

Summary
Central nervous system (CNS) neurovascular units are multicellular complexes consisting of neural cells, blood vessels and a milieu of extracellular matrix (ECM) proteins. ECM-mediated adhesion and signaling events within neurovascular units probably contribute to proper CNS development and physiology; however, the molecular mechanisms that control these events remain largely undetermined. Previous studies from our group and others showed that ablation of the ECM receptor, αvβ8 integrin, in neural progenitor cells (NPCs) of the embryonic mouse brain results in severe developmental neurovascular pathologies and premature death. Here, we have investigated the functions for this integrin in the adult brain by studying mice harboring a homozygous-null β8 gene mutation generated on an outbred background that permits survival for several months. We show that adult β8–/– mice display widespread defects in neurovascular unit homeostasis, including increased numbers of intracerebral blood vessels with pronounced perivascular astrogliosis. Furthermore, in neurogenic regions of the adult brain, where NPCs cluster around blood vessels in neurovascular niches, β8 integrin is essential for normal control of NPC proliferation and survival. Analysis of NPCs cultured ex vivo reveals that the growth and survival defects correlate, in part, with diminished integrin-mediated activation of latent transforming growth factor β1 (TGFβ1), which is an ECM protein ligand for αvβ8 integrin. Collectively, these data identify essential functions for β8 integrin in regulating neurovascular unit physiology in the post-natal mouse brain.
Keywords: Tgfβ, αvβ8 integrin, Angiogenesis, Blood-brain barrier, Extracellular matrix, Neural stem cell
Introduction
The vertebrate CNS contains an elaborate network of blood vessels that intimately associate with neuronal and glial elements to form integrated complexes, or neurovascular units (McCarty, 2005). Neural and vascular cells within such units interact mainly via basement membranes that contain an assortment of growth factors and ECM proteins. Precise regulation of cell-ECM communication within neurovascular units is essential for proper formation and function of the vascular system within the CNS. For example, glial- and neuronal-derived cues regulate blood vessel morphogenesis and blood-brain barrier development (Abbott et al., 2006; Iadecola, 2004). Vascular-derived cues also influence neural cell functions; for example, neural progenitor cells (NPCs) in specific regions of the adult brain preferentially reside near cerebral blood vessels within `neurovascular niches'. Cell-cell contacts as well as niche-derived growth factors and ECM proteins dictate NPC growth, survival and differentiation (Palmer et al., 2000; Wurmser et al., 2004). How perivascular astrocytes and NPCs decipher vascular basement membrane-derived ECM cues, and how these events influence neural cell functions remains enigmatic.
Integrins are heterodimeric cell surface receptors for many ECM proteins. In both mice and humans there are 26 integrin genes: 18 genes encoding α-subunits, and eight genes that encode β-subunits (Hynes, 2002). Many integrins are expressed in post-natal astrocytes and NPCs (Hall et al., 2006; McCarty et al., 2005a), although functional roles for these integrins in adult neurovascular physiology remain obscure. Our group and others have reported that αvβ8 integrin is necessary for normal regulation of neurovascular development in the brain, since deletion of the αv or β8 integrin genes in developing neural progenitors and astrocytes, but not endothelial cells, leads to embryonic and neonatal blood vessel patterning defects and intracerebral hemorrhage (McCarty et al., 2005b; Proctor et al., 2005). αvβ8 integrin-mediated regulation of neurovascular development depends on activation of latent forms of TGFβ, which are secreted from cells as ECM-bound complexes that have minimal bioactivity (Annes et al., 2003). Integrin adhesion to the latent forms of TGFβ1 and TGFβ3 mediates their bioactivation and induces canonical TGFβ receptor signaling (Cambier et al., 2005; Mu et al., 2002; Sheppard, 2004). The importance of integrin-mediated TGFβ activation has been demonstrated in vivo by mutating the integrin-binding motif within the TGFβ1 gene. Mice harboring this mutation die prematurely and display phenotypes that are identical to those observed in TGFβ1 gene knockouts (Yang et al., 2007). Furthermore, mouse embryos harboring this mutated TGFβ1 gene in combination with a TGFβ3–/– gene develop brain-specific neurovascular pathologies that appear identical to the phenotypes in αv and β8 integrin knockouts (Mu et al., 2008).
Here, we have analyzed mice genetically null for β8 integrin to determine functions for this integrin in neural-vascular cell adhesion and communication, primarily within neurogenic regions of the post-natal brain. We show that adult β8–/– mice develop severe deficits in neurogenesis and neurovascular physiology, and that these pathologies are associated with diminished integrin-mediated activation of TGFβs. These data identify β8 integrin as a key regulator of neurovascular physiology in the adult brain, and indicate that integrin-mediated TGFβ activation may be an important component in this process.
Results
Genetic background influences post-natal viability of β8–/– mice
Previous reports have demonstrated that most αv–/– and β8–/– embryos on a C57BL6/129S4 mixed genetic background die by E11 as a result of placental defects (Bader et al., 1998; McCarty et al., 2002; Zhu et al., 2002). αv–/– mice that survive beyond E11 develop progressive intracerebral hemorrhage, a cleft palate, and die in the early neonatal period (Bader et al., 1998). Similar results have been described for β8–/– mice, although the incidence of cleft palate formation was reported to be significantly lower (Zhu et al., 2002). We reasoned that strain-specific genetic modifiers might influence embryonic and post-natal viability in β8–/– mice. Therefore, heterozygous mutant mice (β8+/–) on a C57BL6/129S4 genetic background were backcrossed for two generations with the ICR/CD-1 outbred strain, and F2 progeny were subsequently interbred to generate wild-type control (β8+/+) and homozygous mutant (β8–/–) progeny. Nearly all β8–/– progeny from β8+/– intercrosses were born alive and in nearly the predicted Mendelian distributions: 26 β8–/– mice of 123 total F2 neonates (21%) were identified at birth, as compared with the expected 25%, suggesting a low percentage of in utero lethality. All β8–/– neonates had grossly obvious intracerebral hemorrhage (supplementary material Fig. S1) that was very similar to that reported for C57BL6/129S4 mutants (Zhu et al., 2002). Although β8–/– pups were initially identified based on the intracerebral hemorrhage phenotype, all genotypes were confirmed at the time of weaning using PCR-based methods to amplify β8 gene sequences using genomic DNA isolated from ear snips (data not shown).
