

Abstract
Recent genome-wide scans for colorectal cancer (CRC) have revealed the SMAD7 (mothers against decapentaplegic homolog 7) gene as a locus associated with a modest, but highly significant increase in CRC risk. To identify the causal basis of the association between 18q21 variation and CRC, we resequenced the 17-kb region of linkage disequilibrium and evaluated all variants in 2532 CRC cases and 2607 controls. A novel C to G single nucleotide polymorphism (SNP) at 44,703,563 bp was maximally associated with CRC risk (P = 5.98 × 10−7; ≥1.5-fold more likely to be causal than other variants). Using transgenic assays in Xenopus laevis as a functional model, we demonstrate that the G risk allele leads to reduced reporter gene expression in the colorectum (P = 5.4 × 10−3). Electrophoretic mobility shift assays provided evidence for the role of Novel 1 in transcription factor binding. We propose that the novel SNP we have identified is the functional change leading to CRC predisposition through differential SMAD7 expression and, hence, aberrant TGF-beta signaling.
Genome-wide association (GWA) studies have become a powerful tool to identify susceptibility variants for common diseases (McCarthy et al. 2008). As the single nucleotide polymorphisms (SNPs) (or markers) genotyped during GWA studies are generally not themselves candidates for causality, enumeration of the causal variant at a specific locus generally poses a significant challenge.
We have recently conducted a GWA study of colorectal cancer (CRC) and have shown that common variation defined by the correlated 18q21 SNPs––rs4939827, rs12953717, and rs4464148––influences CRC risk (Broderick et al. 2007). The observation of a relationship between rs4939827 and CRC risk has been replicated in independent studies (Tenesa et al. 2008; Curtin et al. 2009), thereby providing compelling evidence for the association. Although the risk of CRC associated with 18q21 variation is modest (genotypic risk ∼1.4) the contribution of the locus to CRC is highly significant (Broderick et al. 2007).
While the 18q21 SNPs map to SMAD7 (mothers against decapentaplegic homolog 7; MIM602932), the underlying basis of the association is presently unknown. We have systematically interrogated the 18q21 association signal through targeted resequencing, linkage disequilibrium (LD) mapping, and functional analyses to elucidate the causal basis of the association.
Results
The 17-kb genomic region of 18q21 (44,700,221–44,716,898; UCSC May 2006 assembly, NCBI build 36.1) was selected for resequencing based on the observed pattern of LD surrounding rs4939827, rs12953717, and rs4464148 (the most significant 18q21 SNPs represented on the Illumina 550 Hapmap Bead Array (Broderick et al. 2007) in the HapMap CEU samples (n = 60 individuals), such that the causative variant is expected to be identified by this analysis (i.e., the region was chosen such that the likelihood that the disease-linked variant would reside outside the chosen region is very small).
The three SNPs rs4939827, rs12953717, and rs4464148 map to the interval encompassing the 3′ region of intron 3, exon 4, and the 3′ UTR of SMAD7 (between B and C in Figure 1, a–c). Through resequencing we excluded the possibility that the 18q21 association signal is a consequence of a coding sequence change in SMAD7 (Broderick et al. 2007).
Figure 1.

The SMAD7 locus. (a) Single-marker association statistics (as –log10 values) as a function of genomic position (NCBI build 36.1) obtained in the GWAS (Broderick et al. 2007) covering SMAD7 and 100 kb of flanking sequence. All known genes and transcripts in the area are shown (UCSC 2006 March 2006 assembly, NCBI build 36.1). (b) Recombination rate (cM/Mb) across the region derived from HapMap project data (release 21a). (c) The interval between B and C corresponds to the 17-kb region resequenced.
To comprehensively interrogate the 17-kb interval (Fig. 1) for all genetic variation, we resequenced the region in 90 healthy unrelated individuals. Only 722 bp (4%) of the 17 kb was refractory to resequencing, owing to low complexity. In total, we identified 55 variants (Supplemental Table 1); these included 50 SNPs and five insertion/deletion polymorphisms. Of the 55 variants, 43 were common (minor allele frequency [MAF] ≥ 0.05). Of these, 33 common variants had not been genotyped by HapMap. In addition, a SNP caused by a C to G change at 44,703,563 bp (henceforth referred to as Novel 1) with a MAF of 0.47 was unlisted by dbSNP (Build 128; Supplemental Fig. 1A).
