

Abstract
Eukaryotic DNA is wrapped around a histone protein core to constitute the fundamental repeating units of chromatin, the nucleosomes. The affinity of the histone core for DNA depends on the nucleotide sequence; however, it is unclear to what extent DNA sequence determines nucleosome positioning in vivo, and if the same rules of sequence-directed positioning apply to genomes of varying complexity. Using the data generated by high-throughput DNA sequencing combined with chromatin immunoprecipitation, we have identified positions of nucleosomes containing the H2A.Z histone variant and histone H3 trimethylated at lysine 4 in human CD4+ T-cells. We find that the 10-bp periodicity observed in nucleosomal sequences in yeast and other organisms is not pronounced in human nucleosomal sequences. This result was confirmed for a broader set of mononucleosomal fragments that were not selected for any specific histone variant or modification. We also find that human H2A.Z nucleosomes protect only ∼120 bp of DNA from MNase digestion and exhibit specific sequence preferences, suggesting a novel mechanism of nucleosome organization for the H2A.Z variant.
Nucleosomes wrap 147 bp of DNA around an octameric core containing two copies of highly conserved H3, H4, H2A, and H2B histone proteins (Luger et al. 1997; Kornberg and Lorch 1999). Placement of nucleosomes at specific positions in the genome may regulate gene function by altering accessibility of transcription factor binding sites and facilitating formation of higher-order chromatin structures (Wyrick et al. 1999). Because the affinity of the histone core to DNA depends on the nucleotide sequence (Widom 2001), placement of nucleosomes in chromatin may be determined by the genome sequence (Trifonov 1980; Yuan et al. 2005; Johnson et al. 2006; Segal et al. 2006; Albert et al. 2007; Peckham et al. 2007; Kaplan et al. 2009).
One of the most prominent nucleosome positioning signals observed in the DNA sequences is a 10-bp periodicity in the distribution of anisotropically flexible dinucleotides (Trifonov and Sussman 1980; Zhurkin 1985; Ioshikhes et al. 1992). Sequences exhibiting 10-bp periodicity have been shown to position nucleosomes in vitro (Shrader and Crothers 1989; Lowary and Widom 1998). Early studies of the nucleosome positioning sequences from genomic DNA have revealed the preferential placement of short AT- and GC-rich motifs in register with the DNA helical repeat at the sites where nucleosomal DNA is bent into the minor and major grooves, respectively (Satchwell et al. 1986; Ioshikhes et al. 1996). Such sequence organization is consistent with the structural preferences of these motifs (Drew and Travers 1985; Olson et al. 1998) and thus facilitates nucleosome positioning. Recent studies of the large data sets of nucleosome sequences have further emphasized the importance of the 10-bp signal for nucleosome positioning in vivo in yeast and worm (Johnson et al. 2006; Segal et al. 2006; Albert et al. 2007).
Additional factors such as interaction with proteins and crowding effects (Becker and Horz 2002; Rando and Ahmad 2007) may, however, overcome the sequence-directed nucleosome positioning. Since chromatin regulation pathways vary in different organisms, the mechanisms and the overall role of sequence-directed nucleosome positioning may vary as well, especially in evolutionarily distant organisms. In addition, the relationship between sequence and nucleosome positioning within a given organism may depend on the presence of histone variants or modifications.
The essential histone variant H2A.Z, which is enriched near transcription start sites (TSS) and involved in transcriptional activation, was extensively studied in several organisms (Zlatanova and Thakar 2008). The H2A.Z-containing nucleosomes were recently mapped in the entire yeast genome with high resolution using pyrosequencing (Albert et al. 2007). Genome-wide distribution of 20 different histone methylation states and the H2A.Z histone variant in human CD4+ T-cells were assessed by Barski et al. (2007) using the Illumina sequencing platform. With the availability of these data sets, we directly compare the DNA sequences associated with nucleosomes containing the H2A.Z variant in human and yeast chromatin. We also examine nucleosomes bearing the H3K4me3 modification, an epigenetic mark strongly correlated with active transcription and often colocalized with H2A.Z enrichment (Barski et al. 2007). Our analysis reveals striking differences between human and yeast sequences, as well as between sequences of human H2A.Z- and H3K4me3-enriched nucleosomes.
Results
Identification of nucleosomal locations in the human genome
As a first step of our analysis, we determined stable positions of H2A.Z and H3K4me3 nucleosomes in human chromatin based on the sequencing data obtained by Barski et al. (2007). These Illumina measurements identified locations of short (<36 bp) sequence tags corresponding to the ends of DNA fragments obtained by ChIP assay after digestion by micrococcal nuclease (MNase). The distribution of tag occurrences around a stable nucleosome position is expected to exhibit two pronounced peaks resulting from tag alignments on the positive and negative strands (Fig. 1A). To characterize the peak separation, we have calculated cross-correlation between tag density profiles on different strands (see Methods). The H3K4me3 tags show a cross-correlation maximum at 144 bp, a distance close to the expected length of nucleosomal DNA (Fig. 1B). Surprisingly, the cross-correlation profile for the H2A.Z variant exhibits a broad peak with the main maximum at 117 bp (Fig. 1C). This indicates that the size of the MNase-protected region observed in the H2A.Z immunoprecipitation is typically smaller than that expected for the canonical nucleosome (Fu et al. 2008). This does not mean, however, that all H2A.Z nucleosomes are digested to 117 bp; the 147-bp nucleosome fragments still exist in the set.
Figure 1.

Stable nucleosome positions determined from ChIP-Seq data. (A) Tag distribution at a site of strongly positioned nucleosome. (Vertical bars) The number of sequence tags mapping to the positive (red) and negative (blue) strands around the location of a well-positioned H3K4me3-enriched nucleosome. The approximate positions of the ends and center of the nucleosome-protected fragment are estimated based on coarse-grain tag density fit (dashed curves). The position of the fragment center is then refined using fine-grain fit (solid blue and red lines, vertical black dotted line indicates the center of the fine-grain prediction; see Methods). Secondary positions located at multiples of 10 bp can be clearly seen in this example (gray dotted lines). (B) Predominant length of the DNA fragments protected by H3K4me3-enriched nucleosomes, determined from tag density cross-correlation. Pearson linear correlation (y-axis) is calculated between number of tags mapped along positive and negative strands, shifting the positive-strand coordinates relative to the negative in 1-bp increments (x-axis). The cross-correlation maximum observed around 144 bp (red line) corresponds to the predominant fragment length. Secondary peaks are positioned at multiples of 10 bp away from the maximum (pink lines). (C) H2A.Z nucleosomes tend to protected fragments ∼120 bp long, notably shorter than the 147 bp (gray line) expected for a canonical nucleosome. The cross-correlation plot for the H2A.Z tag data shows a broad peak ∼120 bp with a maximum at 117 bp (red line).
