

Abstract
The progressive myoclonus epilepsies, featuring the triad of myoclonus, seizures, and ataxia, comprise a large group of inherited neurodegenerative diseases that remain poorly understood and refractory to treatment. The Cystatin B gene is mutated in one of the most common forms of progressive myoclonus epilepsy, Unverricht–Lundborg disease (EPM1). Cystatin B knockout in a mouse model of EPM1 triggers progressive degeneration of cerebellar granule neurons. Here, we report impaired redox homeostasis as a key mechanism by which Cystatin B deficiency triggers neurodegeneration. Oxidative stress induces the expression of Cystatin B in cerebellar granule neurons, and EPM1 patient-linked mutation of the Cystatin B gene promoter impairs oxidative stress induction of Cystatin B transcription. Importantly, Cystatin B knockout or knockdown sensitizes cerebellar granule neurons to oxidative stress-induced cell death. The Cystatin B deficiency-induced predisposition to oxidative stress in neurons is mediated by the lysosomal protease Cathepsin B. We uncover evidence of oxidative damage, reflected by depletion of antioxidants and increased lipid peroxidation, in the cerebellum of Cystatin B knock-out mice in vivo. Collectively, our findings define a pathophysiological mechanism in EPM1, whereby Cystatin B deficiency couples oxidative stress to neuronal death and degeneration, and may thus provide the basis for novel treatment approaches for the progressive myoclonus epilepsies.
Introduction
The progressive myoclonus epilepsies (PMEs) comprise of a group of inherited diseases that present with myoclonus, epilepsy, and progressive neuronal deterioration (Berkovic et al., 1986; Marseille Consensus Group, 1990). PME of the Unverricht–Lundborg type (EPM1) has the highest incidence among the PMEs worldwide and is characterized by onset at 6–16 years, stimulus-sensitive myoclonus, and tonic-clonic seizures. As EPM1 progresses, patients develop additional neurological symptoms, including ataxia and dysarthria, reflecting widespread neuronal degeneration (Kälviäinen et al., 2008). Loss-of-function mutations in the gene encoding Cystatin B, an inhibitor of lysosomal Cathepsins, are the primary genetic cause of EPM1 (Pennacchio et al., 1996; Lalioti et al., 1997; Joensuu et al., 2008).
Cystatin B knock-out mice recapitulate key clinical features of EPM1 (Pennacchio et al., 1998). Myoclonic seizures in these mice typically occur during sleep and progress from twitching of isolated muscles to spasms affecting the entire body. Electrocorticogram recordings reveal bilaterally synchronous 4–6 Hz repetitive spiking commencing with the myoclonus. In addition, Cystatin B-deficient mice develop progressive ataxia. Importantly, the major feature in Cystatin B-deficient mice is a progressive death of cerebellar granule neurons. These observations suggest that neuronal degeneration plays a critical role in EPM1 pathophysiology. However, the cell biological stimuli and signaling mechanisms by which Cystatin B deficiency triggers neuronal degeneration have remained to be elucidated.
We reasoned that Cystatin B deficiency might trigger a pathophysiological response in neurons, having the potential of inducing hyperexcitability leading to seizures and myoclonus, and neuronal degeneration. Disrupted redox homeostasis resulting from oxidative stress has been linked to diverse disease pathologies including seizure disorders and neurodegenerative diseases (Kunz, 2002; Chong et al., 2005). Increasing evidence suggests that antioxidants may alleviate the progression of epilepsy. Intriguingly, the antioxidant N-acetylcysteine (NAC) reportedly alleviates seizures and myoclonus in EPM1 patients (Hurd et al., 1996; Edwards et al., 2002), suggesting that oxidative stress may contribute to EPM1-induced neuronal hyperexcitability. However, the importance of oxidative stress in neuronal degeneration in EPM1 has remained unexplored.
In this study, we identify a novel function for Cystatin B in defending cerebellar granule neurons from oxidative stress. Our findings define an oxidative stress-responsive Cystatin B–Cathepsin B signaling pathway whose dysregulation couples oxidative stress to neuronal degeneration in EPM1.
Materials and Methods
Mice.
The Cystatin B-deficient mouse strain (129-Cstbtm1Rm/J) was obtained from The Jackson Laboratory (stock #003486). Wild-type offspring from heterozygous matings were used as controls.
Antibodies.
Antibodies to β-galactosidase (Promega), Cystatin B (Biogenesis), DsRed (Clonetech Laboratories), green fluorescent protein (GFP) (Santa Cruz Biotechnology), and β-tubulin and M2-Flag (Sigma) were purchased.
Tissue culture.
Granule neurons prepared from postnatal day 6 (P6) rat or P5/6 mouse cerebella were transfected as described previously (Lehtinen et al., 2006). Neurons were left untreated or treated with H2O2 (50–100 μm) or glutamate (10 mm). To control for the high reactivity of H2O2, the H2O2 concentration used was separately titrated in each set of experiments. The next day, cells were subjected to immunocytochemistry. Neuronal survival was assessed in transfected β-galactosidase-positive neurons based on the integrity of neuronal processes and nuclear morphology (Lehtinen et al., 2006). Approximately 100 cells were counted per treatment condition, per experiment, in a blinded manner and analyzed for statistical significance by ANOVA followed by Fisher's protected least significant difference post hoc test. Statistical analyses represent a minimum of three separate experiments.