We calculated pairwise LD statistics between each of the 52 SNPs and rs4939827, rs12953717, and rs4464148; 22 of these showed evidence of high LD with one or more of the original three SNPs (r2 ≥ 0.50; Supplemental Fig. 2; Supplemental Table 2). These 22 DNA polymorphisms, together with rs4939827, rs12953717, and rs4464148, were genotyped in a series of 2532 CRC cases and 2607 controls. There was no evidence of population stratification in controls with the distribution of genotypes of all DNA polymorphisms showing no significant deviation from Hardy-Weinberg Equilibrium (P > 0.05). All polymorphisms showed evidence of an association with risk of developing CRC at the 5% statistical threshold (Table 1). The strongest evidence for an association between 18q21 variation and CRC risk was provided by Novel 1 (P = 5.98 × 10−7) (Table 1; Fig. 2).
Table 1.
Association between the 25 SNPs and risk of colorectal cancer

Figure 2.

(a) Single marker association statistics (as –log10 values) for each of the 25 SNPs mapping to the 17-kb region sequenced. The five SNPs with the strongest evidence for an association with colorectal cancer are denoted in blue (rs8085824, Novel1, rs34007497, rs4044177, and rs12953717). (b) Pairwise linkage disequilibrium (r2) metrics of the 25 SNPs calculated in Haploview (v4.0) software. The values indicate the LD relationship between each pair of SNPs; the darker the shading, the greater extent of LD. Shown are the two haplotype blocks defined within the region.
We performed logistic regression comparing the log-likelihoods of models based on the maximally associated SNP (Novel 1) and models incorporating Novel 1 and the other 24 candidate DNA polymorphisms. A model based solely on Novel 1 was sufficient to capture all variation at the locus (P > 0.2 for the addition of each SNP to the model; Supplemental Table 3). Computational analysis of the five most significantly associated SNPs––rs8085824, rs34007497, rs4044177, and rs12953717––showed that they are strongly correlated with Novel 1 (r2 > 0.94) (Fig. 2) and constitute a single-risk haplotype in block 1 (Fig. 1; Supplemental Table 4). To further explore the association signal at 18q21, we generated inferred ancestral recombination graphs (ARGs) using the Margarita program (Minichiello and Durbin 2006), inferring ARGs for the 25 SNP haplotypes that span the SMAD7 interval. The strongest evidence for association with CRC risk was provided by Novel 1 (P = 5.2 × 10−6). SNPs rs8085824, rs34007497, rs4044177 and rs12953717, also showed evidence for association (P < 10−4; Supplemental Table 5). For all other SNPs, the permutation P values were >10−4. These observations are consistent with one of these five SNPs being the causal variant. We also calculated Akaike weights to determine the weight of evidence in favor of each variant relative to Novel 1 (Table 1). Novel 1 was 1.5 times more likely to be causal than rs8085824, 1.8 times more likely than rs34007497, 2.1 times more likely than rs12953717, 2.3 times more likely than rs4044177, and >10 times more likely to be causal than the other variants. Collectively, these data strongly support Novel 1 being the variant responsible for the association between 18q21 variation and CRC risk.
We have previously shown lower median mRNA SMAD7 expression associated with CRC risk alleles at rs12953717 in lymphoblastoid cell lines (LD with Novel 1, r2 = 0.94) (Broderick et al. 2007). Predicated on the assertion that the causal variant influences SMAD7 expression in the colorectum, we examined the influence of Novel 1 on expression. To maximize detection of subtle allele-specific differences, we used a Xenopus in vivo model system.
Nucleotides at rs8085824, Novel 1, rs34007497, rs4044177, and rs12953717 are conserved in primates. rs8085824, Novel 1, and rs34007497 are conserved in mouse. 1DN41A sequence conservation in noncoding regions has been shown to be a good predictor of cis-regulatory sequences (Gomez-Skarmeta et al. 2006). Moreover, it has been proposed that variation within evolutionarily-conserved regions is likely to be associated with phenotypic differences that may contribute to expression of traits (Dermitzakis et al. 2005).
Sequence comparison of the intron 3 of SMAD7 of several vertebrate species revealed the presence of a number of highly conserved noncoding regions (HCNRs) in the area. Of particular interest is one HCNR present in all tetrapods, including Xenopus flanked by Novel 1 and rs8085824 (Fig. 3a,b). Moreover, the immediate region surrounding Novel 1 is conserved in all primates (Fig. 3c).
Figure 3.