To confirm the decreased DNA protection by H2A.Z nucleosomes experimentally, we analyzed the lengths of MNase digestion products in H2A- and H2A.Z-enriched samples of chromatin from HeLa cells (Fig. 2A; Supplemental Fig. S1A). Input chromatin was digested to identical levels ensuring that the observed difference in size is the result of the enrichment from the pulldown rather than over-digestion of the sample. We find that H2A.Z nucleosomes protect shorter fragments as compared to H2A nucleosomes. The difference in lengths of the fragments protected by H2A and H2A.Z nucleosomes is 22 ± 8 bp, which agrees with our analysis of the sequencing data. Similar values of the length difference can be obtained for di-nucleosome DNA fragments (upper bands in the gel shown in Fig. 2A). The fact that there is no twofold increase in the difference suggests that only one of the two nucleosomes in a di-nucleosome is shortened by MNase.
Figure 2.

Relative positioning and properties of DNA protection by the H2A.Z and H3K4me3 nucleosomes. (A) Chromatin samples from HeLa cells that express C-terminal Flag-tagged H2A or H2AZ histones were treated with 280 U/mL MNase for 1 h at room temperature. After deproteinization, DNA fragments were run on 7% acrylamide gel. The 1-kb ladder is shown on both sides of the gel. Results are shown for the input samples and for the samples enriched for H2A and H2A.Z nucleosomes (IP). The type of nucleosomes for which the input samples were later enriched is indicated in parentheses. The average difference between the lengths of H2A and H2A.Z fragments in the IP samples (indicated with arrows) at intermediate MNase concentration is estimated as 22 ± 8 bp based on the analysis of seven independent replicates. (B) Distribution of H2A.Z tag density around the positions of H3K4me3 enriched nucleosomes. The black and gray lines represent the tag distributions at positive and negative DNA strands. In addition to the peaks at positions ±73, which correspond to nucleosome termini, there are two inner peaks indicating shortened DNA protection by H2A.Z nucleosomes. The center of the nucleosome fragment is indicated with the dyad symbol. Upper diagram illustrates the possible positioning of 17-bp (gray) fragments relative to the original 147-bp nucleosome sequence (black); the thick gray lines correspond to the predominant digestion scenario; and the thin lines represent positioning of 117-bp fragments that are less likely according to our results. (C) Histogram of the distances between the termini (5′–5′ and 3′–3′ end distances) of the H2A.Z nucleosome fragments predicted based on the 117-bp interpeak distance, and 147-bp H3K4me3 predictions. Dashed lines indicate distances of zero and 30 bp. Two pronounced maxima at distances zero and 30 bp correspond to the predominant digestion scenario depicted in the upper panel of B. (D) Histogram of the distances between 5′–5′ and 3′–3′ ends calculated for the 147-bp H2A.Z and H3K4me3 nucleosome predictions.
To rule out the possibility of partial disassociation of the H2A.Z nucleosomes during chromatin preparation, we repeated digestion experiments varying MNase concentration (Supplemental Fig. S1B). The lengths of the protected fragments in H2A and H2A.Z enrichments were identical for higher and lower concentrations of MNase but different at the intermediate concentration. This confirms that H2A.Z nucleosomes remain intact in the pulldown rather than disassociate to hexa- or tetramers and that the difference exists in organization of H2A.Z nucleosome structure or DNA sequence. In vitro assembled recombinant H2A and H2A.Z nucleosomes show no obvious difference in length of protected DNA upon similar digestion (Supplemental Fig. S1C), suggesting that the increased sensitivity of H2A.Z nucleosomes to MNase is characteristic for in vivo conditions.
The lengths of the protected regions detected for H2A.Z nucleosomes in yeast and Drosophila were close to 147 bp (Albert et al. 2007; Mavrich et al. 2008). In these studies, histones were cross-linked to DNA prior to the digestion, while native chromatin was used in human study, which hinders direct comparison of the results. Our observation that the products of H2A.Z and H2A nucleosome digestion are of the same lengths at lower MNase concentration suggests that the discrepancy between the human and the yeast and Drosophila data may be a function of preparation of the chromatin samples. At the same time, the detected difference in the protected lengths for human H2A.Z and H2A nucleosomes at the same intermediate MNase concentration demonstrates that the shortened protection by H2A.Z nucleosomes is not an experimental artifact. Rather, this phenomenon reflects the difference in the histone–DNA interactions in these nucleosomes (discussed below).
To identify nucleosome positions, we have developed a computational method based on matching of tag density patterns of positive and negative DNA strands. The method extends the strategy used by Albert et al. (2007) to allow for identification of nucleosome positions with variable protected fragment size (see Methods). Using this approach, we determined the locations of 23,787 H2A.Z-associated fragments based on the 117-bp distance between tag density peaks and 28,976 H3K4me3 fragments based on the 147-bp distance. The target false positive rate of the nucleosome position detection is below 1%. Tag renormalization was applied to address sequence bias of MNase digestion (Wingert and von Hippel 1968; Dingwall et al. 1981); however, this correction does not affect the overall results (see Methods).
Properties of DNA protection by H2A.Z nucleosomes
Approximately 40% of the identified H2A.Z calls overlap by at least 50 bp with H3K4me3 predictions. Given such a strong overlap, we used the unambiguous positions of complete 147-bp H3K4me3 fragments as a reference set to further investigate the properties of shortened DNA protection by H2A.Z nucleosomes. Specifically, we examined the distribution of H2A.Z tags around stable H3K4me3 nucleosome positions (Fig. 2B). The observed pattern of the secondary peaks indicates that the 117-bp fragments tend to be aligned with the 147-bp complete fragments on one end and are shorter by 20–30 bp on the other. This is confirmed by direct analysis of the distances between the corresponding termini (5′–5′ and 3′–3′ end distances) of the colocalized H2A.Z and H3K4me3 predictions (Fig. 2C). Our results, therefore, suggest that the main fraction of the 117-bp H2A.Z fragments originates from asymmetric digestion of the 147-bp region on one side, and that there is no notable subpopulation of H2A.Z nucleosomes exhibiting full-length protection.
One may expect that for a given stable nucleosome position, the 30-bp cut would occur randomly on either side of the 147-bp fragment within the cell population. In this case, the tag distribution around the identified 117-bp H2A.Z fragments should exhibit two sets of peaks, corresponding to the two possible orientations of the inner cut relative to a complete 147-bp fragment. The tag distribution, however, does not show secondary peaks expected for the alternative orientation (Supplemental Fig. S2). This indicates that the decreased protection of the H2A.Z fragments tends to appear on the same side for a given nucleosome position in the measured cell population.