Supplemental Methods, available at www.jneurosci.org as supplemental material, include detailed protocols for the following: plasmids; immunoblotting, immunocytochemistry, and luciferase assays; quantitative real-time PCR; Cathepsin B activity; and redox analyses.
Results
We investigated the potential role of Cystatin B in regulating redox homeostasis and oxidative stress responses in cerebellar granule neurons. We first used a plasmid-based method of RNA interference (RNAi) to acutely knockdown Cystatin B. The expression of Cystatin B hairpin RNAs (hpRNAs) dramatically reduced expression of Cystatin B in 293T cells and primary rat cerebellar granule neurons (Fig. 1A,B). We then assessed the functional effect of Cystatin B knockdown on oxidatively stressed neurons. Rat granule neurons were transfected with the Cystatin B hpRNA or control plasmid together with a plasmid encoding β-galactosidase. After transfection, neurons were exposed to oxidative stress, and cell survival was assessed in transfected β-galactosidase-positive neurons based on the integrity of neuronal processes and nuclear morphology. Cystatin B knockdown robustly increased the ability of hydrogen peroxide to induce cell death in granule neurons (Fig. 1C). High concentrations of glutamate (10 mm) induce oxidative stress internally in neurons (Ratan et al., 2002). Cystatin B knockdown neurons were also sensitized to glutamate-induced cell death (Fig. 1D). Together, these data suggest that Cystatin B protects neurons in response to oxidative stress.
Figure 1.
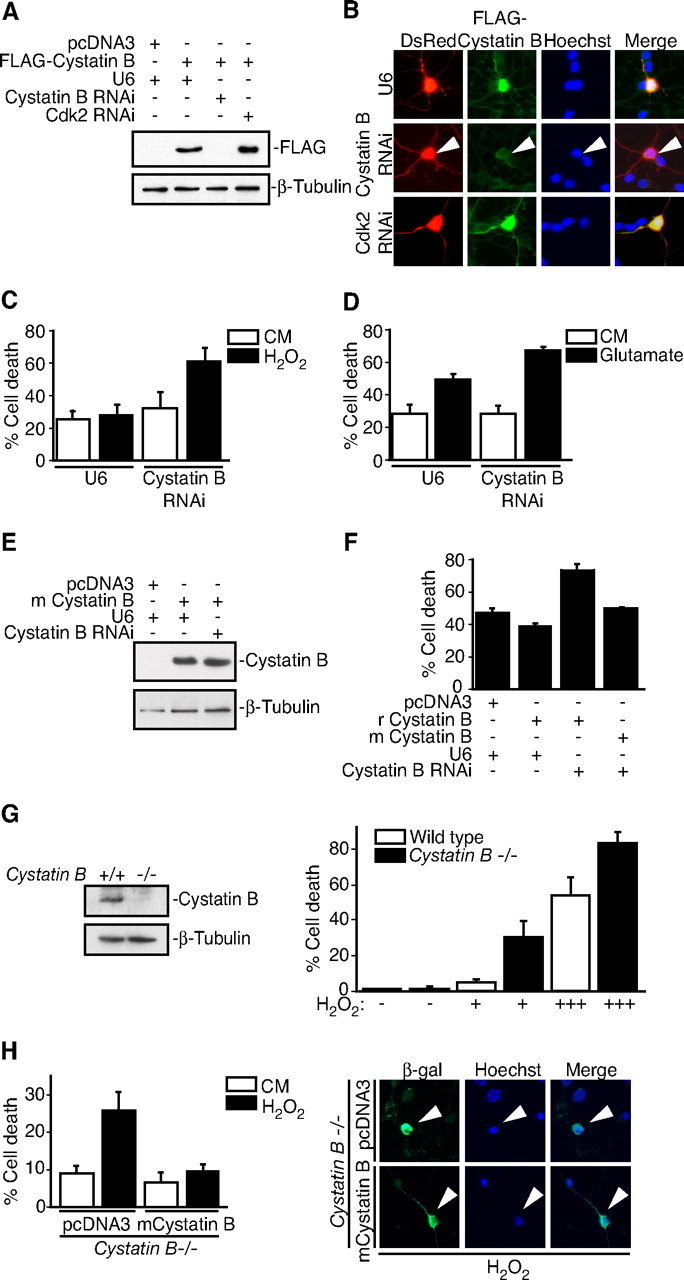
Cystatin B deficiency sensitizes neurons to oxidative stress. A, Lysates of 293T cells transfected with the rat FLAG—Cystatin B expression vector or control plasmid together with the Cystatin B hpRNA, Cdk2 hpRNA, or control U6 plasmid were immunoblotted with the Cystatin B and β-tubulin antibodies. B, Immunocytochemical analysis of rat granule neurons transfected with FLAG—Cystatin B and DsRed expression plasmids together with the Cystatin B hpRNA, Cdk2 hpRNA, or control U6 plasmid. Cystatin B RNAi reduced FLAG–Cystatin B expression in an average of 63.2% of transfected cells (n = 2). C, Rat granule neurons were transfected with the Cystatin B hpRNA or control U6 plasmid together with the β-galactosidase expression plasmid. After 72 h, cultures were treated with H2O2 for 24 h. Percentage cell death in transfected β-galactosidase-positive neurons is represented as mean ± SEM. Cystatin B knockdown sensitized neurons to H2O2-induced cell death (ANOVA; p < 0.05; n = 3). D, Rat granule neurons transfected with the Cystatin B hpRNA or control U6 plasmid together with the β-galactosidase expression plasmid were treated with 10 mm glutamate for 24 h and analyzed as in Figure 1C. Cystatin B RNAi sensitized neurons to glutamate-induced cell death (ANOVA; p < 0.05; n = 3). E, Lysates of 293T cells transfected with the mouse (m) Cystatin B expression vector or control plasmid together with the Cystatin B hpRNA or control U6 plasmid were immunoblotted with the Cystatin B and β-tubulin antibodies. F, Rat granule neurons transfected with the rat (r)FLAG–Cystatin B or mCystatin B expression plasmid and the β-galactosidase expression vector together with the Cystatin B RNAi plasmid were treated with H2O2 and analyzed as in C. Mouse Cystatin B rescued neurons from rat Cystatin B RNAi sensitization to H2O2-induced death (ANOVA; p < 0.001; n = 3). G, Left panels, Lysates of wildtype and Cystatin B−/− mouse granule neurons were immunoblotted with the Cystatin B or β-tubulin antibodies. Right panels, Cultures of Cystatin B−/− and wild-type control granule neurons were treated with increasing amounts