(a) VISTA view of the occurrence of conserved sequence domains in the SMAD7 risk-associated region. Shown from top to bottom are global alignments of human vs. dog, mouse, chicken, and Xenopus tropicalis, respectively. Colored peaks indicate regions of at least 100 bp in length and with 75% sequence similarity. Cyan peaks are UTR, purple peaks are coding regions, and pink peaks are noncoding regions. (b) Detail of the genomic regions used in the Xenopus functional enhancer assays (green and red blocks). Note that this region contains an evolutionarily-conserved noncoding peak. (c) eShadow view (ClustalW multiple sequence alignments presented as percent mismatch) of the SMAD7 construct showing regions of sequence conservation amongst primates (Human, Chimp, Orangutan, Rhesus, and Marmoset). Note that SNP Novel 1 is conserved in primates. (d) The tested regions contain an enhancer that promotes reporter gene expression in the rectal region of Xenopus tadpoles. The bright field image above shows a 5-d tadpole embryo. The rectal region is indicated by a red arrow. The fluorescent image below shows a detail of the rectal region of a Xenopus transgenic embryo in which GFP expression is promoted by the enhancer. The intensity of the rectal expression promoted by the enhancer from the Protective or the Risk haplotypes was measured relative to the signal observed in a fixed area in the muscles region (boxed in gray), which was considered as 100%. The DNA tested contains either the protective or risk variants of both rs8085824 and Novel 1 (green; 1 and 3) or solely Novel 1 (red; 2 and 4). (e) Box-whisker plot of the relative expression observed in transgenic embryos harboring the Protective or the Risk DNA promoting GFP expression. The enhancer from the risk haplotype/allele shows a significantly decreased enhancer activity. (f) EMSA revealing allele-specific binding of unknown nuclear factors at Novel 1 SNP.
We first tested the enhancer potential of a DNA fragment spanning the nucleotide position where these two SNPs map and the HCNR in Xenopus transgenic assays. For this we generated a construct in which this fragment, obtained from a control genomic human DNA sample, was located 5′ of a minimal promoter driving GFP expression (protective construct). Transgenic Xenopus embryos for this construct showed GFP expression in the muscles and in the colorectum at tadpole stages (Fig. 3d). No expression was observed in control embryos transgenic for a similar construct that lacks the human DNA. We next examined the enhancer potential of the same human fragment containing Novel 1 and rs8085824 (risk and protective constructs). As shown in Figure 3e, comparison of the Protective and the Risk enhancer activities indicated that the latter drove GFP expression at significantly reduced levels in the colorectum (median difference in expression levels of 10%; P = 2.5 × 10−4). Following this, we analyzed the impact of a construct in which Novel 1 was solely represented, generated using a shorter construct. Significantly reduced expression of GFP was associated with the risk allele of Novel 1 (median difference in expression levels 11%; P = 5.4 × 10−3). Collectively, these findings demonstrate that the risk variant defined solely by Novel 1 maps to a gut enhancer, and that the risk allele is detrimental for the enhancer's function and leads to reduced expression of SMAD7.
To examine differential binding between C and G alleles of Novel 1, we used electrophoretic mobility shift assays (EMSA) to study protein–DNA interactions in the SW620 CRC cell line. Probes of C and G alleles showed distinct binding patterns. The C allele of Novel 1 was found to form stronger protein–DNA complexes with nuclear extracts compared with the disease-associated G allele (Fig. 3f).
To investigate SMAD7 expression in colorectal carcinogenesis, we studied a series of 36 adenomas and 43 rectal cancers in which we had determined 18q21 copy number status (Fig. 4). Expression was significantly lower in cancers than adenomas irrespective of 18q21 copy number status (P = 5.1 × 10−6), therefore providing a basis for the association between 18q21 variation and CRC through differential SMAD7 expression. Limited study power, given the variation in expression in tissues, made us unable to demonstrate a significant relationship between genotype and expression after adjustment for copy number change and histology.
Figure 4.

SMAD7 expression in 36 rectal adenomas and 43 carcinomas. Vertical axis present normalized relative SMAD7 gene expression (Log2-scale). Expression of SMAD7 was significantly lower in carcinomas than adenomas, irrespective of 18q21 copy-number status. (Difference of expression between tumor groups: 1 vs. 2, P = 0.524; 3 vs. 4, P = 0.34; 1 vs. 3, P = 0.13; 2 vs. 4, P = 4.0 × 10−4; 1,3 vs. 2,4, P = 0.06; 1,2 vs. 3,4, P = 5.1 × 10−6)
Allelic imbalance at the 8q24 variant rs6983267 has been reported (Tuupanen et al. 2008). To examine whether there is selection of the 18q21 risk alleles in somatic CRC evolution, we correlated genotypes with copy number status. No evidence of a significant relationship was observed (data not shown).
