glass DA, 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Goldring SR, Goldring MB. Eating bone or adding it: the Wnt pathway decides. Nat Med. 2007;13:133–134. doi: 10.1038/nm0207-133. [DOI] [PubMed] [Google Scholar]
- Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- Guo X, Day TF, Jiang X, Garrett-Beal L, Topol L, Yang Y. Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004;18:2404–2417. doi: 10.1101/gad.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann C. A Wnt canon orchestrating osteoblastogenesis. Trends Cell Biol. 2006;16:151–158. doi: 10.1016/j.tcb.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, Tian Q, Zeng X, He X, Wiedemann LM, et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat Genet. 2004;36:1117–1121. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- Hens JR, Wilson KM, Dann P, Chen X, Horowitz MC, Wysolmerski JJ. TOPGAL mice show that the canonical Wnt signaling pathway is active during bone development and growth and is activated by mechanical loading in vitro. J Bone Miner Res. 2005;20:1103–1113. doi: 10.1359/JBMR.050210. [DOI] [PubMed] [Google Scholar]
- Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, Deng L, Clemens TL, Williams BO. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. 2005;280:21162–21168. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- Itasaki N, Jones CM, Mercurio S, Rowe A, Domingos PM, Smith JC, Krumlauf R. Wise, a context-dependent activator and inhibitor of Wnt signalling. Development. 2003;130:4295–4305. doi: 10.1242/dev.00674. [DOI] [PubMed] [Google Scholar]
- Itoh K, Udagawa N, Katagiri T, Iemura S, Ueno N, Yasuda H, Higashio K, Quinn JM, Gillespie MT, Martin TJ, et al. Bone morphogenetic protein 2 stimulates osteoclast differentiation and survival supported by receptor activator of nuclear factor-kappaB ligand. Endocrinology. 2001;142:3656–3662. doi: 10.1210/endo.142.8.8300. [DOI] [PubMed] [Google Scholar]
- Kalajzic I, Kalajzic Z, Kaliterna M, Gronowicz G, Clark SH, Lichtler AC, Rowe D. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J Bone Miner Res. 2002;17:15–25. doi: 10.1359/jbmr.2002.17.1.15. [DOI] [PubMed] [Google Scholar]
- Kamiya N, Ye L, Kobayashi T, Lucas DJ, Mochida Y, Yamauchi M, Kronenberg HM, Feng JQ, Mishina Y. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J Bone Miner Res. 2008 doi: 10.1359/JBMR.080809. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–1209. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, Thesleff I, Itoh N. Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J Biol Chem. 2003;278:24113–24117. doi: 10.1074/jbc.M301716200. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70:11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Tang Y, Qiu T, Cao X, Clemens TL. A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells. J Biol Chem. 2006;281:17156–17163. doi: 10.1074/jbc.M513812200. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M. Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol. 2008;40:46–62. doi: 10.1016/j.biocel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Maes C, Kobayashi T, Kronenberg HM. A novel transgenic mouse model to study the osteoblast lineage in vivo. Ann N Y Acad Sci. 2007;1116:149–164. doi: 10.1196/annals.1402.060. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 1995;9:3027–3037. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Hanks MC, Miura S, Tallquist MD, Behringer RR. Generation of Bmpr/Alk3 conditional knockout mice. Genesis. 2002;32:69–72. doi: 10.1002/gene.10038. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Starbuck MW, Gentile MA, Fukuda T, Kasparcova V, Seedor JG, Hanks MC, Amling M, Pinero GJ, Harada S, et al. Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J Biol Chem. 2004;279:27560–27566. doi: 10.1074/jbc.M404222200. [DOI] [PubMed] [Google Scholar]
- Ohyama Y, Nifuji A, Maeda Y, Amagasa T, Noda M. Spaciotemporal association and bone morphogenetic protein regulation of sclerostin and osterix expression during embryonic osteogenesis. Endocrinology. 2004;145:4685–4692. doi: 10.1210/en.2003-1492. [DOI] [PubMed] [Google Scholar]
- Okamoto M, Murai J, Yoshikawa H, Tsumaki N. Bone morphogenetic proteins in bone stimulate osteoclasts and osteoblasts during bone development. J Bone Miner Res. 2006;21:1022–1033. doi: 10.1359/jbmr.060411. [DOI] [PubMed] [Google Scholar]