of H2O2 and analyzed as in C. Cell death significantly increased in Cystatin B−/− neurons during H2O2 treatment compared with control (ANOVA; p < 0.0001; n = 3). H, Left panels, Cystatin B−/− granule neurons transfected with the mouse Cystatin B or control plasmid together with the β-galactosidase expression plasmid were treated with H2O2 and analyzed as in C. Cystatin B expression in Cystatin B−/− neurons rescued neurons from oxidative stress sensitivity (ANOVA; p < 0.01; n = 3). Right panels, Arrowheads denote representative images of neurons scored for survival.
We next performed a rescue experiment in which we expressed mouse Cystatin B in the background of rat Cystatin B RNAi. The Cystatin B hpRNAs target a sequence in rat Cystatin B mRNA that differs from the mouse Cystatin B mRNA by five base pairs such that mouse Cystatin B was resistant to Cystatin B RNAi (Fig. 1E). Expression of mouse Cystatin B in rat granule neurons reversed the ability of Cystatin B RNAi to sensitize neurons to stress-induced cell death (Fig. 1F). These results indicate that Cystatin B RNAi sensitizes neurons to hydrogen peroxide via specific knockdown of Cystatin B rather than off-target effects of Cystatin B RNAi or nonspecific activation of the RNAi machinery.
We also tested whether primary granule neurons isolated from Cystatin B-deficient mice are sensitive to oxidative stress. Consistent with the RNAi experiments in rat granule neurons (Fig. 1A–F), hydrogen peroxide induced significantly greater levels of cell death in Cystatin B−/− than wild-type mouse granule neurons (Fig. 1G). Importantly, Cystatin B expression rescued Cystatin B−/− neurons from oxidative stress-induced death, suggesting that the sensitivity of these neurons to oxidative stress is not attributable to nonspecific alteration in Cystatin B knock-out neurons independent of their genotype (Fig. 1H). Together, these results establish an essential function for Cystatin B in protecting neurons from oxidative stress.
Since Cystatin B deficiency sensitized granule neurons to oxidative stress, we reasoned that Cystatin B-deficient cerebella might incur oxidative damage in vivo that contributes to the pathogenesis of EPM1. To test this hypothesis, we performed an extensive redox analysis of Cystatin B−/− and wild-type control brains. We measured the levels of antioxidants including superoxide dismutase (SOD), glutathione (GSH), and catalase, the deregulation of which have been associated with neurodegeneration (Chong et al., 2005). SOD dismutates superoxide to hydrogen peroxide, whereas GSH and catalase reduce hydroperoxides (Lehtinen and Bonni, 2006). We observed a significant reduction in SOD activity and GSH levels specifically in the cerebellum but not in other regions of the brain, including the telencephalon and diencephalon, in Cystatin B knock-out animals compared with wild-type animals (Table 1; supplemental Table 1, available at www.jneurosci.org as supplemental material). Accordingly, the ratio of oxidized to reduced glutathione (GSSG:GSH) was increased in the cerebellum, a hallmark of oxidative damage (Table 1; supplemental Table 1, available at www.jneurosci.org as supplemental material). No differences in the overall glutathione reductase activity were observed in the brains of Cystatin B−/− mice compared with control (data not shown), suggesting that decreased GSH levels were not attributable to impaired glutathione reductase activity. No change in catalase activity was found in the cerebellum or other brain regions in Cystatin B knock-out animals (data not shown). Together, these data demonstrate that Cystatin B deficiency leads to impaired antioxidant capacity in the cerebellum.