Discussion
Recent GWA studies have robustly demonstrated that common genetic variation contributes to the risk of developing cancer, and an increasing number of genomic regions have been shown to be associated with cancer risk. However, because the majority of GWA studies are based on an analysis of tagging SNPs that enable common genetic variation to be interrogated, the genotyped SNPs associated with cancer risk are not necessarily the functional variants.
Here, we identified through sequencing all polymorphisms within the 17-kb region of the 18q21 locus, which, on the basis of LD, harbors the disease-causing variant responsible for the SMAD7–18q21 association with CRC. Characterizing all DNA polymorphisms within the region was a necessary prerequisite to large-scale fine mapping, as it allowed us to establish the correlations among all genetic variants, including those not previously documented. This facilitated the rapid nomination of the variants for functional analysis that were most highly correlated with the tagging SNPs most significantly associated with CRC risk.
Twenty-nine per cent of the DNA polymorphisms we identified mapping to the 17-kb region of LD at 18q21 were not cataloged by dbSNP. Furthermore, for 60% of all identified DNA polymorphisms there was no pre-existing HapMap genotype data. Our data underscores the value of resequencing analysis in order to generate a full catalog of variants for further genotyping and nomination for functional analyses.
Through cataloging of all DNA polymorphisms and genotyping of a large case control series, we were able to fine map the 17-kb region of association and provide statistical evidence to implicate a novel SNP as the basis of the observed association between SMAD7–18q21 variation and CRC risk.
SMAD7 acts as an intracellular antagonist of TGF-beta signaling by binding stably to the receptor complex and blocking activation of downstream signaling events (ten Dijke and Hill 2004). Perturbation of SMAD7 expression has been previously documented to influence CRC progression (Levy and Hill 2006), and loss of chromosome 18q21 encompassing SMAD7 is common in CRC (Gaasenbeek et al. 2006). Our observation that SMAD7 expression is lower in cancers than adenomas irrespective of 18q copy number status makes a bystander effect unlikely and suggests a direct role for SMAD7 in carcinogenesis. Hence, allele-specific expression of SMAD7 is likely to be the biological basis for CRC predisposition associated with 18q21 variation.
Demonstrating a relationship between expression and genotype provides one means of establishing a causal basis. However, given that any single variant is unlikely to have more than a subtle effect on expression of transcripts such as SMAD7 in cancers, coupled with the fact that cancers are genetically heterogeneous, makes searching for such relationships, as demonstrated herein, inherently problematic. Moreover, prohibitively large numbers of samples may be required to be analyzed in order to attain statistical significance. In view of this, it is likely there will be increasing reliance on animal model systems to demonstrate subtle differences in allele-specific gene expression.
In our study we based the functional analysis of Novel 1 on a Xenopus model system to maximize detection of a subtle functional effect. Using this transgenic system, we were able to demonstrate that possession of the G allele of Novel 1 is associated with a reduced expression of SMAD7 in the colorectum. The magnitude of the effect observed is in keeping with risk of CRC associated with 18q21 variation. A direct role for Novel 1 on transcription factor binding was supported by our EMSA data.
The BMP/TGF-beta pathway FGF, WNT, and Notch signaling pathways are the major stem-cell signaling networks in the development of CRC (Katoh and Katoh 2006). The SMAD7 colorectal enhancer to which Novel 1 maps harbors the predicted control module mod052296 (Supplemental Fig. 1B). Intriguingly, this domain contains putative transcription factor binding sites for SRY, RUNX3 (also known as AML), and PAX4. Given SOX17 (a SRY-box containing gene) is a negative regulator of beta-catenin signaling (Paoni et al. 2003) and inactivation of RUNX3 is a feature of CRC (Ku et al. 2004), there is strong biological plausibility of the mechanism of association we have identified. Furthermore, the G allele of Novel 1 potentially disrupts an EP300 (also known as p300) transcription-binding site (Supplemental Fig. 1C) which, through buffering regulation of TCF-beta-cat/ARM binding (Li et al. 2007), may directly impact on the development of CRC.
Here, we have shown one mechanism by which common variation influences CRC risk through differential expression of a regulatory protein. DNA polymorphisms affecting gene expression have long been postulated to contribute to disease predisposition (Hudson 2003). However, the knowledge of many regulatory networks remains sparse and only a restricted number of such polymorphisms have been identified (De Gobbi et al. 2006; Steidl et al. 2007). On the basis of sequence data relating to the other CRC loci 8q23.3 (Tomlinson et al. 2008), 8q24 (Zanke et al. 2007), 10p14 (Tomlinson et al. 2008), 11q23 (Tenesa et al. 2008) and 15q13 (Jaeger et al. 2008), 14q22, 16q22, 19q13, and 20q12 (Houlston et al. 2008), it is probable that some of the causal variants underlying these associations will also influence CRC risk through differential gene expression.