Distribution of the H2A.Z and H3K4me3 nucleosomes in the genome
Although the H2A.Z DNA is often cut to 117 bp (Fig. 1C), it was possible to retrieve the positions of the full-length 147-bp sequences from the tag distribution owing to nonzero tag density beyond the 117-bp fragments (see Methods). This set includes 17,667 positions and overlaps by 93% with the set of 117-bp H2A.Z fragments. We note that while predicted positions of many H2A.Z and H3K4me3 nucleosomes coincide (17% of H2A.Z nucleosomes are within 5 bp of H3K4me3 positions; Fig. 2D), we cannot conclude from our analysis whether the H2A.Z variant and H3K4me3 modification are present in the same nucleosomes because the measurements reflect the average nucleosome occupancy in the cell population.
The identified nucleosome positions in the human genome exhibit several features previously observed in other analyses (Albert et al. 2007; Ozsolak et al. 2007; Schones et al. 2008). Secondary positions located at multiples of 10 bp away from the predominant position are found for many nucleosomes (Fig. 1A; Supplemental Fig. S3), which have been attributed to multiple translational settings of a nucleosome restricted to a single rotational setting (Shrader and Crothers 1989). The average spacing of H2A.Z nucleosomes in the TSS-proximal regions (185 bp) is smaller than that observed for H2A.Z throughout the rest of the genome (200 bp) (Supplemental Fig. S4). The H3K4me3 nucleosomes are found almost exclusively in the TSS-proximal regions and show the average spacing equal to that of the TSS-proximal H2A.Z nucleosomes. We find a stable pattern of nucleosome positions near annotated transcriptional start sites (Fig. 3). The H2A.Z nucleosomes preferentially occupy positions flanking the nucleosome-free regions around the TSS, in agreement with the H2A.Z distribution observed in yeast (Guillemette et al. 2005; Raisner et al. 2005; Albert et al. 2007). The H3K4me3 nucleosome arrays extend further downstream from the TSS. This stable pattern of nucleosome positioning around the TSS is found regardless of the presence of CpG islands or TATA or INR motifs (Supplemental Fig. S5).
Figure 3.

Distribution of predicted nucleosome positions around transcription start sites (TSS). The overall density of determined H3K4me3- and H2A.Z-enriched nucleosome positions is shown relative to the TSS position, with the direction of transcription oriented from left to right. Distribution of RNA Pol II binding sites is shown for reference. The nucleosome density is calculated using a Gaussian kernel with bandwidth of 5 bp. Both H2A.Z- and H3K4me3-enriched nucleosomes show preferred stable positions flanking the nucleosome-free region around TSS, with H3K4me3-enriched nucleosome arrays extending further downstream.
Comparative analysis of human and yeast nucleosomal sequences
The nucleosome positions detected in our study were used to compare the sequence organization of the DNA fragments associated with the H2A.Z nucleosomes in yeast and human genomes at mono- and dinucleotide levels. The sequences differ in the average GC content—54% in human compared to 39% in yeast. This difference reflects the presence of CpG islands and increased GC content in the vicinity of many human genes (Lander et al. 2001), where ∼60% of the H2A.Z nucleosomes are located (Table 1).
Table 1.
Occurrences of H2A.Z and H3K4me3 predicted nucleosomes in the genomic regions

Borders of CpG islands were determined according to the annotation of the UCSC Genome Browser (Karolchik et al. 2003). Coding regions are defined as CDS of protein-coding genes. Promoters comprise 2 kb upstream of TSS of the genes, and TSS proximal regions comprise ±2 kb around transcription starts.
As previously reported (Segal et al. 2006; Albert et al. 2007), the nucleosome-associated sequences in yeast exhibit characteristic regularity in the distribution of AT- and GC-rich dimers, which tend to occur every 10 bp in counterphase. Surprisingly, the 10-bp periodicity is barely seen in the average profile for the human sequences, while it is strongly pronounced for the yeast sequences (Fig. 4A).
Figure 4.

Properties of dinucleotide distributions in yeast and human nucleosomal DNA. (A) Frequency distribution of the AT-rich (WW = AA, TT, AT, TA) and GC-rich (SS = GG, CC, CG, GC) dinucleotides in yeast and human H2A.Z nucleosomal sequences. The yeast sequence set comprises approximately 8000 “well-positioned” H2A.Z nucleosomes identified by pyrosequencing (Albert et al. 2007). Both direct and reverse sequences orientations were considered and the distributions were smoothed with a 3-bp running window. (B,C) Periodograms showing spectral density for the WW and SS dinucleotide autocorrelation functions are shown for (B) human and (C) yeast H2A.Z nucleosomes. Nucleosomal sequences from position −70 to position +70 relative to the dyad were taken for the analysis. The red lines in the periodograms represent the power spectral density for nucleosomal sequences; solid and dashed blue lines represent the statistical significance levels P = 0.001 and P = 0.05 as estimated based on the 10,000 randomizations (not corrected for multiple hypothesis testing; see Methods).
The absence of this periodicity in the average profile can potentially be a result of poor alignment of sequences. To rule out such a possibility, we applied our nucleosome detection procedure to the yeast data and observed that the 10-bp period can be readily seen in the average dinucleotide profile (Supplemental Fig. S6). We also plotted the average profiles for much larger sets of human and yeast H2A.Z sequences treating the 5′-ends of all the sequenced tags as 5′-termini of independent nucleosomes (Supplemental Fig. S7). The sequence alignment is entirely determined by the experimental cut site for this set. The 10-bp pattern is still not as pronounced in this set of human sequences as it is in the yeast nucleosomes. The 10-bp pattern in the average dinucleotide profiles is more apparent for another set of human sequences considered in our analysis—H3K4me3 nucleosomes—and for a broader class of nucleosomes detected in a recent study (Schones et al. 2008) not selected for a specific histone variant or modification, “bulk” nucleosomes (Supplemental Fig. S8).
It is, however, critical to note that presence of the periodic pattern in the average profile does not necessarily imply that the 10-bp periodicity can be found in individual sequences. The periodicity in average profile can reflect the structural organization of the nucleosome core particle rather than the signal in genomic sequences (see Discussion). Therefore, we applied autocorrelation analysis, which focuses on the dinucleotide distributions in the individual sequences and does not depend on the alignment procedure (see Methods). The dinucleotide autocorrelation function counts the number of pairs of the same dinucleotides separated by specified distances. Thus, if the 10-bp periodicity is seen in the average autocorrelation profile calculated for a certain dinucleotide in a set of sequences, it means that this set is enriched in the sequences containing at least two dinucleotides in question separated by the distances multiple to 10 bp.