- Patel MS, Karsenty G. Regulation of bone formation and vision by LRP5. New Engl J Med. 2002;346:1572–1574. doi: 10.1056/NEJM200205163462011. [DOI] [PubMed] [Google Scholar]
- Piccolo S, Agius E, Leyns L, Bhattacharyya S, Grunz H, Bouwmeester T, De Robertis EM. The head inducer Cerberus is a multifunctional antagonist of Nodal, BMP and Wnt signals. Nature. 1999;397:707–710. doi: 10.1038/17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–1844. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- Rawadi G, Vayssiere B, Dunn F, Baron R, Roman-Roman S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res. 2003;18:1842–1853. doi: 10.1359/jbmr.2003.18.10.1842. [DOI] [PubMed] [Google Scholar]
- Rossert J, Eberspaecher H, de Crombrugghe B. Separate cis-acting DNA elements of the mouse pro-alpha 1(I) collagen promoter direct expression of reporter genes to different type I collagen-producing cells in transgenic mice. J Cell Biol. 1995;129:1421–1432. doi: 10.1083/jcb.129.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–26775. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, Gardner JC, Galas D, Schatzman RC, Beighton P, et al. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110:144–152. doi: 10.1002/ajmg.10401. [DOI] [PubMed] [Google Scholar]
- Sutherland MK, Geoghegan JC, Yu C, Winkler DG, Latham JA. Unique regulation of SOST, the sclerosteosis gene, by BMPs and steroid hormones in human osteoblasts. Bone. 2004;35:448–454. doi: 10.1016/j.bone.2004.04.019. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Krause C, de Gorter DJ, Lowik CW, van Bezooijen RL. Osteocyte-derived sclerostin inhibits bone formation: its role in bone morphogenetic protein and Wnt signaling. J Bone Joint Surg Am. 2008;90 (Suppl 1):31–35. doi: 10.2106/JBJS.G.01183. [DOI] [PubMed] [Google Scholar]
- Thomas JT, Lin K, Nandedkar M, Camargo M, Cervenka J, Luyten FP. A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat Genet. 1996;12:315–317. doi: 10.1038/ng0396-315. [DOI] [PubMed] [Google Scholar]
- Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L, Einhorn T, Tabin CJ, Rosen V. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet. 2006;38:1424–1429. doi: 10.1038/ng1916. [DOI] [PubMed] [Google Scholar]
- Urist MR. Bone: formation by autoinduction. Science. 1965;150:893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- Usui M, Xing L, Drissi H, Zuscik M, O’Keefe R, Chen D, Boyce BF. Murine and chicken chondrocytes regulate osteoclastogenesis by producing RANKL in response to BMP2. J Bone Miner Res. 2008;23:314–325. doi: 10.1359/JBMR.071025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Lowik CW. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–814. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bezooijen RL, Svensson JP, Eefting D, Visser A, van der Horst G, Karperien M, Quax PH, Vrieling H, Papapoulos SE, ten Dijke P, et al. Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res. 2007;22:19–28. doi: 10.1359/jbmr.061002. [DOI] [PubMed] [Google Scholar]
- Van Wesenbeeck L, Cleiren E, Gram J, Beals RK, Benichou O, Scopelliti D, Key L, Renton T, Bartels C, Gong Y, et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet. 2003;72:763–771. doi: 10.1086/368277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–6276. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DG, Sutherland MS, Ojala E, Turcott E, Geoghegan JC, Shpektor D, Skonier JE, Yu C, Latham JA. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J Biol Chem. 2005;280:2498–2502. doi: 10.1074/jbc.M400524200. [DOI] [PubMed] [Google Scholar]
- Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9:2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA. 2005;102:5062–5067. doi: 10.1073/pnas.0500031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bradley A. Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development. 1996;122:2977–2986. doi: 10.1242/dev.122.10.2977. [DOI] [PubMed] [Google Scholar]
- Zhang J, He XC, Tong WG, Johnson T, Wiedemann LM, Mishina Y, Feng JQ, Li L. BMP signaling inhibits hair follicle anagen induction by restricting epithelial stem/progenitor cell activation and expansion. Stem Cells. 2006;24:2826–2839. doi: 10.1634/stemcells.2005-0544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.