GWA studies are identifying an increasing number of susceptibility SNPs for complex diseases such as CRC. The future challenge will be to unravel the causative mechanism by which variants influence disease risk. Moving from an association signal to identification of the causative variant is far from straightforward. As we have demonstrated, such analysis will, for many loci, be contingent on a combination of genetic and functional-based assays. Using this combinational strategy, we propose that the novel SNP we have identified is the functional change leading to CRC predisposition through differential SMAD7 expression and, hence, aberrant TGF-beta signaling.
Methods
Participants
Resequencing/SNP discovery panel
A total of 90 healthy unrelated individuals from the Royal Marsden Hospital Trust/Institute of Cancer Research Family History and DNA Registry (50 males, 40 females; mean age at sampling 59 yr; SD ± 11). None had a personal history of malignancy at time of ascertainment and all were British Caucasians.
Regenotyping cohort
A total of 2532 CRC cases (1336 males, 1196 females; mean age at diagnosis 58.2 yr; SD ± 7.97) ascertained through two initiatives at the Institute of Cancer Research—The National Study of Colorectal Cancer Genetics (NSCCG) (Penegar et al. 2007) and an existing series; Royal Marsden Hospital Trust/Institute of Cancer Research Family History and DNA Registry. A total of 2607 healthy individuals were recruited as part of ongoing National Cancer Research Network genetic epidemiological studies, NSCCG (1200), and the Genetic Lung Cancer Predisposition Study (GELCAPS) (1999–2004; n = 771), the Royal Marsden Hospital Trust/Institute of Cancer Research Family History and DNA Registry (1999–2004; n = 636). These controls (1074 males, 1533 females; mean age 56.7 yr; SD ± 8.95) were the spouses or unrelated friends of patients with malignancies. None had a personal history of malignancy at time of ascertainment. All cases and controls were British caucasians, and there were no obvious differences in the demography of cases and controls in terms of place of residence within the UK.
In all cases, CRC was defined according to the ninth revision of the International Classification of Diseases (ICD) by codes 153–154, and all cases had pathologically proven colorectal adenocarcinoma.
Resequencing
Sequence changes in the interval chromosome 18: 44,700,221–44,716,898 (UCSC May 2006 assembly, NCBI build 36.1) encompassing the 3′ end of SMAD7 intron 3, exon 4, and the 3′ UTR were identified by direct sequencing. PCR and sequencing primers were designed by Primer3 software (Primer sequences available on request). Amplicons were sequenced by ABI chemistry (BigDye v3.1; Applied Biosystems) and platform (ABI 3730xl DNA analyzer; Applied Biosystems). Sequence reads were analyzed using Mutation Surveyor software (v3.10; Softgenetics).
Genotyping
DNA was extracted from samples using conventional methodologies and quantified using PicoGreen (Invitrogen). Genotyping was conducted by competitive allele-specific PCR KASPar chemistry (KBiosciences Ltd) or Illumina iSelect Arrrays (details available on request). Genotyping quality control was tested using duplicate DNA samples within studies and SNP assays together with direct sequencing of subsets of samples to confirm genotyping accuracy. For all SNPs, >99.9% concordant results were obtained.
Statistical analysis
Statistical analyses were undertaken in Stata v8 or R v2.6.2 software. A P-value of 0.05 was considered to be significant in all analyses and P-values were two-sided.
Genotype data were used to search for duplicates and closely related individuals amongst all samples. Identity by state values was calculated for each pair of individuals, and for any pair with allele sharing >80%, the sample generating the lowest call rate was removed from further analysis.
Deviation of the genotype frequencies in the controls from those expected under Hardy-Weinberg equilibrium (HWE) was assessed by χ2 test (1 degree of freedom [df]). The risks associated with each SNP were estimated by per allele, heterozygous and homozygous odds ratios (OR), and associated 95% confidence intervals (CIs) were calculated in each case. Haplotype analysis was conducted using Haploview software (v4.0). The haplotypes are estimated using an accelerated EM algorithm similar to the partition/ligation method described in Qin et al. (2002) and tested for association via a likelihood ratio test. Linkage disequilibrium statistics were calculated using Haploview software (v4.0).
Relationships between multiple SNPs showing association with CRC risk in the region were investigated using logistic regression analysis. The impact of additional SNPs from the same region was assessed by a likelihood-ratio test.