The autocorrelation analysis shows an absence of statistically significant 10-bp periodicity in human H2A.Z sequences, while such signal is clearly seen in the yeast sequences (Fig. 4B,C). The prominent 3-bp signal—a known signature of protein-coding sequences (Trifonov and Sussman 1980)—is present in both organisms (Fig. 4B,C). This reflects the fact that a fraction of H2A.Z nucleosomes is found in the coding regions (Table 1) and demonstrates the ability of our analysis to detect hidden periodic signals.
In yeast, the 10-bp periodicity is most pronounced for AT-rich dinucleotides (AA:TT and TA, in particular) (Segal et al. 2006). Previous studies reported that sequences positioning human nucleosomes may exhibit periodicity in GG/CC dinucleotides (Kogan et al. 2006). We therefore examined the autocorrelations for individual dinucleotides and found that the 10-bp signal does not reach the selected threshold of statistical significance P = 0.001 for any of the dinucleotides in human nucleosomes (Supplemental Figs. S9–S14). We note, however, that weak 10-bp oscillations can be seen in the autocorrelations of GG/CC, AG/CT, and GA/TC dinucleotides when the complementary dinucleotides are considered independently, particularly in the case of “bulk” nucleosomes (Supplemental Fig. S11). This observation is in agreement with previous analyses of human nucleosomes (Kogan et al. 2006; Salih et al. 2008b). The signal is within 1%–2% of the absolute value of the autocorrelation and remains below the selected level of statistical significance.
We further validated these findings using larger sets of all the sequenced tags and various subsets of nucleosome sequences (Supplemental Figs. S15–S21; discussion in Supplemental Material). In particular, we applied the autocorrelation analysis to the subsets of the nucleosome sequence associated with high number of sequenced tags or located in the vicinity of genes stratified by the presence of CpG islands, TATA-box, INR motif, or by overall GC content near TSS. This analysis reveals weak 10-bp peaks in some subsets (e.g., low GC-content H2A.Z sequences; Supplemental Fig. S19B) that nevertheless remain below the P = 0.001 significance level. We additionally confirmed that similar results can be obtained for the entire set of “bulk” nucleosomes and for the subsets associated with transcriptionally active and silent genes (Supplemental Figs. S14 and S21I,J).
Thus, our findings suggest that the pronounced 10-bp periodicity present in yeast is not a general characteristic for the human chromatin. The above assertion is further supported by the absence of 10-bp signal in the nucleosomal sequences detected in a microarray study of human HOX clusters (Kharchenko et al. 2008). Although we cannot exclude that the strong 10-bp periodicity may be present in some subsets of human sequences or as a very sparse signal in the entire population of the sequences, our results demonstrate that it is far less pronounced than in yeast and unlikely to play a universal role in nucleosome positioning in human genome.
Specific sequence organization of H2A.Z-associated DNA
We next investigated whether the asymmetric protection of H2A.Z nucleosomes is reflected in the underlying DNA sequences. The GC profile of the human H2A.Z sequences with expected 147-bp fragment length has two noticeable features: a high-amplitude outer variation in the GC content located at the very end of the nucleosomal fragment (positions ±70–80) and a low-amplitude inner variation in the GC content located at positions ±40–60 (Fig. 5B). The outer variation has been consistently observed in most nucleosomal sequences and is attributed to MNase sequence bias (Johnson et al. 2006; Albert et al. 2007). It is also present in the H3K4me3 sequences (Fig. 5A) and parallels the GC profile observed around all sequence tags (Supplemental Fig. S22).
Figure 5.

GC profiles of nucleosomal sequences. (A–C) GC profiles, representing the combined fraction of guanines and cytosines at each position along the center-aligned human H3K4me3 sequences (A), human H2A.Z sequences (B), and yeast H2A.Z sequences (C). Both direct and reverse orientations of the sequences were taken into account. The mean GC profile for all the nucleosomal sequences in the corresponding set is shown for positions from −100 to 0 relative to the nucleosome dyad position. Thick black lines denote 11-bp smoothing of the initial data (thin gray lines). Black dashed lines give reference to 117- and 147-bp fragment lengths. Note that the decrease in the GC profile of human H2A.Z sequences at positions ±50–60 corresponds to the similar variation in the yeast profile and, moreover, is colocalized with the most pronounced oscillation in 10-bp periodicity pattern (black arrows). (D,E) The inner variation in the GC profile is not explained by the MNase bias. (D) Tag density and (E) GC profile for the human H2A.Z nucleosomes that are shown for the H2A.Z nucleosome sequences that contain no more than three tags in total at positions from ±68 to ±18.
The inner variation, however, is absent from the GC profile of the H3K4me3 sequences, suggesting that it is characteristic of the H2A.Z nucleosomes. Consistent with that, we find that the inner variation is only slightly visible in the GC profile of the H2A.Z nucleosomes that are colocalized with the K4me3 nucleosomes. Yet, the inner variation is pronounced for the subset of the H2A.Z nucleosomes shifted 30–50 bp from the H3K4me3 positions (Supplemental Fig. S23). Similar variation, albeit less pronounced, is present in the GC profile of yeast H2A.Z sequences (Fig. 5C). The presence of inner variation in GC content was further verified using the subsets of the H2A.Z sequences that contain inner tags at either only one side of the distinctive 147-bp fragment or at both sides of the 147-bp fragment as well as for the entire set of sequenced tags extended by 200 bp toward the 3′-end (Supplemental Figs. S24–S26; discussion in Supplemental Material).
The inner variation exhibits a GC profile similar to that of the MNase bias and may therefore be a consequence of shortened protection of H2A.Z DNA against MNase digestion. To check this, we generated a GC profile for a subset of H2A.Z nucleosomes that are not associated with the increased tag density around the inner variation (Fig. 5D,E). The inner variation remains pronounced in such a subset, indicating that the observed feature is not a direct consequence of the MNase bias. However, sequencing of the full-length 147-bp fragments from nonover-digested H2A.Z chromatin would be required to confidently determine the extent to which MNase bias may contribute to the observed inner variation.