Nearly 90% of β8–/– neonates (23 of 26 β8–/– mutants identified) survived beyond the first post-natal week (Fig. 1A), although most β8–/– mice were distinguishable phenotypically from wild-type littermates. Mutant animals were generally smaller than control littermates (Fig. 1B) with an abnormal posture (Fig. 1D). Approximately 50% of β8–/– mice that survived beyond the first week died between post-natal days 21 and 30 (Fig. 1A). The remaining mutants survived for up to five months but developed progressively more severe neurological phenotypes, including ataxia and paresis that eventually led to an abnormal gait (Fig. 1E). β8 integrin protein expression was analyzed by immunoblotting tissue homogenates prepared from various adult brain regions including the subventricular zone (SVZ) of the lateral ventricle, the olfactory bulbs, as well as the hippocampal dentate gyrus and cerebellum. As shown in Fig. 1F, β8 integrin protein was expressed at similar levels in all brain regions analyzed from adult wild-type mice; by contrast, β8 integrin protein was not detected in brain lysates from adult β8–/– mice.
Fig. 1.

Genetic background influences post-natal survival of β8–/– mice. (A) Kaplan-Meier survival plot using wild-type (n=40) and β8–/– (n=39) mice. Note that approximately 60% of β8–/– mice die by post-natal day 30 (P30) whereas the remaining animals survive for up to 5 months. (B) Weight analysis using P30 and P60 control and mutant male mice (n=7-10 males per genotype, although similar results were observed with female mice). Note that β8–/– mice weigh significantly less than wild-type littermates. *P<0.0001 compared with wild-type group. (C,D) Images of 4-month-old (P120) wild-type (C) and β8–/– (D) littermates. β8–/– mice have a hunched posture and limb paresis; these phenotypes develop with 100% penetrance in mutant mice that survive to adulthood. (E) Footprint analysis performed with P90 wild-type (upper panel) and β8–/– (lower panel) animals with hind paws painted blue and fore paws painted red. Note that the β8–/– mouse (like all other β8–/– mutants examined) displays an abnormal gait characterized by pronounced dragging of the limbs. Arrow indicates direction of movement. (F) Brain tissue homogenates were prepared from P60 wild-type and β8–/– mice. Detergent-soluble protein lysates were immunoblotted with an anti-β8 integrin polyclonal antibody. Note that β8 integrin protein is expressed in the various brain regions of wild-type mice, but is not detected in β8–/– animals. SVZ, subventricular zone of lateral ventricle; OB, olfactory bulbs; DG, hippocampal dentate gyrus; CB, cerebellum.
Defects in neurovascular unit homeostasis in adult β8–/– mice
Analysis of serial brain sections from β8–/– mice older than 4 weeks of age showed the absence of obvious intracerebral hemorrhage (supplementary material Fig. S1). This appears to be the result of resolution of hemorrhage that was present at birth. A similar situation has been found by our group and others in conditional αv and β8 integrin knockout mice (McCarty et al., 2005b; Proctor et al., 2005).
Next, we quantified blood vessel densities within the cerebral cortices of adult wild-type and β8–/– animals using an anti-laminin antibody to highlight vascular basement membranes. In comparison to wild-type controls (Fig. 2A) approximately twofold more laminin-positive blood vessels were detected within cerebral cortices of β8–/– adult mice (Fig. 2B). Many of the mutant vessels showed more intense laminin immunoreactivity (Fig. 2B), suggesting higher levels of endogenous laminin protein expression. In order to determine if the increased blood vessel densities in adult β8–/– brains were due to ongoing vascular cell proliferation, we intraperitoneally injected the thymidine analog bromodeoxyuridine (BrdU) into adult wild-type and β8–/– mice. BrdU incorporation in vascular endothelial cells was analyzed by double fluorescence staining with anti-BrdU and anti-laminin antibodies. However, we did not detect obvious differences in BrdU incorporation in control versus β8–/– blood vessels (data not shown). These data suggest that the increased cerebral blood vessel densities in adult β8–/– mice are probably not due to pathological angiogenesis, but may be secondary remnants of vascular damage that occurred in the developing brain (supplementary material Fig. S1).
Fig. 2.

Adult β8–/– mice have widespread neurovascular pathologies. (A,B) Coronal sections through cerebral cortices of wild-type (A) and β8–/– (B) mice were immunostained with anti-laminin to reveal cerebral blood vessels. (C) Quantification of cerebral blood vessels based on anti-laminin immunoreactivity of P60 wild-type (n=5) and β8–/– (n=5) mice, *P=0.002 as compared with wild-type group. (D,E) Double immunofluorescence analysis using anti-CD34 (green) and anti-GFAP (red) reveals increased numbers of astrocytes in β8–/– brains. (F) Quantification of GFAP-expressing astrocytes in sections from wild-type (n=4) and β8–/– (n=4) mice, *P<0.001 as compared with wild-type group. (G,H) Transmission electron microscopy reveals neurovascular unit pathologies in β8–/– mice. Note the increased vessel coverage by perivascular astrocyte endfeet in the mutant (H) versus control (G) samples (arrows). (I) Quantification of cerebral blood vessel coverage by astrocytes within cerebral cortices of wild-type and β8–/– mice. Coronal sections through the cerebral cortices of P60 wild-type (n=5) and β8–/– (n=5) brains were labeled with both anti-lamina and anti-GFAP. The percentages of lamina-expressing blood vessels contacting more than one GFAP+ astrocytes were quantified per ×400 field, *P=0.002 as compared with the wild-type group.
Analysis of control (Fig. 2D) and mutant (Fig. 2E) cerebral cortices with an anti-GFAP (glial fibrillary acidic protein) antibody (an astrocyte marker) revealed nearly five times more GFAP-expressing astrocytes in adult β8–/– mice (Fig. 2F), and similar patterns were observed in most other brain regions (data not shown). To determine if the elevated numbers of GFAP-expressing cells were the result of increased astrocyte proliferation, we analyzed GFAP-expressing cells following intraperitoneal injection of BrdU using double immunofluorescence labeling. Cells that were positive for both GFAP and BrdU were not detected (data not shown), suggesting that the increased numbers of GFAP cells were not the result of astrocyte hyperplasia. Many of these GFAP-expressing cells in the β8–/– brains were closely associated with cerebral blood vessels (Fig. 2E). Ultrastructural analyses also revealed increased astrocyte coverage of cerebral blood vessels in adult β8–/– mice (Fig. 2H), as opposed to wild-type controls (Fig. 2G). Astrocyte coverage of blood vessels was also quantified using anti-GFAP and anti-laminin double immunofluorescence staining, which revealed nearly threefold more cerebral blood vessels coverage by astrocyte endfeet in β8–/– mice (Fig. 2I).