The weight of evidence in favor of each SNP being causal was quantified by calculating Akaike weights for each SNP model:
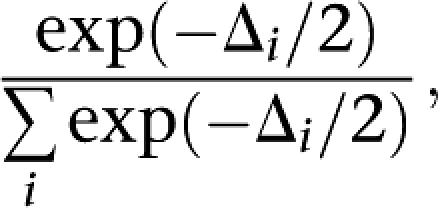 |
where Δi is the difference in log-likelihood between model i and the best fitting model.
Using the Margarita program (Minichiello and Durbin 2006), we inferred ARGs for the 25 SNP haplotypes spanning the SMAD7 interval. For every ARG, a putative risk mutation was placed on the marginal genealogy at each SNP position by maximizing the association between the mutation and disease status. The significance of associations was evaluated through 106 permutations.
Xenopus transgenic assays
Xenopus laevis transgenic embryos were generated using the I-SceI method recently described (Ogino et al. 2006). A 0.70-kb fragment containing the conserved region from the protective or risk human samples containing both variants of rs8085824 and Novel 1 were amplified with the following primers:
5′-GCTACCTTAACAAAGCTTCCTCC-3′ and 5′-CGCCTGTAAAAGTTGGAGC-3′ (Supplemental Fig. 1B). A 771-bp fragment containing the conserved region from the protective or risk human samples containing only variants of Novel 1 were amplified with the following primers: 5′-CCTCTCTCCCCTCCTTCC-3′ and 5′-CGCCTGTAAAAGTTGGAGC-3′ (Supplemental Fig. 1B). The PCR products were cloned in a TOPO T/A vector (Invitogen), sequenced, and then inserted using the gateway technology 5′ of a Xenopus 663-bp Gata2 minimal promoter driving GFP in a transgenesis vector (Invitogen). This vector contains two I-SceI sites flanking the expression cassette. Thirty-five and 42 transgenic embryos were generated for the Risk and Protective constructs, respectively. In each embryo, using the histogram function of Adobe Photoshop (Adobe), the intensity of the rectal expression was measured relative to the signal in a fixed area in the muscles, which served as the internal reference. Differences in the distribution of levels of expression between genotypes were compared using the Mann-Whitney test.
EMSA
Biotin end-labeled and unlabeled complementary oligonucleotide probes (5′-TCAGGGCCTTGCCCTCGCTCCCTGCAGCCCC-3-biotin and 5′-TCAGGGCCTTGCCCTGGCTCCCTGCAGCCCC-3′) were purchased from Invitrogen and annealed together to form double-stranded EMSA probes. Nuclear protein was extracted from the colorectal cancer cell line SW620 by use of the NE-PER nuclear and cytoplasmic extraction kit (Thermo scientific). EMSAs were performed by use of the Lightshift Chemiluminescent EMSA Kit (Pierce). Briefly, each 20-uL binding reaction contained 20 fmols of biotin end-labeled target DNA, 10× binding buffer, 50 ng of Poly(dI.dC), 2.5% glycerol, 0.05% NP-40, and ∼5 ug of nuclear protein extract. After a 20-min incubation, the samples were electrophoresed for 1 h at 100 V in a 6% polyacrylamide gel (0.5% TBE running buffer), followed by electroblotting for 1 h at 30 V. Chemiluminescent detection of the biotin end-labeled DNA was performed with a strepdavidin-horseradish peroxidase conjugate captured onto X-ray film and developed according to the manufacturer's instructions. Control reactions consisting of lanes omitting nuclear extract and the addition unlabeled probes (1000-fold excess) were also performed.
SMAD7 expression and 18q copy number analysis in colorectal cancer
Snap-frozen rectal adenomas and carcinomas from patients who had not received radiotherapy or other adjuvant therapy were evaluated for SMAD7 expression and 18q copy number. Frozen tissue sections were reviewed by a pathologist (HM), the degree of dysplasia scored, and tumor percentage assessed (50%–80%). Tumors were macrodissected in a cryostat, removing all of surrounding healthy tissue, guided by a 4-μm H&E section before the first and after the 10th and 20th section. DNA was isolated from tumors using the Genomic Wizard kit (Promega) and DNA quality was checked on a 1% agarose gel. GeneChip Mapping 10 K 2.0 arrays (Affymetrix, Inc.) were hybridized at the Leiden Genome Technology Center, and copy numbers were analyzed as described previously (Lips et al. 2007). RNA was isolated from tumors using the Qiagen RNeasy mini kit with Dnase I digestion (Qiagen Science) and quality checked by lab-on-a-chip (Agilent Technologies). A total of 2 ug of RNA was labeled using Ambion's Amino Allyl MessageAm aRNA kit (Ambion, Inc.) and hybridized to human 35 K oligo microarrays from the CMF of the Netherlands Cancer Institute as previously described (de Bruin et al. 2005). The Limma (linear models for microarray data) package of Bioconductor was used for importing the data, normalizing the arrays, and data analysis. Data were corrected for local background (method normexp) and normalized within arrays by print-tip loess normalization and between arrays by quantile normalization. Comparison of the difference in expression levels was assessed using the Mann-Whitney test.