Table 1.
Cystatin B deficiency disrupts cerebellar redox homeostasis at 6 months of age
| Redox assay | Cerebellum |
||
|---|---|---|---|
| Cystatin B+/+ | Cystatin B−/− | p value* | |
| SODa (units/mg protein) | 632.1 ± 35.8 | 482.4 ± 20.6 | <0.05 |
| GSHb (nmol/mg tissue) | 1.8 ± 0.004 | 1.0 ± 0.142 | <0.05 |
| GSSG:GSHc | 0.061 ± 0.003 | 0.085 ± 0.006 | <0.05 |
| 8-epi PGF2αd (pg/mg) | 1.0 ± 0.09 | 5.6 ± 1.1 | <0.005 |
| Glutathione peroxidasee (units/mg protein) | 24.4 ± 1.8 | 30.1 ± 1.3 | <0.05 |
*p values are determined by Student's t test. In the last column, p values corresponding to the following redox analyses are compared as follows.
aOne unit of SOD activity (units per milligrams of protein) is defined as 50% inhibition of formazan production. SOD activity was significantly reduced in Cystatin B−/− cerebella compared with wild-type controls [n = 5 (+/+), n = 4 (−/−)].
bGSH was significantly reduced in Cystatin B−/− cerebella compared with wild-type controls [n = 4 (+/+), n = 4 (−/−)].
cGSSG:GSH was significantly increased in Cystatin B−/− cerebella compared with wild-type controls [n = 4 (+/+), n = 4 (−/−)].
dLipid peroxidation was assayed based on competition between 8-epi–PGF2α and an 8-epi–PGF2α-acetylcholinesterase conjugate. Lipid peroxidation was significantly increased in Cystatin B−/− cerebella compared with wild-type controls [n = 3 (+/+), n = 5 (−/−)].
eGlutathione peroxidase activity was assayed by NADPH oxidation. Glutathione peroxidase activity increased significantly in Cystatin B−/− cerebella compared with wild-type controls [n = 3 (+/+), n = 5 (−/−)].
Accumulating free radicals can promote damage to cells via reactions with macromolecules including lipids, proteins, and DNA (Chong et al., 2005). Although we did not observe significant changes in oxidative damage to proteins or DNA in Cystatin B knock-out animals (data not shown), we found a dramatic increase in oxidative damage to lipids in Cystatin B knock-out animals using an assay for 8-epi prostaglandin Fα (8-epi-PGF2α), an established and specific marker of lipid peroxidation. Lipid peroxidation was increased in the cerebellum but not the telencephalon or diencephalon of Cystatin B−/− mice when compared with wild-type mice (Table 1; supplemental Table 1, available at www.jneurosci.org as supplemental material) (data not shown). Importantly, lipid peroxidation accelerated with age, increasing from baseline at 2 months, to nearly fivefold in 6-month-old mice compared with wild-type controls (Table 1; supplemental Table 1, available at www.jneurosci.org as supplemental material) (data not shown). Consistent with these observations, the activity of glutathione peroxidase, an enzyme that reduces lipid hydroperoxides, was increased in Cystatin B−/− cerebella compared with wild-type controls (Table 1), suggesting a cellular adaptation to oxidative lipid damage in these animals. Our findings suggest that Cystatin B deficiency increases cerebellar susceptibility to oxidative damage and specifically to lipid peroxidation.
Identification of a function for Cystatin B in protecting neurons against oxidative stress led us to determine the mechanisms regulating Cystatin B-mediated neuronal survival. We first measured Cystatin B levels in neurons in response to oxidative stress. Exposure of granule neurons to hydrogen peroxide stimulated a robust increase in Cystatin B protein (Fig. 2A) and RNA levels (expressed as fold change: control = 1.0; H2O2 = 1.8 ± 0.4; t test: p < 0.01; n = 3). The hydrogen peroxide-induced upregulation of Cystatin B was blocked by the transcriptional inhibitor actinomycin D (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), suggesting that oxidative stress induces Cystatin B transcription.
Figure 2.