The increased numbers of perivascular astrocytes suggested that cerebral blood vessels in adult β8–/– mice might have compromised blood-brain barrier properties. Hence, blood-brain barrier permeability was analyzed using a reactive derivative of biotin (EZLink-NHS-Biotin). This small molecule (approximately 500 Da) is water-soluble, membrane impermeable, and covalently couples to free amine groups commonly present on many protein and macromolecules. Upon pathological disruption of the blood-brain barrier this tracer labels components of the brain parenchyma (Nitta et al., 2003). As shown in Fig. 3A,C, in both wild-type and β8–/– brains, passage of reactive biotin into parenchymal regions surrounding cortical cerebral blood vessels was not detected. This is in contrast to periventricular regions of wild-type and β8–/– mouse brains, which displayed robust passage of amine-reactive biotin into the brain parenchyma (Fig. 3B,D). These data are consistent with a recent report showing that blood vessels within the SVZ and other periventricular regions of the adult mouse brain display enhanced blood-brain barrier permeability (Tavazoie et al., 2008). To further analyze the blood-brain barrier in wild-type and adult β8–/– mice, we immunofluorescently stained brain sections with an anti-zona occludens protein 1 (ZO1) antibody to label endothelial cell tight junctions that comprise the blood-brain barrier (Wolburg and Lippoldt, 2002). However, we did not detect obvious differences in the expression patterns of ZO1 in cerebral blood vessels in adult wild-type or β8–/– brains (Fig. 3E,F). We also immunoblotted brain protein lysates using antibodies directed against occludin and claudin 5, proteins expressed in tight junctions that form the blood-brain barrier (Nitta et al., 2003; Wolburg and Lippoldt, 2002). As shown in Fig. 3G, similar amounts of occludin and claudin 5 proteins were detected in homogenates from wild-type and β8–/– cerebral cortices.
Fig. 3.

Lack of obvious blood-brain barrier disruption in adult β8–/– mice. (A-D) P60 wild-type (n=3) or β8–/– (n=5) mice were perfused with amine-reactive biotin, and sagittal brain sections were labeled with fluorescently conjugated streptavidin. In both wild-type (A) and mutant (C) cortices, biotin labeled cerebral endothelial cells (arrows), but did not cross the blood-brain barrier and label the brain parenchyma. By contrast, vessels within the choroid plexus (CP) and nearby periventricular (PV) regions of wild-type (B) and mutant (D) mice are highly permeable to the amine-reactive biotin, although β8-integrin-dependent differences were not apparent. (E,F) Frozen coronal brain sections from adult wild-type (E) and mutant (F) mice were immunofluorescently labeled with anti-zona occludin 1 (ZO1), a marker for endothelial cell tight junctions that form the blood-brain barrier. No apparent difference in ZO1 protein expression was detected in wild-type and β8–/– cerebral blood vessels (arrows in E,F). (G) Detergent-soluble brain lysates from the cerebral cortices of P60 wild-type and β8–/– mice were immunoblotted with anti-occludin, anti-claudin 5 or anti-actin antibodies. Note that both occludin and claudin 5 are expressed in β8–/– brains.
Olfactory bulb size reduction in adult β8–/– mice
Gross and microscopic examination of wild-type (n=3) and mutant (n=3) brains (Fig. 4A,B) revealed approximately 15% reduction in brain size in all β8–/– mice [brain weight (mg): wild type, 537.85±16.43; β8–/–, 444.59±18.25]. Microscopic analyses of adult wild-type and β8–/– brains revealed that most mutant animals displayed varying degrees of hydrocephalus (supplementary material Fig. S1). Additionally, in comparison to wild-type littermates, olfactory bulbs in all adult β8–/– mice examined were nearly 50% smaller [olfactory bulb weight (mg): wild type, 25.16±1.60; β8–/–, 13.76±1.89]. Microscopic examination of β8–/– olfactory bulbs revealed an apparently normal cytoarchitecture (Fig. 4C); however, in comparison to control mice (Fig. 4D) β8–/– olfactory bulbs (Fig. 4E) had reduced numbers of neuroblasts, as detected by doublecortin (DCX) expression within the rostral migratory stream (RMS). After migrating tangentially along the RMS to the olfactory bulbs, DCX+ neuroblasts migrate radially and form functional interneurons within the olfactory bulb granule cell layers (Sawamoto et al., 2006). Analysis of granule cell layers, however, did not reveal obvious differences in numbers of DCX+ interneurons in wild-type or β8–/– olfactory bulbs (arrows in Fig. 4D,E).
Fig. 4.

Size-reduced olfactory bulbs in adult β8–/– mice. (A,B) P90 brains from wild-type (A) and β8–/– (B) littermates that were killed and cardiac-perfused with Bouin's fixative. In comparison to the wild-type mouse (arrow in A), note the smaller olfactory bulbs in the β8–/– animal (arrow in B). (C) Images of H&E-stained coronal sections through olfactory bulbs of P90 wild-type and β8–/– mice. Note that β8–/– olfactory bulbs are significantly smaller, but have an apparently normal cytoarchitecture. GLO, glomerular layer; EPL, external plexiform layer; MCL, mitral cell layer; IPL, internal plexiform layer; GCL, granule cell layer; RMS, rostral migratory stream. (D,E) Coronal sections through olfactory bulbs taken from P90 wild-type (D) and β8–/– (E) mice were immunofluorescently labeled with anti-DCX (green) to visualize neuroblasts and differentiated neurons and anti-GFAP (red) to identify astrocytes. Note that fewer DCX+ neuroblasts (green) are present in the RMS of β8–/– olfactory bulb than in the wild-type control (arrowheads in D,E). However, similar numbers of GFAP+ astrocytes (red) and differentiated neurons (green) are present in the olfactory bulbs of β8–/– mice in comparison to wild-type control mice (arrows in D,E). RMS, rostral migratory stream; DCX, doublecortin; GFAP, glial fibrillary acidic protein.