Discussion
Our results demonstrate that the nucleosome-associated DNA in the human genome does not follow the sequence organization models established for lower eukaryotes. The 10-bp dinucleotide periodicity reported for yeast is not pronounced in human nucleosome sequences. It is possible that the positions in which human nucleosomes were captured in vivo are not thermodynamically favorable (e.g., displaced by RNA polymerase in the course of transcription) and sequences exhibiting 10-bp signal are present elsewhere. Two lines of evidence indicate that this is not the case. First, the significant reduction of the expected 10-bp signal is also observed on the broad set of nucleosomes not selected for any specific modification or variant, in particular, those associated with silent genes (Schones et al. 2008). This indicates that these findings are not explained by transcription-driven nucleosome repositioning. Second, the disparity in the presence of 10-bp periodicity between humans and yeast is also observed for the gene-proximal regions in general (Supplemental Fig. S27), indicating that selective pressure to maintain such periodicity is not pronounced in humans. It was previously reported that the 10-bp periodicity is operational in positioning of only a fraction of nucleosomes in yeast, and that the strength of the 10-bp signal is further diminished in Drosophila (Peckham et al. 2007; Mavrich et al. 2008). Our analysis indicates paucity of the 10-bp signal in all the examined sets of the human genomic sequences, which can be potentially attributed to the presence of a more capable chromatin-remodeling machinery in humans than those in lower organisms.
A couple of methodological issues need to be mentioned. First, our results reflect primarily euchromatic regions, as they tend to be more susceptible to MNase digestion. In addition, the heterochromatic regions of the genome are enriched in repetitive sequences and are poorly assembled, which hinders unambiguous tag alignment to those regions. At the same time, at least some repeats can position nucleosomes. The nucleosome positioning was demonstrated in Alu repeats in vitro, and the role of DNA sequence for such positioning was highlighted in the recent work of Salih et al. (2008a).
Second, we note that the absence of the pronounced 10-bp periodicity in the nucleosome DNA sequences is not contradictory to the presence of the 10-bp periodicity either in the profile of the tag density around nucleosome positions (Supplemental Fig. S3) or in the average dinucleotide profiles (Supplemental Fig. S8). Restricted rotational setting of a nucleosome may arise as a consequence of higher-order chromatin structures (Thoma 1992), which would be reflected in tag density distribution. Periodic patterns in average profiles could potentially arise owing to alignment procedures or antiselection of certain rotational settings as a result of MNase sequence bias (McGhee and Felsenfeld 1983). Also, the presence of a strongly positioning sequence that does not exhibit 10-bp periodicity may give rise to periodic patterns both in tag density and average dinucleotide profiles.
The latter mechanism can be described as follows. The histone core imposes severe deformations on the DNA duplex, bending it into the minor and major grooves with periodicity of ∼10 bp (Luger et al. 1997). The energy required to deform DNA dinucleotide steps at the sites of such deformations varies significantly depending on the sequence of the step (Tolstorukov et al. 2007). It is likely that the histone core would shift a few base pairs on the genomic sequence to achieve the setting, which is the most favorable even relative to one or two sequence motifs with pronounced structural preferences in the otherwise “structurally neutral” sequence. Such a genomic location would exhibit rotational positioning of nucleosome. Periodic enrichment in certain dinucleotides in average profiles at the sites where DNA bends into the minor or major groove would originate from different sequences organized in this way present in a large set (Supplemental Figs. S28 and S29; corresponding discussion in Supplemental Material). It is important to note that we have used autocorrelation analysis in the present study. This analysis identifies periodicities present within individual sequences rather than periodic patterns found only in the average profiles.
It is possible that in humans sequence patterns other than 10-bp periodicity account for thermodynamically favorable positioning in vivo. For a broad class of nucleosomes identified in the CD4+ T-cells, we observe a pronounced 10% increase in GC content of the DNA region that is in direct contact with the histone core as compared to the linker regions (Supplemental Fig. S30). Since GC rich sequence exhibits increased bendability (Olson et al. 1998), such a pattern should contribute to nucleosome positioning (Lee et al. 2007; Peckham et al. 2007; Kharchenko et al. 2008). The overall nucleosome occupancy profiles observed in yeast in vivo can be reproduced by reconstituting nucleosomes on bare DNA in vitro (Kaplan et al. 2009). At the same time, the recent study on worm showed that exact nucleosome positioning is not preserved in cell population on single-nucleotide scale (Valouev et al. 2008). Our results, suggesting the diminished role of the 10-bp periodicity in humans, are in line with the second observation. It is yet to be determined if nucleosome positioning signals encoded in the human genomic DNA, such as the increase in GC content discussed above, are sufficient to predict the nucleosome occupancy over sizable fragments of chromatin.
The sequence properties may be of great importance for “fine-tuning” positioning of specific nucleosomes in humans. Our results suggest that the organization of DNA in H2A.Z nucleosomes is distinct from that of other nucleosomes in the human genome. The H2A.Z nucleosomes protect only ∼120 bp of DNA and the shortening of the nucleosome-associated DNA fragment occurs in an asymmetric way. The specific sequence preferences of the H2A.Z nucleosomes and/or MNase may explain why DNA shortening occurs on one side of the H2A.Z fragment in human chromatin. Since genomic sequence would rarely contain two H2A.Z-positioning sequence motifs separated by the appropriate spacing, one side of the nucleosome should be more susceptible to MNase attack because of enzyme sequence or structural bias. Another possible mechanism responsible for the asymmetry in the H2A.Z digestion may be related to the fact that H2A.Z can form hybrid nucleosomes with major H2A histone variants as was demonstrated both in vitro and in vivo (Chakravarthy et al. 2004; Viens et al. 2006). The asymmetry in the histone core structure of such hybrid nucleosomes would be reflected in their DNA protection properties. We note that these two mechanisms are not mutually exclusive but could, rather, complement each other.
Analysis of the H2A.Z nucleosome structure (Suto et al. 2000) shows that the 30-bp cut occurs at a nucleosomal location structurally favorable for endonuclease attack, where the DNA minor groove faces outward from the histone core (Fig. 6). The N-terminal tail of H2A.Z interacts with the minor groove at this site. It was shown that reducing the positive net charge of this tail in the H2A.Z nucleosomes through acetylation or mutations is essential for viability of Tetrahymena thermophila cells (Ren and Gorovsky 2001). These observations raise the possibility that a mechanism involving modification of the H2A.Z tail contributes to the asymmetric digestion observed in human cells. Modification of the tail, resulting in less tight binding of the tail to DNA, might facilitate the accessibility at this site for regulatory factors that promote an active chromatin structure. Another H2A variant associated with active chromatin, H2A.Bbd, protects only 118 bp of DNA from MNase digestion in vitro (Fan et al. 2002). This variant was shown to cause partial unwrapping of DNA from the histone octamer. While it is not clear if a similar mechanism might cause shortened protection by H2A.Z, which is more similar to H2A than is H2A.Bbd, loosened contacts in this region could be a characteristic of more than one H2A variant.
Figure 6.