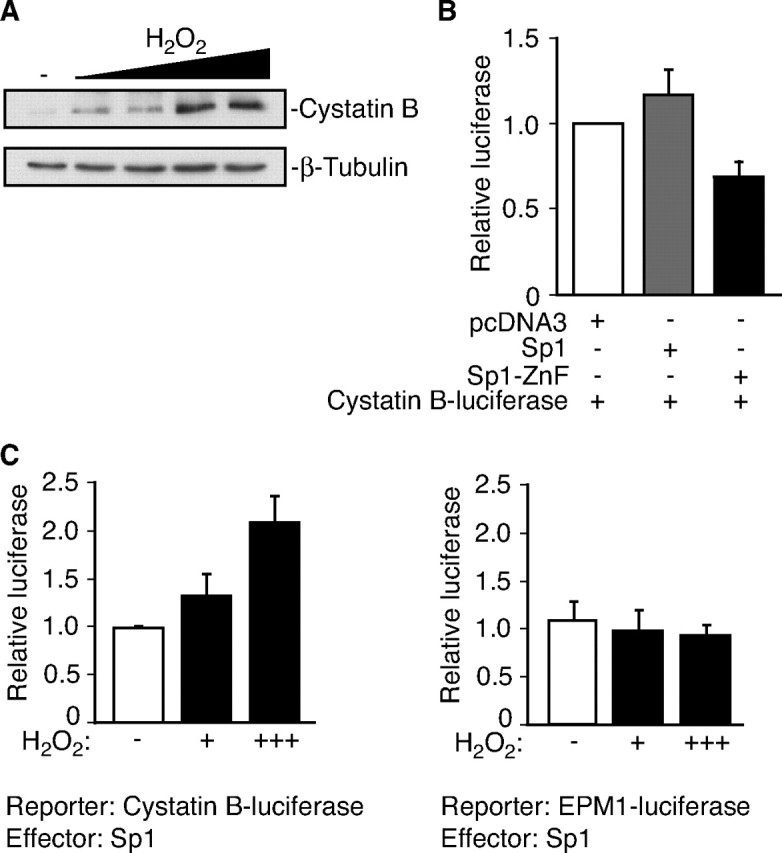
Oxidative stress induces Cystatin B expression. A, Lysates of rat granule neurons treated with increasing concentrations of H2O2 (50–80 μm) for 24 h were immunoblotted with Cystatin B and β-tubulin antibodies. B, Granule neurons were transfected with an expression vector encoding Sp1, the dominant interfering form of Sp1, Sp1–ZnF, or control vector together with the Cystatin B firefly luciferase gene. A Renilla luciferase reporter gene was cotransfected to control for transfection efficiency. Sp1–ZnF inhibited Cystatin B luciferase activity compared with control and Sp1 (ANOVA; p < 0.05; n = 5). C, Oxidative stress stimulates Sp1-mediated Cystatin B transcription. Left panel, Granule neurons were transfected with an expression vector encoding Sp1 together with the Cystatin B firefly luciferase gene and a Renilla luciferase reporter gene. Granule neurons were left untreated or treated with H2O2 for 16 h. Data are expressed as (Cystatin B luciferase plus Sp1)/(Cystatin B luciferase plus control). H2O2 induced Cystatin B (ANOVA; p < 0.01; n = 3). Right panel, Granule neurons were transfected with an Sp1 expression vector together with a Cystatin B firefly luciferase gene harboring an EPM1-like dodecamer repeat expansion and treated as in the left panel. Data are expressed as (EPM1 luciferase plus Sp1)/(EPM1 luciferase plus control). H2O2 failed to induce EPM1-linked Cystatin B.
The transcription factor Sp1 binds the Cystatin B promoter (Alakurtti et al., 2000). Expression of a dominant interfering form of Sp1 containing the DNA-binding domain but lacking the transactivation domain (Sp1-ZnF) in neurons impaired the expression of a luciferase reporter gene controlled by the Cystatin B promoter (Fig. 2B) (Chapman and Perkins, 2000; Ryu et al., 2003), suggesting that Sp1 drives Cystatin B transcription. Since Sp1 has been implicated in oxidative stress-induced transcriptional responses (Dunah et al., 2002; Ryu et al., 2003), we tested if oxidative stress might stimulate Sp1-mediated Cystatin B transcription. Hydrogen peroxide induced the ability of Sp1 to activate the expression of the Cystatin B-luciferase reporter gene (Fig. 2C). The predominant mutation that predisposes patients to EPM1 is the expansion of a dodecamer repeat (5′-ccccgccccgcg-3′) in the Cystatin B promoter (Lafrenière et al., 1997; Lalioti et al., 1997). In contrast to Sp1 induction of the wild-type Cystatin B promoter during exposure to hydrogen peroxide, Sp1 failed to activate a Cystatin B promoter harboring an EPM1-related dodecamer repeat expansion (Alakurtti et al., 2000) in granule neurons (Fig. 2C). These results suggest that oxidative stress induces Sp1-dependent Cystatin B transcription in neurons, and the EPM1-linked dodecamer repeat expansion in the Cystatin B promoter impairs the oxidative stress-induced Cystatin B transcriptional response.
Cystatin B inhibits the activity of the lysosomal protease Cathepsin B (Turk and Bode, 1991). Cathepsin B activity is upregulated in EPM1 patients (Rinne et al., 2002). In agreement with this result, Cathepsin B activity is enhanced in cultures of Cystatin B-deficient cerebellar granule neurons [relative Cathepsin B activity: 34.4 ± 3.07 (Cystatin B+/+); 42.6 ± 1.97 (Cystatin B−/−); t test: p < 0.05; n = 4 (+/+), n = 3 (−/−)]. Since granule neuron degeneration is reduced in Cystatin B–Cathepsin B double knock-out mice compared with Cystatin B knock-out mice (Houseweart et al., 2003), these observations raised the possibility that Cystatin B might protect neurons against oxidative stress-induced cell death by inhibiting Cathepsin B. Consistent with the hypothesis that Cathepsin B plays an intimate role in oxidative stress responses in neurons, hydrogen peroxide robustly induced the expression of Cathepsin B RNA (expressed as fold change: Control = 1.0; H2O2 = 1.96 ± 0.1; t test: p < 0.01; n = 3) and protein in neurons (Fig. 3A). Importantly, hydrogen peroxide stimulated the enzymatic activity of Cathepsin B in neurons (Fig. 3B). We next assessed Cathepsin B function in neurons. Overexpression of Cathepsin B sensitized neurons to hydrogen peroxide-induced death (Fig. 3C). Conversely, Cathepsin B knockdown protected neurons from oxidative stress-induced death (Fig. 3D; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Importantly, whereas Cystatin B deficiency sensitized neurons to oxidative stress-induced death (Figs. 1C,D,G, 3E), Cathepsin B RNAi rescued Cystatin B knockdown neurons from hydrogen peroxide-induced death (Fig. 3E). Together, these findings suggest that Cathepsin B may signal downstream of Cystatin B to promote neuronal death under oxidative stress conditions.