Adult β8–/– mice have abnormalities in the SVZ and RMS
Various mouse mutants with abnormalities in SVZ neurogenesis and/or RMS migration develop smaller olfactory bulbs than wild-type mice (Belvindrah et al., 2007; Chazal et al., 2000). Therefore, sections from the SVZ and RMS of wild-type and β8–/– mice were analyzed using Hematoxylin and Eosin (H&E) staining and immunofluorescence to identify DCX+ neuroblasts and GFAP+ astrocytes. The RMS cytoarchitecture in wild-type mice consisted of DCX+ neuroblasts forming closely bundled cell chains (Fig. 5A), with GFAP+ astrocytes forming distinct `glial tubes' that ensheath the RMS (Fig. 5A). By contrast, the RMS cytoarchitectures in β8–/– mice were abnormally organized (Fig. 5B). GFAP+ astrocytes that normally make up glial tubes were intermingled with DCX+ cells (Fig. 5B), and β8–/– neuroblasts did not form closely bundled chains, resulting in increased RMS diameters as determined by measuring the boundaries defined by the migrating DCX+ cells (Fig. 5C). Analysis of H&E-stained brain sections from the SVZ of wild-type (Fig. 5D) and β8–/– mice (Fig. 5E) showed an abnormal cytoarchitecture in the SVZ of mutant animals. Immunofluorescence quantification revealed nearly threefold more DCX+ cells in the β8–/– SVZ, in comparison to wild-type control mice (Fig. 5F). β8–/– DCX+ cells were often clustered in the SVZ, further supporting integrin-dependent abnormalities in neuroblast migration.
Fig. 5.

Rostral migratory stream and subventricular zone abnormalities in adult β8–/– mice. (A,B) Sagittal brain sections from P90 wild-type (A) and β8–/– (B) mice that were H&E stained to reveal the RMS cytoarchitecture. Sections were also immunofluorescently labeled with an anti-DCX antibody (green) to label migrating neuroblasts and an anti-GFAP antibody (red) to label astrocytes in glial tubes that ensheath the RMS. Note the apparently normal DCX+ neuroblast cell chains and the GFAP+ glial tubes in wild-type brain sections (arrows in A). By contrast, the β8–/– RMS is wider with displaced DCX+ cells and intermingled GFAP+ astrocytes (arrows in B). (C) Rostral migratory stream widths were quantified in wild-type (n=3) and β8–/– (n=3) sagittal brain sections that were immunofluorescently labeled with anti-doublecortin antibody to visualize migrating neuroblasts, *P=0.004 compared with wild-type samples. (D,E) Sagittal brain sections from P90 wild-type (D) and β8–/– (E) mice were H&E stained to reveal the cytoarchitecture of the SVZ. Sections were also immunofluorescently labeled with an anti-DCX antibody (green) to identify neuroblasts and anti-GFAP (red) to identify NPCs and astrocytes. Note the abnormal cytoarchitecture of the SVZ, as revealed by H&E staining in β8–/– samples. Additionally, more DCX+ neuroblasts (arrows in E), often found in clusters, were present in the SVZ of β8–/–mice. (F) DCX+ cells were quantified within the SVZ of control (n=6) and mutant mice (n=6) by analyzing sagittal brain sections immunofluorescently labeled with anti-DCX, *P=0.0001 compared with wild-type samples. v, ventricle; SCJ, striatocortical junction; DCX, doublecortin; GFAP, glial fibrillary acidic protein.
To determine possible functions for β8 integrin in NPC proliferation in vivo, we intraperitoneally injected BrdU into post-natal day 60 (P60) wild-type (n=5) and β8–/– mutant (n=5) mice once per day for two consecutive days. Twenty-four hours after the second injection, animals were killed and brains were fixed by intracardiac perfusion, sagitally sliced, and embedded in freezing medium. Mitotic cells were quantified after immunostaining sections with an anti-BrdU monoclonal antibody. As shown in Fig. 6A-C, in comparison to wild-type mice, there was only a third of the number of BrdU+ cells in the SVZ of β8–/– mutants. Some BrdU+ cells were detected in the RMS and olfactory bulbs of mutant animals (data not shown), suggesting that neuroblast migration from the SVZ is not completely defective in the absence of β8 gene expression.
Fig. 6.

Neural cell proliferation and survival defects in the β8–/– subventricular zone. (A,B) Sagittal brain sections from P60 wild-type (A) and β8–/– (B) mice that were killed after receiving intraperitoneal injections of BrdU, were immunofluorescently labeled with anti-BrdU antibody. Dashed white lines denote ventricle boundaries. (C) Brain sections from wild-type (n=6) and β8–/– (n=11) mice were immunofluorescently labeled with anti-BrdU antibody and BrdU+ cells were quantified by analyzing serial sections. Note the significant reduction in the numbers of proliferating cells in the SVZ of β8–/– mutant mice, *P<0.001 compared with wild-type samples. (D,E) Ultrastructural analyses of SVZ regions dissected from P90 wild-type (D) and β8–/– (E) mice. Unlike the wild-type SVZ (D), which contains distinct units of a, b and c cells, the SVZ in β8–/– mice has an abnormal cytoarchitecture with increased numbers of apoptotic cells (arrows in E). (F) Serial coronal sections through the SVZ of P60 wild-type (n=7) and β8–/– (n=9) mice were immunofluorescently labeled to identify TUNEL+ cells and these cells were quantified. Note the higher numbers of apoptotic cells in the SVZ of β8–/– mice. *P<0.001 compared with wild-type samples. V, ventricle; BrdU, bromodeoxyuridine; a, SVZ type a neuroblast; b, SVZ neural stem cell; c, SVZ transit amplifying cell.
Next, the ultrastructure of the adult β8–/– SVZ was analyzed using transmission electron microscopy. As shown in Fig. 6D, the SVZ in wild-type mice consisted of distinct units of type A, B and C cells arranged near the ependymal cell lining of the brain ventricles. By contrast, β8–/– mice displayed cytoarchitectural abnormalities in the SVZ, with obvious disorganization of the A, B, and C cell units (Fig. 6E). Electron microscopy also identified increased numbers of apoptotic cells in subventricular regions of β8–/– mice (Fig. 6E). Apoptosis in the SVZ of wild-type (n=4) and β8–/– adult mice (n=6) was quantified by measuring DNA fragmentation with fluorescent terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL). As shown in Fig. 6F, in comparison to wild-type littermates there was nearly a tenfold increase in apoptotic cells numbers in the SVZ of β8–/– mice. Increased numbers of TUNEL+ cells were not detected in the corpus putamen or in the cerebral cortices of adult β8–/– mice (data not shown), indicating that apoptosis in the β8–/– brain occurs more selectively in the SVZ.