Structural context of the H2A.Z nucleosome over-digestion. (A) Nucleosome core particle structure containing H2A.Z variant (PDB_ID 1f66) (Suto et al. 2000) with only half of nucleosomal DNA (cyan) and one set of the histone proteins (white and magenta) shown for clarity. The major H2A histone (dark blue) from the best resolved nucleosome structure (PDB_ID 1kx5) (Davey et al. 2002) is superimposed onto the H2A.Z variant (magenta) for comparison. Base pair at position −43 (yellow) marks the 30-bp shortening of the protected DNA fragment. (B) Detailed view of the H2A.Z structure fragment marked with the box in A. Note that the N-terminal tail of the H2A.Z histone was not included in the published structure as too disordered (Suto et al. 2000).
The incorporation of the H2A.Z-containing nucleosomes has been previously shown to affect nucleosome positioning in vitro and in vivo (Fan et al. 2002; Guillemette et al. 2005; Schones et al. 2008). The nucleosome repositioning induced by the H2A.Z deposition has also been associated with chromatin activation in vivo (Schones et al. 2008). The in vitro studies demonstrate that the replacement of the histone H2A with H2A.Z may cause nucleosome sliding to a new stable position even without chromatin-remodeling proteins (Guillemette et al. 2005). One can therefore speculate that the sequence specificity of the H2A.Z-containing nucleosomes observed in our study is an important regulatory mechanism that facilitates nucleosome repositioning and alteration of larger chromatin structure. Detailed analysis of the nucleosome positioning in the chromatin from the cells at different developmental and physiological stages will be needed to address this question.
Methods
Cross-correlation and autocorrelation analysis
For each chromosome c, the tag count vector nsc(x) was calculated to give the number of tags whose 5′-end maps to the position x on the strand s. Strand cross-correlation was then calculated as
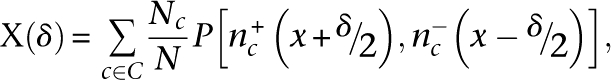 |
where P[a,b] is the Pearson linear correlation coefficient between vectors a and b, C is the set of all chromosomes, Nc is the number of tags mapped to a chromosome c, and N is the total number of tags. Autocorrelation was calculated as
 |
where Nsc is the number of tags mapping to strand s on a chromosome c.
Nucleosome detection
Similarly to the analysis of H2A.Z nucleosomes in yeast (Albert et al. 2007), stable nucleosome positions in the human genome were determined in two steps for each of the three sets of the sequenced tags: (1) enriched for H3K4me3 modification, (2) enriched for H2A.Z histone variant, and (3) not selected for any specific histone variant or modification (Barski et al. 2007; Schones et al. 2008). The chromosomes were first scanned to determine regions exhibiting statistically significant signal (coarse-grain positions). For a given chromosome, the density of mapped sequence tags was calculated for each strand s as a sum of Gaussian contributions:
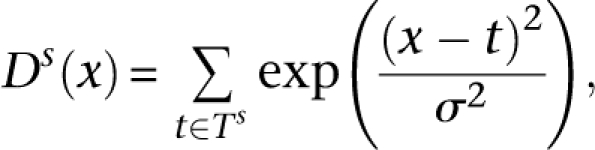 |
where Ts is a set of all tag coordinates mapped to a strand s, and σ = 15 bp. The joint density was calculated as a product of shifted strand densities:
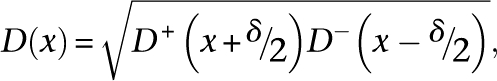 |
where δ is the size of the protected region (147 bp or 117 bp). Centers of the regions where joint density exceeds threshold θ for more than 2 bp were recorded as coarse-grained positions. The value of the threshold θ was chosen so that no more than 100 coarse-grained positions are found in the genome for randomized tag data (E-value of 100). The tags were randomized in blocks of 100 bp, independently for each strand.
Positions were then refined to obtain more precise coordinates (fine-grain positions). To this end, the tag density in a 147-bp region around each coarse-grained position was recalculated using σ = 2 bp. The joined density was calculated using protected region size δ′, chosen to maximize cross-correlation between individual strand densities and bounded by |δ′ − δ| ≤ 10 bp. Local maxima of the resulting joint density were recorded as fine-grained positions, with primary position corresponding to the highest peak within 105 bp.
In the case of H2A.Z DNA, two sets of nucleosome predictions were generated based on the interstrand distances δ = 117 bp and δ = 147 bp. To confirm that the predictions based on the 147-bp distance correctly identify the positions of the full-length nucleosome fragments, we verified that the distributions of fragment termini accurately align with respect to the overlapping H3K4me3 and 117-bp H2A.Z nucleosome predictions (Supplemental Fig. S31).
Addressing MNase sequence bias
The MNase digestion used by Barski et al. (2007) preferentially cuts the unprotected internucleosomal DNA. However, the cleavage pattern is also biased by the underlying nucleotide sequence (Wingert and von Hippel 1968; Dingwall et al. 1981). To examine the extent to which potential MNase bias influences our analysis, we generated a series of alternative nucleosome predictions weighting the contributions of individual tags. The tags were weighted by the relative abundance of the n-mers occurring at the cut sites as compared to their occurrence genome-wide. Several renormalizations based on n = 2–7 were applied. We find that such renormalizations consistently reduce the number of pronounced nucleosome positions; however, the overall results and conclusions presented in this analysis are not affected (see Supplemental Fig. S32, showing the results obtained for the tag contributions weighted by the relative occurrence of 5-mers), and the results for the initial non-normalized tag set are presented in the paper.
Sequence analysis
The dinucleotide autocorrelations were calculated for DNA fragments centered at the predicted nucleosome position as previously described (Trifonov and Sussman 1980; Zhurkin 1981; Tolstorukov et al. 2005). The 147-bp nucleosome sequences were trimmed by 3 bp from each side to exclude influence of the MNase sequence bias on the results. The mean autocorrelation profiles were calculated for each set of nucleosomal sequences by averaging the individual profiles over all the sequences in the set. The power spectral density was calculated for the mean autocorrelation profiles. In such a way, the periodic signal could be detected even in the case of poorly aligned sequences that contain stretches of periodically placed dinucleotides.
For each set of nucleosome predictions, we generated 10,000 sets of random sequences, each containing the same number of sequences as the initial set of nucleosome predictions. Random sequences were generated so that their dinucleotide compositions in three nonoverlapping 47-bp windows along the sequence were the same as those in the corresponding windows in the original sequences. Averaging dinucleotide composition in three separate windows rather than over the entire sequence allowed accounting for the nonmonotonic GC-content profile of the nucleosome sequences. Then the dinucleotide autocorrelations and power spectral density were calculated for the random sequences in the same way as for the nucleosome predictions. The statistical significance levels, P = 0.001 and P = 0.05, of the spectral density for each period were estimated as the 10th and 500th largest density values, respectively, observed at a given period in the spectra of 10,000 random sets.