Figure 3.
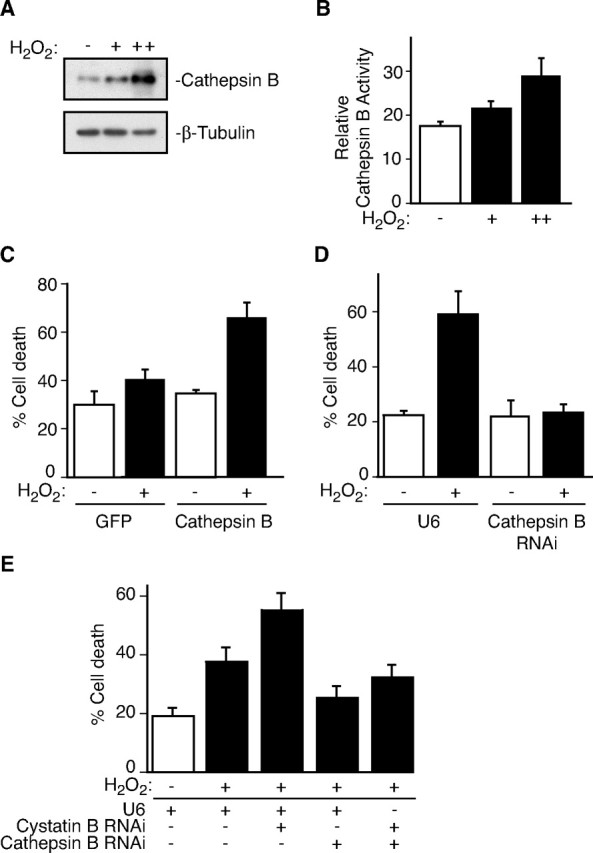
Cathepsin B mediates oxidative stress-induced neuronal death downstream of Cystatin B. A, Lysates of granule neurons treated with increasing concentrations of H2O2 (25–50 μm) for 18 h were immunoblotted with Cathepsin B and β-tubulin antibodies. B, Granule neurons treated with H2O2 for 14 h were assayed for Cathepsin B activity. H2O2 promoted Cathepsin B activity (ANOVA; p < 0.01; n = 5). C, Neurons transfected with Cathepsin B or control plasmid together with the β-galactosidase expression vector were treated with H2O2 and analyzed as in Figure 1C. Cathepsin B sensitized neurons to H2O2-induced death (ANOVA; p < 0.005; n = 3). D, Granule neurons transfected with the Cathepsin B hpRNA or control U6 plasmid together with the β-galactosidase expression plasmid were treated and analyzed as in Figure 1C. Cathepsin B RNAi protected neurons from H2O2-induced cell death (ANOVA; p < 0.001; n = 4). E, Granule neurons transfected with the Cystatin B hpRNA, Cathepsin B hpRNA, and/or control U6 plasmid together with the β-galactosidase expression plasmid were treated and analyzed as in Figure 1C. Cathepsin B RNAi protected neurons from Cystatin B–RNAi-induced H2O2 sensitivity (ANOVA; p < 0.001; n = 6 and n = 4, respectively).
Discussion
Our data reveal an oxidative stress-responsive Cystatin B-signaling pathway that protects neurons from oxidative stress and whose dysregulation contributes to the pathophysiology of EPM1. The major findings of our study are as follows: (1) Cystatin B protects cerebellar granule neurons from oxidative stress, (2) Cystatin B deficiency leads to the accumulation of oxidative damage in the cerebellum reflected in both a reduced antioxidant capacity and a marked susceptibility to lipid peroxidation, (3) oxidative stress induces the expression of Cystatin B, (4) Cathepsin B expression and enzymatic activity are regulated by oxidative stress, and (5) Cystatin B-deficient neurons are rescued from oxidative stress-induced death by a concurrent decrease in Cathepsin B levels. Collectively, our findings suggest that dysregulation of Cystatin B–Cathepsin B signaling may serve as a critical mechanism coupling oxidative stress to neuronal degeneration and death in EPM1.