Ethics
Medical ethical committee approval for this study was obtained from the relevant study center (UK MREC protocol no. MREC 02/0/97, Netherlands, LUMC/CME protocol no. P04.124).
Acknowledgments
Cancer Research UK (C1298/A8362 supported by the Bobby Moore Fund) provided principal funding for the study. Additional funding was provided by the European Union (CPRB LSHC-CT-2004-503465). J.L.G.-S. and F.C. acknowledge grants from the Spanish Ministry of Education and Science (BFU2007-60042/BMC, BFU2006-00349/BMC, CSD2007-00008) and Junta de Andalucía (CVI 00260 and CVI 2658). H.M. and T.v.W. acknowledge support by Dutch Cancer Society grant UL2003-2807. Finally, we thank all of the patients and individuals for their participation.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.092668.109.
References
- Broderick P., Carvajal-Carmona L., Pittman A.M., Webb E., Howarth K., Rowan A., Lubbe S., Spain S., Sullivan K., Fielding S., et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat. Genet. 2007;39:1315–1317. doi: 10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- Curtin K., Lin W.Y., George R., Katory M., Shorto J., Cannon-Albright L.A., Bishop D.T., Cox A., Camp N.J. Meta association of colorectal cancer confirms risk alleles at 8q24 and 18q21. Cancer Epidemiol. Biomarkers Prev. 2009;18:616–621. doi: 10.1158/1055-9965.EPI-08-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin E.C., van de Pas S., Lips E.H., van Eijk R., van der Zee M.M., Lombaerts M., van Wezel T., Marijnen C.A., van Krieken J.H., Medema J.P., et al. Macrodissection versus microdissection of rectal carcinoma: Minor influence of stroma cells to tumor cell gene expression profiles. BMC Genomics. 2005;6:142. doi: 10.1186/1471-2164-6-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gobbi M., Viprakasit V., Hughes J.R., Fisher C., Buckle V.J., Ayyub H., Gibbons R.J., Vernimmen D., Yoshinaga Y., de Jong P., et al. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science. 2006;312:1215–1217. doi: 10.1126/science.1126431. [DOI] [PubMed] [Google Scholar]
- Dermitzakis E.T., Reymond A., Antonarakis S.E. Conserved non-genic sequences - an unexpected feature of mammalian genomes. Nat. Rev. Genet. 2005;6:151–157. doi: 10.1038/nrg1527. [DOI] [PubMed] [Google Scholar]
- Gaasenbeek M., Howarth K., Rowan A.J., Gorman P.A., Jones A., Chaplin T., Liu Y., Bicknell D., Davison E.J., Fiegler H., et al. Combined array-comparative genomic hybridization and single-nucleotide polymorphism-loss of heterozygosity analysis reveals complex changes and multiple forms of chromosomal instability in colorectal cancers. Cancer Res. 2006;66:3471–3479. doi: 10.1158/0008-5472.CAN-05-3285. [DOI] [PubMed] [Google Scholar]
- Gomez-Skarmeta J.L., Lenhard B., Becker T.S. New technologies, new findings, and new concepts in the study of vertebrate cis-regulatory sequences. Dev. Dyn. 2006;235:870–885. doi: 10.1002/dvdy.20659. [DOI] [PubMed] [Google Scholar]
- Houlston R.S., Webb E., Broderick P., Pittman A.M., Di Bernardo M.C., Lubbe S., Chandler I., Vijayakrishnan J., Sullivan K., Penegar S., et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat. Genet. 2008;40:1426–1435. doi: 10.1038/ng.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson T.J. Wanted: Regulatory SNPs. Nat. Genet. 2003;33:439–440. doi: 10.1038/ng0403-439. [DOI] [PubMed] [Google Scholar]
- Jaeger E., Webb E., Howarth K., Carvajal-Carmona L., Rowan A., Broderick P., Walther A., Spain S., Pittman A., Kemp Z., et al. Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. Nat. Genet. 2008;40:26–28. doi: 10.1038/ng.2007.41. [DOI] [PubMed] [Google Scholar]
- Katoh Y., Katoh M. Hedgehog signaling pathway and gastrointestinal stem cell signaling network (review) Int. J. Mol. Med. 2006;18:1019–1023. [PubMed] [Google Scholar]