The GC profiles were calculated by averaging the fraction of guanines and cytosines position-wise over all the sequences in the set. Profiles for individual dinucleotides representing the fraction of the corresponding dinucleotide at each position were calculated in the same way.
Gene sets
A nucleosome was considered to belong to a certain gene if its position was within the gene boundaries or no more than 2 kb upstream of the gene 5′-end. The gene expression status was estimated for the resting CD4+ T-cells based on published microarray data (GEO accession number GSE10437) using the Affymetrix Microarray Suite 5 algorithm (Hubbell et al. 2002; Liu et al. 2002). Only the genes that were unambiguously called Present (Absent) for all the probe sets corresponding to them were defined as expressed (unexpressed) genes. Annotation from the UCSC Genome Browser was used to determine the locations and boundaries of CpG islands (Karolchik et al. 2003). The subsets of the genes containing TATA or INR elements in their promoters were defined according to the results of the recent computational analysis of the human genome (Yang et al. 2007).
Experimental analysis of the DNA fragments protected against nuclease digestion
Nuclei were isolated from stable HeLa S3 cell lines expressing C-terminal Flag-tagged H2A or H2A.Z using standard procedures (Foltz et al. 2006). Nuclei were digested with indicated amounts of MNase (Roche) for 1 h at room temperature. Immunoprecipitation was performed using M2-Flag beads (Sigma) in the presence of 300 mM KCl and 0.1% Tween and eluted with 1 mg/mL Flag peptide. After proteinase K treatment and phenol/choloroform extraction, DNA was run on 7% acrylamide in 0.25× TAE buffer and post-stained with EtBr. Size measurements were made using ImageQuant.
Acknowledgments
We are grateful to Kami Ahmad, Vasily Studitsky, and Keji Zhao for valuable discussions and to Mitzi Kuroda, Marnie Gelbart, Erika Larschan, and Gavin Schnitzler for critical reading of the manuscript and many insightful comments. This work was supported by grants from the National Institutes of Health to P.J.P.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.084830.108.
References
- Albert I., Mavrich T.N., Tomsho L.P., Qi J., Zanton S.J., Schuster S.C., Pugh B.F. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Becker P.B., Horz W. ATP-dependent nucleosome remodeling. Annu. Rev. Biochem. 2002;71:247–273. doi: 10.1146/annurev.biochem.71.110601.135400. [DOI] [PubMed] [Google Scholar]
- Chakravarthy S., Bao Y., Roberts V.A., Tremethick D., Luger K. Structural characterization of histone H2A variants. Cold Spring Harb. Symp. Quant. Biol. 2004;69:227–234. doi: 10.1101/sqb.2004.69.227. [DOI] [PubMed] [Google Scholar]
- Davey C.A., Sargent D.F., Luger K., Maeder A.W., Richmond T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- Dingwall C., Lomonossoff G.P., Laskey R.A. High sequence specificity of micrococcal nuclease. Nucleic Acids Res. 1981;9:2659–2673. doi: 10.1093/nar/9.12.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew H.R., Travers A.A. DNA bending and its relation to nucleosome positioning. J. Mol. Biol. 1985;186:773–790. doi: 10.1016/0022-2836(85)90396-1. [DOI] [PubMed] [Google Scholar]
- Fan J.Y., Gordon F., Luger K., Hansen J.C., Tremethick D.J. The essential histone variant H2A.Z regulates the equilibrium between different chromatin conformational states. Nat. Struct. Biol. 2002;9:172–176. doi: 10.1038/nsb767. [DOI] [PubMed] [Google Scholar]
- Foltz D.R., Jansen L.E., Black B.E., Bailey A.O., Yates J.R., III, Cleveland D.W. The human CENP-A centromeric nucleosome-associated complex. Nat. Cell Biol. 2006;8:458–469. doi: 10.1038/ncb1397. [DOI] [PubMed] [Google Scholar]
- Fu Y., Sinha M., Peterson C.L., Weng Z. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 2008;4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette B., Bataille A.R., Gevry N., Adam M., Blanchette M., Robert F., Gaudreau L. Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol. 2005;3:e384. doi: 10.1371/journal.pbio.0030384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell E., Liu W.M., Mei R. Robust estimators for expression analysis. Bioinformatics. 2002;18:1585–1592. doi: 10.1093/bioinformatics/18.12.1585. [DOI] [PubMed] [Google Scholar]
- Ioshikhes I., Bolshoy A., Trifonov E.N. Preferred positions of AA and TT dinucleotides in aligned nucleosomal DNA sequences. J. Biomol. Struct. Dyn. 1992;9:1111–1117. doi: 10.1080/07391102.1992.10507982. [DOI] [PubMed] [Google Scholar]
- Ioshikhes I., Bolshoy A., Derenshteyn K., Borodovsky M., Trifonov E.N. Nucleosome DNA sequence pattern revealed by multiple alignment of experimentally mapped sequences. J. Mol. Biol. 1996;262:129–139. doi: 10.1006/jmbi.1996.0503. [DOI] [PubMed] [Google Scholar]
- Johnson S.M., Tan F.J., McCullough H.L., Riordan D.P., Fire A.Z. Flexibility and constraint in the nucleosome core landscape of Caenorhabditis elegans chromatin. Genome Res. 2006;16:1505–1516. doi: 10.1101/gr.5560806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N., Moore I.K., Fondufe-Mittendorf Y., Gossett A.J., Tillo D., Field Y., Leproust E.M., Hughes T.R., Lieb J.D., Widom J., et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D., Baertsch R., Diekhans M., Furey T.S., Hinrichs A., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko P.V., Woo C.J., Tolstorukov M.Y., Kingston R.E., Park P.J. Nucleosome positioning in human HOX gene clusters. Genome Res. 2008;18:1554–1561. doi: 10.1101/gr.075952.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan S.B., Kato M., Kiyama R., Trifonov E.N. Sequence structure of human nucleosome DNA. J. Biomol. Struct. Dyn. 2006;24:43–48. doi: 10.1080/07391102.2006.10507097. [DOI] [PubMed] [Google Scholar]
- Kornberg R.D., Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lee W., Tillo D., Bray N., Morse R.H., Davis R.W., Hughes T.R., Nislow C. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Genet. 2007;39:1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- Liu W.M., Mei R., Di X., Ryder T.B., Hubbell E., Dee S., Webster T.A., Harrington C.A., Ho M.H., Baid J., et al. Analysis of high density expression microarrays with signed-rank call algorithms. Bioinformatics. 2002;18:1593–1599. doi: 10.1093/bioinformatics/18.12.1593. [DOI] [PubMed] [Google Scholar]