Identification of a function for Cystatin B in oxidative stress responses in neurons has important implications for our understanding of EPM1 pathophysiology. Cystatin B deficiency in mice triggers progressive degeneration of cerebellar granule neurons and ataxia (Pennacchio et al., 1998). Our findings define impaired redox homeostasis as a mechanism by which Cystatin B deficiency promotes neuronal degeneration. Oxidative stress sensitivity and progressive cerebellar granule neuron degeneration are also principal features of a mouse model of progressive ataxia in which expression of the mitochondrial oxidoreductase gene apoptosis-inducing factor (Aif) is dramatically reduced (Klein et al., 2002). The shared oxidative stress-associated pathologies and cellular responses to oxidative stress in granule neurons in Cystatin B- and Aif-deficient mice raise the interesting possibility that lysosomally derived proteases and mitochondrially derived oxidoreductases may converge on a common mechanism to govern redox homeostasis and neuronal survival.
Beyond granule neuron degeneration, it will be important to determine if Cystatin B regulation of redox homeostasis also contributes to the other cardinal features of EPM1, seizures, and myoclonus. The absence of overt oxidative damage in the cerebrum in Cystatin B knock-out mice suggests that oxidative damage in other regions of the brain might be limited spatially, have delayed onset, or occur at lower levels than in the cerebellum.
Remarkably, several case reports suggest that treating EPM1 patients with the antioxidant NAC alleviates ataxia and seizures (Hurd et al., 1996; Edwards et al., 2002). Differences in EPM1 patient responses to NAC treatment might be accounted by distinct underlying mechanisms mediating oxidative stress responses together with genetic and environmental factors. NAC represents just one type of antioxidant available in the clinic. Our findings suggest that in addition to NAC, therapeutic intervention targeting lipid peroxidation might provide further benefit.
Cystatin B deficiency-induced sensitization of granule neurons to oxidative stress is rescued by concurrent Cathepsin B loss (Fig. 3E). Consistent with these findings, cerebellar granule neuron degeneration is reduced in Cystatin B–Cathepsin B double knock-out mice compared with Cystatin B knock-out mice (Houseweart et al., 2003). However, the double knock-out mice retain myoclonic seizures and ataxia (Houseweart et al., 2003). Thus, oxidative stress and Cystatin B may regulate neuronal excitability in EPM1 independently of deregulated Cathepsin B activity.
The sensitivity of Cystatin B deficient neurons to oxidative stress suggests links in EPM1 pathogenesis with other neurodegenerative disorders (Chong et al., 2005), suggesting that Cystatin B–Cathepsin B signaling might play a more widespread role in neurodegenerative diseases. Conversely, our understanding of the common neurodegenerative diseases may provide insights into EPM1 pathogenesis. Sp1 function is impaired in models of Huntington's disease, whereby mutant Huntingtin sequesters Sp1 from its target promoters (Dunah et al., 2002). Analogously, the EPM1 dodecamer repeat expansion might disrupt the proper assembly of the transcriptional machinery at the Cystatin B promoter, compromising the neuronal response to stress. Since Sp1 mediates oxidative stress-induced transcriptional responses (Ryu et al., 2003), these findings raise the possibility that diverse neurodegenerative mechanisms may deregulate Sp1 function.
The PMEs are refractory to most available conventional therapies (Marseille Consensus Group, 1990). Aberrant oxidative stress extends beyond EPM1 and includes other PMEs where, for example, mutations in genes encoding mitochondrial tRNAs disrupt oxidative phosphorylation (Kunz, 2002). Thus, our study suggests the exciting possibility that specific antioxidant therapeutic approaches might prove useful in the larger set of PMEs as well.
Footnotes
This work was supported by a Sigrid Jusélius Fellowship to M.K.L., the Finnish Graduate School of Neuroscience to S.T., National Institutes of Health Grants to A.B. (NS047188 and NS41021), and the Folkhälsan Research Foundation, the Sigrid Jusélius Foundation, and the Academy of Finland (NEURO Research Programme 2006–2009 and Center of Excellence Programme 2006–2011) to A.-E.L. We thank E. Andermann and F. Andermann for helpful discussions and critical reading of this manuscript; A. Liberman, M. Taipale, and P. Uusen for technical assistance; A. Baici for the human Cathepsin B-GFP plasmid; K. Brix for the rat Cathepsin B–GFP plasmid; G. Suske for the Sp1 plasmid; N. Perkins for the Sp1–ZnF plasmid; and E. Weber for the Cathepsin B antibody. Hyman M. Schipper has served as consultant to Osta Biotechnologies, TEVA Neurosciences, and Caprion Pharmaceuticals. He holds stock options in Osta and equity in Molecular Biometrics LLC. Anna-Elina Lehesjoki has served as an independent speaker for the symposium “European Forum Addressing Challenges in Epilepsy Care (FACE)” sponsored by Pfizer Limited. Anna-Elina Lehesjoki is the coordinator of a Finnish research consortium “EPIMEC” on Progressive Myoclonic Epilepsy (EPM1). The consortium receives support from UCB Pharma for the project's clinical part for which another investigator is the primary investigator.