β8 integrin is necessary for neurosphere proliferation and survival in vitro
We next cultured neurospheres from SVZ regions of P60 wild-type and β8–/– mice (n=3 per genotype). In comparison to wild-type neurospheres (Fig. 7A), β8–/– neurospheres (Fig. 7B) were noticeably smaller and this was confirmed by measuring sphere diameters (Fig. 7C). Antibodies directed against the αv and β8 integrin subunits were also used to determine integrin protein expression in neurospheres cultured from wild-type and β8–/– mice (Fig. 7D). αv and β8 integrin proteins were detected in wild-type mouse neurospheres (Fig. 7D). There was no detectable β8 integrin protein in adult neurospheres isolated from the β8–/– SVZ (Fig. 7D). αv integrin protein expression was not significantly reduced in β8–/– neurospheres (Fig. 7D). β8 integrin only heterodimerizes with the αv subunit, but αv integrin can also heterodimerize with β1, β3, β5 and β6 subunits (Hynes, 2002). We did not detect differences in expression of the β1, β3, β5, integrin subunits in β8–/– neurospheres. β6 integrin was not detected in wild-type or β8–/– neurospheres (data not shown). Our results demonstrating αv and β8 integrin protein expression in adult mouse NPCs are consistent with previous reports showing αv and β8 integrin mRNA expression in human fetal SVZ NPCs (Hall et al., 2006) and αv, β5 and β8 integrin protein expression in neonatal rat brain neurospheres (Jacques et al., 1998). Lastly, it is probable that the CNS defects in β8–/– mice are due to loss of αvβ8 integrin expression and function, since αv is the only integrin subunit that is reported to heterodimerize with β8 integrin (Hynes et al., 2002; Nishimura et al., 1998). αvβ5 integrin is also expressed in adult NPCs (Fig. 7D); however, ablation of the mouse β5 integrin gene does not influence NPC growth and survival in the SVZ or neuroblast migration in the RMS (Belvindrah et al., 2007).
Fig. 7.
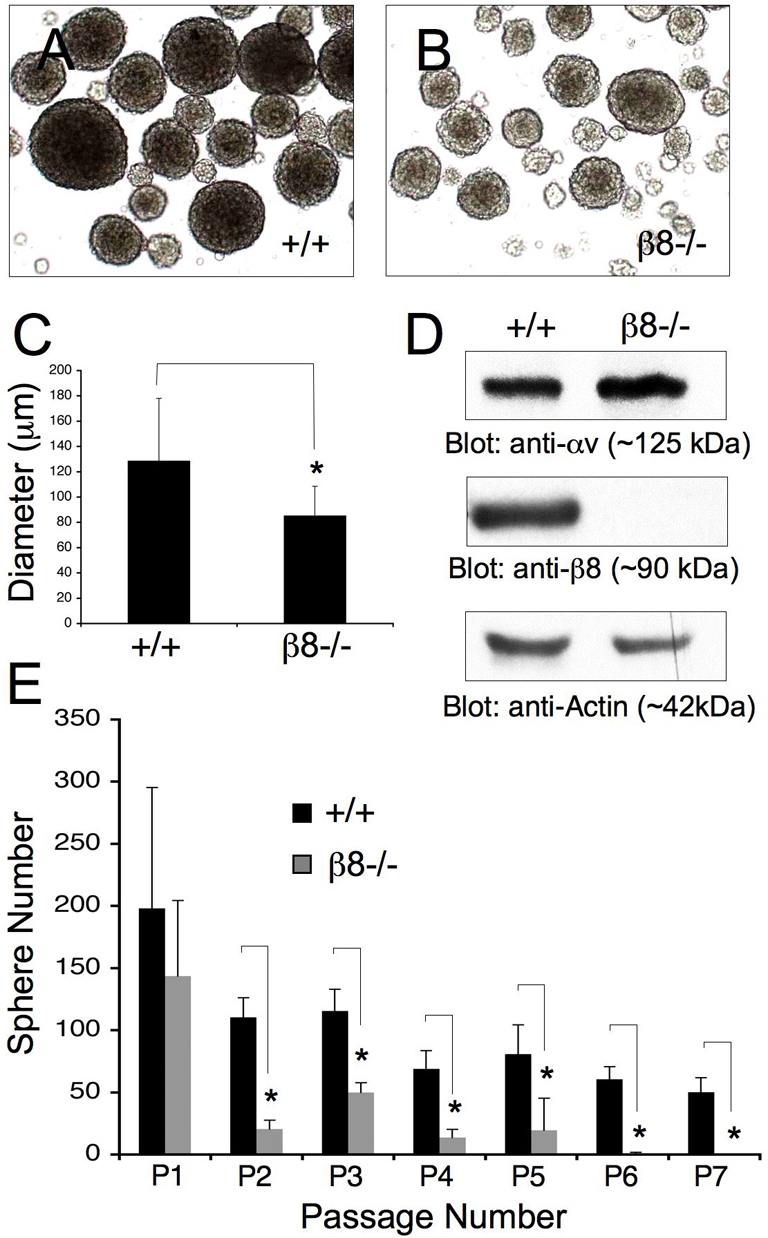
β8 integrin promotes NPC proliferation and self-renewal in vitro. (A,B) Neural progenitors were cultured from the SVZs of P60 wild-type (A) and β8–/– (B) adult mice. NPCs proliferate to form free-floating neurospheres in serum-free medium containing EGF and bFGF. (C) Diameters of 300 randomly selected neurospheres from P60 wild-type mice (n=3) or β8–/– mice (n=3) were measured. Note the reduced diameters of β8–/– neurospheres, *P=0.0006 compared with wild-type neurospheres. (D) Detergent-soluble protein lysates from wild-type or β8–/– neurospheres were immunoblotted with anti-αv and -β8 integrin polyclonal antibodies, or anti-actin to control for protein loading. (E) A proliferation and self-renewal assay in which individual neurospheres from each of two wild-type and two β8–/– mice (n=12 neurospheres per genotype) were physically dissociated and the total numbers of newly formed spheres were quantified over seven sequential passages. Note the progressive reduction in new sphere formation in cells genetically null for β8 integrin, *P<0.001 for passage two to passage seven compared with wild-type neurospheres.
We further analyzed possible β8 integrin-dependent proliferation and/or self-renewal functions in primary neurospheres cultured from the SVZ of P60 adult wild-type and β8–/– mice (n=3 per genotype). Selected neurospheres with similar diameters (∼75 μm) were physically dissociated and their ability to regenerate new neurospheres was assayed over multiple passages. In comparison to wild-type samples (Fig. 7E), there was a reduction in the numbers of neurospheres generated from individual β8–/– neurospheres. These defects were more pronounced after sequential passaging, suggesting progressive depletion of cells within β8–/– neurospheres with self-renewing and/or long-term proliferative capacities. These findings suggest that β8 integrin plays a critical role in regulating NPC proliferation and self-renewal in vitro, and also support the in vivo data showing reduced cell proliferation in the SVZ of adult β8–/– mice (Fig. 6).