- Lowary P.T., Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Mavrich T.N., Jiang C., Ioshikhes I.P., Li X., Venters B.J., Zanton S.J., Tomsho L.P., Qi J., Glaser R.L., Schuster S.C., et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453:358–362. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee J.D., Felsenfeld G. Another potential artifact in the study of nucleosome phasing by chromatin digestion with micrococcal nuclease. Cell. 1983;32:1205–1215. doi: 10.1016/0092-8674(83)90303-3. [DOI] [PubMed] [Google Scholar]
- Olson W.K., Gorin A.A., Lu X.J., Hock L.M., Zhurkin V.B. DNA sequence-dependent deformability deduced from protein–DNA crystal complexes. Proc. Natl. Acad. Sci. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F., Song J.S., Liu X.S., Fisher D.E. High-throughput mapping of the chromatin structure of human promoters. Nat. Biotechnol. 2007;25:244–248. doi: 10.1038/nbt1279. [DOI] [PubMed] [Google Scholar]
- Peckham H.E., Thurman R.E., Fu Y., Stamatoyannopoulos J.A., Noble W.S., Struhl K., Weng Z. Nucleosome positioning signals in genomic DNA. Genome Res. 2007;17:1170–1177. doi: 10.1101/gr.6101007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisner R.M., Hartley P.D., Meneghini M.D., Bao M.Z., Liu C.L., Schreiber S.L., Rando O.J., Madhani H.D. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell. 2005;123:233–248. doi: 10.1016/j.cell.2005.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando O.J., Ahmad K. Rules and regulation in the primary structure of chromatin. Curr. Opin. Cell Biol. 2007;19:250–256. doi: 10.1016/j.ceb.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Ren Q., Gorovsky M.A. Histone H2A.Z acetylation modulates an essential charge patch. Mol. Cell. 2001;7:1329–1335. doi: 10.1016/s1097-2765(01)00269-6. [DOI] [PubMed] [Google Scholar]
- Salih F., Salih B., Kogan S., Trifonov E.N. Epigenetic nucleosomes: Alu sequences and CG as nucleosome positioning element. J. Biomol. Struct. Dyn. 2008a;26:9–16. doi: 10.1080/07391102.2008.10507219. [DOI] [PubMed] [Google Scholar]
- Salih F., Salih B., Trifonov E.N. Sequence structure of hidden 10.4-base repeat in the nucleosomes of C. elegans. J. Biomol. Struct. Dyn. 2008b;26:273–282. doi: 10.1080/07391102.2008.10531241. [DOI] [PubMed] [Google Scholar]
- Satchwell S.C., Drew H.R., Travers A.A. Sequence periodicities in chicken nucleosome core DNA. J. Mol. Biol. 1986;191:659–675. doi: 10.1016/0022-2836(86)90452-3. [DOI] [PubMed] [Google Scholar]
- Schones D.E., Cui K., Cuddapah S., Roh T.Y., Barski A., Wang Z., Wei G., Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E., Fondufe-Mittendorf Y., Chen L., Thastrom A., Field Y., Moore I.K., Wang J.P., Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrader T.E., Crothers D.M. Artificial nucleosome positioning sequences. Proc. Natl. Acad. Sci. 1989;86:7418–7422. doi: 10.1073/pnas.86.19.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto R.K., Clarkson M.J., Tremethick D.J., Luger K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat. Struct. Biol. 2000;7:1121–1124. doi: 10.1038/81971. [DOI] [PubMed] [Google Scholar]
- Thoma F. Nucleosome positioning. Biochim. Biophys. Acta. 1992;1130:1–19. doi: 10.1016/0167-4781(92)90455-9. [DOI] [PubMed] [Google Scholar]
- Tolstorukov M.Y., Virnik K.M., Adhya S., Zhurkin V.B. A-tract clusters may facilitate DNA packaging in bacterial nucleoid. Nucleic Acids Res. 2005;33:3907–3918. doi: 10.1093/nar/gki699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstorukov M.Y., Colasanti A.V., McCandlish D.M., Olson W.K., Zhurkin V.B. A novel roll-and-slide mechanism of DNA folding in chromatin: Implications for nucleosome positioning. J. Mol. Biol. 2007;371:725–738. doi: 10.1016/j.jmb.2007.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifonov E.N. Sequence-dependent deformational anisotropy of chromatin DNA. Nucleic Acids Res. 1980;8:4041–4053. doi: 10.1093/nar/8.17.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifonov E.N., Sussman J.L. The pitch of chromatin DNA is reflected in its nucleotide sequence. Proc. Natl. Acad. Sci. 1980;77:3816–3820. doi: 10.1073/pnas.77.7.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A., Ichikawa J., Tonthat T., Stuart J., Ranade S., Peckham H., Zeng K., Malek J.A., Costa G., McKernan K., et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18:1051–1063. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viens A., Mechold U., Brouillard F., Gilbert C., Leclerc P., Ogryzko V. Analysis of human histone H2AZ deposition in vivo argues against its direct role in epigenetic templating mechanisms. Mol. Cell. Biol. 2006;26:5325–5335. doi: 10.1128/MCB.00584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widom J. Role of DNA sequence in nucleosome stability and dynamics. Q. Rev. Biophys. 2001;34:269–324. doi: 10.1017/s0033583501003699. [DOI] [PubMed] [Google Scholar]
- Wingert L., von Hippel P.H. The conformation dependent hydrolysis of DNA by micrococcal nuclease. Biochim. Biophys. Acta. 1968;157:114–126. doi: 10.1016/0005-2787(68)90270-0. [DOI] [PubMed] [Google Scholar]
- Wyrick J.J., Holstege F.C., Jennings E.G., Causton H.C., Shore D., Grunstein M., Lander E.S., Young R.A. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature. 1999;402:418–421. doi: 10.1038/46567. [DOI] [PubMed] [Google Scholar]
- Yang C., Bolotin E., Jiang T., Sladek F.M., Martinez E. Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene. 2007;389:52–65. doi: 10.1016/j.gene.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G.C., Liu Y.J., Dion M.F., Slack M.D., Wu L.F., Altschuler S.J., Rando O.J. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- Zhurkin V.B. Periodicity in DNA primary structure is defined by secondary structure of the coded protein. Nucleic Acids Res. 1981;9:1963–1971. doi: 10.1093/nar/9.8.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhurkin V.B. Sequence-dependent bending of DNA and phasing of nucleosomes. J. Biomol. Struct. Dyn. 1985;2:785–804. doi: 10.1080/07391102.1985.10506324. [DOI] [PubMed] [Google Scholar]
- Zlatanova J., Thakar A. H2A.Z: View from the top. Structure. 2008;16:166–179. doi: 10.1016/j.str.2007.12.008. [DOI] [PubMed] [Google Scholar]