References
- Alakurtti K, Virtaneva K, Joensuu T, Palvimo JJ, Lehesjoki AE. Characterization of the cystatin B gene promoter harboring the dodecamer repeat expanded in progressive myoclonus epilepsy, EPM1. Gene. 2000;242:65–73. doi: 10.1016/s0378-1119(99)00550-8. [DOI] [PubMed] [Google Scholar]
- Berkovic SF, Andermann F, Carpenter S, Wolfe LS. Progressive myoclonus epilepsies: specific causes and diagnosis. N Engl J Med. 1986;315:296–305. doi: 10.1056/NEJM198607313150506. [DOI] [PubMed] [Google Scholar]
- Chapman NR, Perkins ND. Inhibition of the RelA(p65) NF-kappaB subunit by Egr-1. J Biol Chem. 2000;275:4719–4725. doi: 10.1074/jbc.275.7.4719. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75:207–246. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- Edwards MJ, Hargreaves IP, Heales SJ, Jones SJ, Ramachandran V, Bhatia KP, Sisodiya S. N-acetylcysteine and Unverricht-Lundborg disease: variable response and possible side effects. Neurology. 2002;59:1447–1449. doi: 10.1212/wnl.59.9.1447. [DOI] [PubMed] [Google Scholar]
- Houseweart MK, Pennacchio LA, Vilaythong A, Peters C, Noebels JL, Myers RM. Cathepsin B but not cathepsins L or S contributes to the pathogenesis of Unverricht-Lundborg progressive myoclonus epilepsy (EPM1) J Neurobiol. 2003;56:315–327. doi: 10.1002/neu.10253. [DOI] [PubMed] [Google Scholar]
- Hurd RW, Wilder BJ, Helveston WR, Uthman BM. Treatment of four siblings with progressive myoclonus epilepsy of the Unverricht-Lundborg type with N-acetylcysteine. Neurology. 1996;47:1264–1268. doi: 10.1212/wnl.47.5.1264. [DOI] [PubMed] [Google Scholar]
- Joensuu T, Lehesjoki AE, Kopra O. Molecular background of EPM1-Unverricht-Lundborg disease. Epilepsia. 2008;49:557–563. doi: 10.1111/j.1528-1167.2007.01422.x. [DOI] [PubMed] [Google Scholar]
- Kälviäinen R, Khyuppenen J, Koskenkorva P, Eriksson K, Vanninen R, Mervaala E. Clinical picture of EPM1-Unverricht-Lundborg disease. Epilepsia. 2008;49:549–556. doi: 10.1111/j.1528-1167.2008.01546.x. [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Kunz WS. The role of mitochondria in epileptogenesis. Curr Opin Neurol. 2002;15:179–184. doi: 10.1097/00019052-200204000-00009. [DOI] [PubMed] [Google Scholar]
- Lafrenière RG, Rochefort DL, Chrétien N, Rommens JM, Cochius JI, Kälviäinen R, Nousiainen U, Patry G, Farrell K, Söderfeldt B, Federico A, Hale BR, Cossio OH, Sørensen T, Pouliot MA, Kmiec T, Uldall P, Janszky J, Pranzatelli MR, Andermann F, et al. Unstable insertion in the 5′ flanking region of the cystatin B gene is the most common mutation in progressive myoclonus epilepsy type 1, EPM1. Nat Genet. 1997;15:298–302. doi: 10.1038/ng0397-298. [DOI] [PubMed] [Google Scholar]
- Lalioti MD, Scott HS, Buresi C, Rossier C, Bottani A, Morris MA, Malafosse A, Antonarakis SE. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature. 1997;386:847–851. doi: 10.1038/386847a0. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Bonni A. Modeling oxidative stress in the central nervous system. Curr Mol Med. 2006;6:871–881. doi: 10.2174/156652406779010786. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villén J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- Marseille Consensus Group. Classification of progressive myoclonus epilepsies and related disorders. Ann Neurol. 1990;28:113–116. doi: 10.1002/ana.410280129. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Lehesjoki AE, Stone NE, Willour VL, Virtaneva K, Miao J, D'Amato E, Ramirez L, Faham M, Koskiniemi M, Warrington JA, Norio R, de la Chapelle A, Cox DR, Myers RM. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1) Science. 1996;271:1731–1734. doi: 10.1126/science.271.5256.1731. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Bouley DM, Higgins KM, Scott MP, Noebels JL, Myers RM. Progressive ataxia, myoclonic epilepsy and cerebellar apoptosis in cystatin B-deficient mice. Nat Genet. 1998;20:251–258. doi: 10.1038/3059. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Ryu H, Lee J, Mwidau A, Neve RL. In vitro model of oxidative stress in cortical neurons. Methods Enzymol. 2002;352:183–190. doi: 10.1016/s0076-6879(02)52018-8. [DOI] [PubMed] [Google Scholar]
- Rinne R, Saukko P, Järvinen M, Lehesjoki AE. Reduced cystatin B activity correlates with enhanced cathepsin activity in progressive myoclonus epilepsy. Ann Med. 2002;34:380–385. doi: 10.1080/078538902320772124. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R, Ratan RR. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci. 2003;23:3597–3606. doi: 10.1523/JNEUROSCI.23-09-03597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk V, Bode W. The cystatins: protein inhibitors of cysteine proteinases. FEBS Lett. 1991;285:213–219. doi: 10.1016/0014-5793(91)80804-c. [DOI] [PubMed] [Google Scholar]