Diminished TGFβ1 activation in β8–/– neurospheres
Neurospheres cultured from SVZ regions of P60 wild-type and β8–/– adult mice (n=3 per genotype) were used to analyze β8 integrin-mediated TGFβ activation. Conditioned media from wild-type or β8–/– neurospheres was transferred to HEK293T cells transfected with a plasmid containing the plasminogen activator inhibitor promoter fused to luciferase cDNA (PAI1-luciferase) (Abe et al., 1994) and luciferase activity was assayed using a luminometer. Alternatively, purified latent TGFβ1 (LAP-TGFβ1) was added to wild-type or β8–/– neurospheres and then transferred to control or transfected HEK293T cells. The use of HEK293T cells in this assay provides a means of quantifying TGFβ-induced signaling events that are minimally influenced by endogenous β8 integrin expression.
As shown in Fig. 8A, conditioned medium from wild-type neurospheres that had been pre-incubated with exogenous LAP-TGFβ1 induced robust luciferase activity (Fig. 8A). Conditioned medium from β8–/– neurospheres treated with exogenous LAP-TGFβ1 induced statistically significantly lower levels of luciferase activity (Fig. 8A). Adult rodent neurospheres also express αvβ1, αvβ3 and αvβ5 integrins (Jacques et al., 1998), and activation of exogenous LAP-TGFβ1 in β8–/– neurospheres may occur via one or more of these integrins. We also detected a statistically significant increase in luciferase activity using conditioned media from both wild-type and β8–/– neurospheres not treated with LAP-TGFβ1, suggesting that one or more TGFβs produced endogenously by neurospheres are activated via integrin-independent mechanisms. This may be due to activation of LAP-TGFβ2, which does not contain an RGD binding motif, but is expressed in brain neurospheres (Roussa et al., 2006).
Fig. 8.

Autocrine TGFβ signaling does not promote NPC proliferation and self-renewal. (A) Quantification of β8 integrin-mediated TGFβ activation using PAI1-lucferase reporter assays. The results are from two independent experiments using conditioned media from neurospheres cultured from P60 wild-type (n=3 per experiment) or β8–/– (n=3 per experiment) mice. Neurosphere conditioned medium (+/– exogenous LAP-TGFβ1) was transferred to transiently transfected HEK293T cells, followed by quantification of luciferase activity. Wild-type neurospheres induce a robust TGFβ-mediated luciferase activity; however, note the significant decrease in luciferase activity using conditioned medium from β8–/– neurospheres, *P=0.002 compared with non-conditioned media; **P<0.0001 compared with wild-type cells; ***P<0.0001 compared with wild-type cells. (B) Exogenous TGFβ1 does not rescue β8–/– neurosphere proliferation and self-renewal defects. Ten SVZ neurospheres of similar sizes (∼75 μm diameter) from wild-type mice (n=2) or β8–/– mice (n=2) were dissociated, and new neurosphere formation was quantified over four passages. Addition of active TGFβ1 (10 ng/ml) significantly inhibits neurosphere formation in both wild-type and β8–/– samples in each passage. The experiment was stopped after P4 because of the absence of new neurospheres generated in the β8–/– samples treated with TGFβ1. *P<0.0001 at all passages compared with untreated wild-type samples; **P<0.001 for P2 and **P=0.001 for P3 and P4 compared with untreated β8–/– samples. (C) Antibody-mediated inhibition of TGFβ does not inhibit wild-type neurosphere self-renewal and proliferation. Five individual SVZ neurospheres cultured from each of two wild-type mice (n=10 total neurospheres) were dissociated and allowed to reform new spheres over three sequential passages, either in the presence of 5 μM isotype control IgG, 5 μM anti-TGFβ, and/or active TGFβ1 (10 ng/ml). Note that inhibition of TGFβ alone does not influence neurosphere self-renewal, but does largely reverse the growth inhibitory effects of bioactive TGFβ1, †P=0.002 compared with untreated P2 samples; ††P<0.0001 compared with untreated P3 samples; *P=0.0004 compared with untreated P1 samples; **P<0.001 compared with untreated P2 samples.
To determine if integrin-activated TGFβs function in an autocrine manner to promote NPC proliferation and survival, we added bioactive TGFβ1 to wild-type neurospheres. Exogenous TGFβ1 was also added to β8–/– neurospheres in an attempt to `rescue' the self-renewal and proliferation defects. As shown in Fig. 8B, addition of active TGFβ1 (10 ng/ml) resulted in robust inhibition of self-renewal in both wild-type and β8–/– neurospheres over multiple passages. We also observed similar neurosphere growth inhibition at lower TGFβ1 concentrations, ranging from 0.01 to 1.0 ng/ml (data not shown). These data are consistent with other in vitro and in vivo results showing that exogenous TGFβs inhibit NPC self-renewal and proliferation (Battista et al., 2006; Falk et al., 2008; Wachs et al., 2006). Lastly, addition of an antibody that neutralized activities of TGFβ1, TGFβ2 and TGFβ3 (Dasch et al., 1989) did not impair wild-type neurosphere self-renewal (Fig. 8C), suggesting that TGFβ autocrine signaling pathways are not essential for NPC proliferation and self-renewal.
Discussion
Does β8 integrin differentially regulate blood vessel functions in the embryonic and adult brain?
Our group and others have used various conditional gene ablation strategies to demonstrate that αvβ8 integrin in perivascular NPCs regulates blood vessel morphogenesis in the embryonic mouse brain; yet, the severe brain-specific pathologies in αv and β8 integrin mutant mice resolve by adulthood (McCarty et al., 2005b; Proctor et al., 2005). We initially attributed resolution of the vascular pathologies to mosaic Cre transgene expression and heterogeneous integrin gene deletion in neural progenitor cells. However, in the present study we also observe resolution of hemorrhage in adult mice that are genetically null for β8 integrin in all cells. Also, while this manuscript was in the advanced stages of revision Munger and colleagues reported neurological phenotypes and premature death in the absence of ongoing hemorrhage in adult β8–/– mice (Aluwihare et al., 2009). These results significantly negate our original hypothesis that neural cells expressing αvβ8 integrin, which had `escaped' Cre-mediated gene deletion, contribute to repair of the hemorrhagic phenotypes. It remains possible that other pathways are activated in integrin mutant mice, and these may compensate for loss of normal β8 integrin functions. However, an alternative mechanism that we now favor is that β8 integrin in perivascular neural cells regulates active phases of blood vessel growth, similar to those that occur during embryonic and neonatal brain development. Angiogenesis in most regions of the rodent brain largely terminates by post-natal day 30 (Plate, 1999), which coincides with the reduction of neurovascular pathologies in adult αv and β8 integrin mutant mice (Fig. 4; supplementary material Fig. S1). By contrast, BrdU labeling experiments in rodents have shown that endothelial cells in neurogenic regions of the brain (also termed neurovascular niches) retain angiogenic characteristics (Kerever et al., 2007; Mercier et al., 2002; Palmer et al., 2000). Hence, it is enticing to speculate that similar to its function in embryonic brain NPCs, β8 integrin in adult NPCs continues to promote interactions with blood vessels in neurovascular niches and these events are essential for NPC growth, survival and migration to the olfactory bulbs.