- Ku J.L., Kang S.B., Shin Y.K., Kang H.C., Hong S.H., Kim I.J., Shin J.H., Han I.O., Park J.G. Promoter hypermethylation downregulates RUNX3 gene expression in colorectal cancer cell lines. Oncogene. 2004;23:6736–6742. doi: 10.1038/sj.onc.1207731. [DOI] [PubMed] [Google Scholar]
- Levy L., Hill C.S. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Li J., Sutter C., Parker D.S., Blauwkamp T., Fang M., Cadigan K.M. CBP/p300 are bimodal regulators of Wnt signaling. EMBO J. 2007;26:2284–2294. doi: 10.1038/sj.emboj.7601667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips E.H., de Graaf E.J., Tollenaar R.A., van Eijk R., Oosting J., Szuhai K., Karsten T., Nanya Y., Ogawa S., van de Velde C.J., et al. Single nucleotide polymorphism array analysis of chromosomal instability patterns discriminates rectal adenomas from carcinomas. J. Pathol. 2007;212:269–277. doi: 10.1002/path.2180. [DOI] [PubMed] [Google Scholar]
- McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P., Hirschhorn J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- Minichiello M.J., Durbin R. Mapping trait loci by use of inferred ancestral recombination graphs. Am. J. Hum. Genet. 2006;79:910–922. doi: 10.1086/508901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino H., McConnell W.B., Grainger R.M. High-throughput transgenesis in Xenopus using I-SceI meganuclease. Nat. Protocols. 2006;1:1703–1710. doi: 10.1038/nprot.2006.208. [DOI] [PubMed] [Google Scholar]
- Paoni N.F., Feldman M.W., Gutierrez L.S., Ploplis V.A., Castellino F.J. Transcriptional profiling of the transition from normal intestinal epithelia to adenomas and carcinomas in the APCMin/+ mouse. Physiol. Genomics. 2003;15:228–235. doi: 10.1152/physiolgenomics.00078.2003. [DOI] [PubMed] [Google Scholar]
- Penegar S., Wood W., Lubbe S., Chandler I., Broderick P., Papaemmanuil E., Sellick G., Gray R., Peto J., Houlston R. National study of colorectal cancer genetics. Br. J. Cancer. 2007;97:1305–1309. doi: 10.1038/sj.bjc.6603997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z.S., Niu T., Liu J.S. Partition-ligation-expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am. J. Hum. Genet. 2002;71:1242–1247. doi: 10.1086/344207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidl U., Steidl C., Ebralidze A., Chapuy B., Han H.J., Will B., Rosenbauer F., Becker A., Wagner K., Koschmieder S., et al. A distal single nucleotide polymorphism alters long-range regulation of the PU.1 gene in acute myeloid leukemia. J. Clin. Invest. 2007;117:2611–2620. doi: 10.1172/JCI30525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Dijke P., Hill C.S. New insights into TGF-beta-Smad signalling. Trends Biochem. Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Tenesa A., Farrington S.M., Prendergast J.G., Porteous M.E., Walker M., Haq N., Barnetson R.A., Theodoratou E., Cetnarskyj R., Cartwright N., et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat. Genet. 2008;40:631–637. doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson I.P., Webb E., Carvajal-Carmona L., Broderick P., Howarth K., Pittman A.M., Spain S., Lubbe S., Walther A., Sullivan K., et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat. Genet. 2008;40:623–630. doi: 10.1038/ng.111. [DOI] [PubMed] [Google Scholar]
- Tuupanen S., Niittymaki I., Nousiainen K., Vanharanta S., Mecklin J.P., Nuorva K., Jarvinen H., Hautaniemi S., Karhu A., Aaltonen L.A. Allelic imbalance at rs6983267 suggests selection of the risk allele in somatic colorectal tumor evolution. Cancer Res. 2008;68:14–17. doi: 10.1158/0008-5472.CAN-07-5766. [DOI] [PubMed] [Google Scholar]
- Zanke B.W., Greenwood C.M., Rangrej J., Kustra R., Tenesa A., Farrington S.M., Prendergast J., Olschwang S., Chiang T., Crowdy E., et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat. Genet. 2007;39:989–994. doi: 10.1038/ng2089. [DOI] [PubMed] [Google Scholar]