It is possible that the neurovascular unit defects we report herein are related, in part, to previous embryonic neurovascular pathologies, or to the post-hemorrhagic hydrocephalus that develops in the adult brain. Indeed, similar to what we observe in β8–/– mice, intraventricular hemorrhage in the human neonatal brain can lead to hydrocephalus and long-term neurological deficits (Roland and Hill, 2003). Also, studies in rats with experimentally induced hydrocephalus have shown increases in reactive astrocytes throughout the brain (Del Bigio and Zhang, 1998). Hence, the β8–/– mice may serve as a potentially useful model for studying neurodegeneration related to post-hemorrhagic hydrocephalus. Based on the following experimental data, however, we also argue that β8 integrin provides essential NPC-intrinsic growth and survival functions in the SVZ: (i) β8 integrin protein is expressed in the SVZ in vivo and in SVZ NPCs cultured ex vivo (Figs 1 and 7); (ii) β8–/– NPCs isolated from the adult brain and cultured in medium that partly mimics the SVZ microenvironment have abnormal growth and survival behaviors (Fig. 7); (iii) adult mice with mild or severe hydrocephalus show similar NPC defects in the SVZ in vivo and in ex vivo cultures; and (iv) hydrocephalus can develop as a result of cortical atrophy as a result of impaired developmental and adult neurogenesis (Tomita et al., 2003).
A β8 integrin-TGFβ paracrine signaling axis in adult neurovascular niches?
αvβ8 integrin is a receptor for latent forms of TGFβ1 and TGFβ3, and some pathologies in αv and β8 integrin mutant mice have been linked to reduced integrin-mediated activation of TGFβs (Lacy-Hulbert et al., 2007; Travis et al., 2007). In this report we have shown that β8–/– NPCs have a modest, yet statistically significant reduction in activation of latent TGFβ1 (Fig. 8A), suggesting that the brain pathologies we detect in β8–/– mice may be caused by diminished TGFβ activation and signaling. However, much of our experimental data are contrary to an important role for integrin-activated TGFβs acting in an autocrine manner to promote NPC growth and survival. First, we have not been able to `rescue' NPC proliferation and self-renewal defects by exogenous addition of bioactive TGFβ1 to β8–/– neurosphere cultures (Fig. 8B), suggesting other pathways are involved. Second, a blocking antibody directed against bioactive TGFβs does not induce growth defects in wild-type NPCs similar to those that develop in β8–/– NPCs (Fig. 8C), suggesting that TGFβs play limited physiological roles in neurosphere growth. Lastly, genetic ablation of TGFβ receptor II signaling in embryonic neural cells using a nestin-Cre transgene (J.S. and J.H.M., unpublished data), or in adult SVZ NPCs using a GLAST-CreERT2 transgene (Colak et al., 2008), does not impair brain development or post-natal neurogenesis. Hence, we propose a model in which β8-integrin-activated TGFβs may act in a paracrine manner to regulate endothelial cell or pericyte functions, in turn leading to the production of vascular-derived factors that promote NPC growth and survival (Fig. 9). Other reports have suggested similar TGFβ-dependent paracrine mechanisms for astrocyte-mediated regulation of endothelial cell functions (Cambier et al., 2005; Garcia et al., 2004). It is also possible, and indeed probable, that β8 integrin serves as a receptor for other ECM ligands within adult neurovascular niches.
Fig. 9.

A model for αvβ8 integrin adhesion and signaling at the neural-vascular interface. αvβ8 integrin is expressed in perivascular neural cells where it mediates interactions with multiple ECM ligands, including latent TGFβs (LAP-TGFβ), present within vascular basement membranes. Integrin-mediated adhesion induces signaling pathways in cerebral blood vessels via canonical TGFβ receptors (RI/RII). Activation of these pathways in endothelial cells leads to the production of growth factors, and probably other vessel-derived survival cues, which subsequently modulate NPC behaviors. Genetic ablation of β8 integrin causes loss of integrin-mediated adhesion to ECM ligands, which uncouples the neural-vascular cell connections and leads to imbalances in NPC proliferation and survival.
Functions for β8 integrin outside of the subventricular zone?
Although these studies demonstrate necessary roles for β8 integrin in adult neurovascular units and SVZ neurogenesis, this integrin may also be important in other populations of neural cells that contribute to brain physiology. Indeed, we detected abnormal development of the hippocampus and dentate gyrus in adult β8–/– mice (supplementary material Fig. S1), suggesting more general functions for β8 integrin within other neurogenic brain regions. Oligodendroglial progenitor cells give rise to oligodendrocytes that myelinate axons in various regions of the adult brain (Menn et al., 2006). Conditional αv integrin knockout mice develop progressive neurodegenerative defects and die prematurely as a result of cerebellar white matter demyelination (McCarty et al., 2005b). Interestingly, these cerebellar pathologies are not apparent in adult β8–/– mice (supplementary material Fig. S2), suggesting that multiple αv-containing integrins regulate oligodendroglial progenitor cell functions. αvβ6 integrin is also a receptor for latent TGFβs (Munger et al., 1999) and is expressed in the adult mouse brain (Chan et al., 2003). Neurodegeneration has not been reported in β6–/– mice, however, indicating that defective adhesion and signaling in αv conditional knockouts may be due to combined loss of β6 and β8 functions. Hence, β8 integrin provides the necessary adhesion and signaling roles within multiple CNS neural cell types to promote normal brain development and physiology.
Materials and Methods
Experimental mice
ICR/CD-1 mice were purchased from Jackson Laboratories. β8+/– mice (Zhu et al., 2002) were obtained from the Mutant Mouse Regional Resource Center. The genotypes of embryonic, neonatal and adult progeny were confirmed using genomic DNA and PCR-based methodologies as previously described (Zhu et al., 2002). Primer sequences used for the β8 PCR are as follows: 5′-ATTATCTGGTTGATGTGTCAGC-3′, 5′-GGAGGCATACAGTCTAAATTGT-3′, 5′-AGAGGCCACTTGTGTAGCGCCAAG-3′ and 5′-AGAGAGGAACAAATATCCTTCCC-3′. PCR amplifies a wild-type product of 330 base pairs and mutant product of 450 base pairs, with both products amplified in heterozygous animals.
Antibodies and immunostaining
Adult mice were anesthetized and brains were fixed by cardiac perfusion with ice-cold 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Brains were removed and post-fixed overnight in 4% PFA-PBS and then processed for standard paraffin embedding and 7 μm sections were cut. Sections were analyzed using a Zeiss Axio Imager.Z1 microscope. The following antibodies were purchased from commercial sources and used for immunofluorescence on tissue sections: rabbit anti-laminin (1:100, Sigma), mouse anti-GFAP monoclonal antibody (mAb; 2.5 μg/ml, Chemicon), rabbit anti-GFAP polyclonal antibody (pAb; 10 μg/ml, DAKO), rat anti-BrdU (Accurate Chemical), rabbit anti-DCX (Chemicon) and anti-ZO1 (Zymed). Prior to immunofluorescence staining with anti-DCX and anti-GFAP, paraffin sections were processed for heat-based antigen retrieval at pH 9 according to the manufacturer's protocol (DAKO). For anti-ZO1 immunofluorescence staining, brains were removed without prior fixation and embedded in OCT for frozen section preparation. TUNEL fluorescence was performed on paraformaldehyde-fixed and paraffin-embedded brain sections using the manufacturer protocols (Promega). Secondary antibodies used for immunofluorescence were goat anti-rabbit and goat anti-mouse conjugated to Alexa Fluor 488 or Alexa Fluor 594 (Molecular Probes). The following antibodies used for immunoblotting were purchased from commercial sources: rabbit anti-β-actin antibody (Sigma-Aldrich), rabbit anti-occludin and rabbit anti-claudin 5 antibodies (Zymed). The anti-αv antibody was generated against a synthetic peptide (CKRVRPPQEEQEREQLQPHENGEGTSEA) corresponding to a region of the chicken αv cytoplasmic tail. The anti-β8 integrin antibodies used for immunoblotting have been described previously (McCarty et al., 2005a; McCarty et al., 2005b).
Neurosphere cultures
NPCs were cultured from the SVZ of individual adult mice using standard protocols (Rietze and Reynolds, 2006). Briefly, SVZ regions were microdissected and tissue was trypsinized for 10 minutes and triturated with a polished glass pipette. Cells were cultured in the following growth media: DME-F12 (Mediatech), 20 ng/ml EGF and FGF (Biosource), B27 supplement (Gibco) and one unit per ml penicillin-streptomycin (Sigma-Aldrich). After 7-10 days, primary neurospheres were passaged by mechanical disruption using a 1 ml syringe and a 23-gauge needle, and dissociated cells were re-plated in fresh growth medium. For secondary sphere formation assays, single spheres with similar diameters (∼75 μm) were mechanically dissociated and plated in one well of a 24-well plate. After intervals of 7 days (for multiple passages) the number of newly formed spheres was counted. Alternatively, neurosphere self-renewal was quantified in the presence of exogenously added TGFβ1 (10 ng/ml) and/or anti-TGFβ antibody (5 μM; both from R&D Systems). For neurosphere immunoblotting, cell lysates were prepared in RIPA Buffer (10 mM Tris, pH 7.4, 1% NP40, 0.5% deoxycholate, 0.1% SDS, 150 mM NaCl and 1 mM EDTA) and protein concentrations were determined (BCA Assay, Thermo Scientific). Protein lysates were mixed with an equal volume of 2× sample buffer, heated at 100°C, resolved by SDS-PAGE, and then immunoblotted with the various antibodies (see above).
BrdU incorporation assays
BrdU (BD Pharmingen) was injected at 65 μg/g body weight into mice once daily for two consecutive days. 24 hours after the last injection mice were cardiac-perfused with 4% paraformaldehyde and the brains were removed and post-fixed overnight. Brains were cryopreserved with 10% and 20% sucrose in PBS at 4°C, then embedded in Tissue Tek OCT compound (Sakura, Inc). Frozen sections (7 μm thickness) were cut and at least eight serial sections per brain were analyzed to quantify BrdU incorporation. Briefly, genomic DNA was denatured in 2 M HCl for 1 hour at 37°C and then neutralized with 0.1 M sodium borate buffer for 30 minutes at room temperature. Sections were then blocked with 10% goat serum (Vector Labs) in PBS for 30 minutes and incubated in primary antibody overnight at 4°C, followed by incubation with Alexa-Fluor-488-conjugated goat anti-rat secondary antibodies.
Luciferase reporter assays
HEK293T cells were plated into poly-lysine-coated six-well plates at 4×105 cells per well and allowed to attach overnight. Cells were then transfected with a PAI1-luciferase plasmid (Abe et al., 1994) using Effectene reagents (Qiagen). After 24 hours the transfection medium was removed and serum free medium was added for an additional 24 hours. Meanwhile, 100 wild-type or β8–/– adult neurospheres of similar diameters (∼75 μm) were added to six-well dishes containing 2 ml of neurosphere growth medium. LAP-TGFβ1 (SLC, R&D Systems) at 10 ng/ml (or PBS as a negative control) was added to the neurospheres. After 16 hours conditioned medium was collected, passed through a sterile 0.2 μm filter, and added to transfected HEK293T cells (see above) for 16 hours. As a control to confirm PAI1-luciferase plasmid transfection, bioactive TGFβ1 (R&D Systems) was added at 5 ng/ml to transfected HEK293T cells and incubated for 4 hours. Cell lysates were prepared and luciferase substrates were added according to the Enhanced Luciferase Assay kit instructions (BD Biosciences). Lysates were plated in duplicate wells in a 96-well plate and luciferase activities were determined using a Lumistar Galaxy Luminometer (BMG Lab Technologies).
Biotin perfusions
Post-natal day 60 (P60) control and mutant animals were deeply anesthetized with Avertin (250 mg/kg) and a 20 gauge needle was inserted into the left ventricle of the heart. Animals were first perfused with 20 ml of PBS to flush the circulation so that no residual cells or serum proteins were labeled with reactive biotin. Animals were then perfused for five minutes continuously with 1 mg/ml EZLink-NHS-Biotin (Pierce Chemicals, Inc.) dissolved in PBS (approximately 7.5 ml in total per animal). Animals were then perfused with 4% paraformaldehyde-PBS fixative to preserve brain morphology. Brains were removed and coronally sliced into 1 mm segments followed by cryopreservation in 30% sucrose-PBS, embedded in OCT, and cut in 7 μm sections. Frozen brain sections were rehydrated in PBS and blocked with 10% goat serum in PBS and visualized using streptavidin conjugated to Alexa Fluor 488 fluorophore (Molecular Probes).
Electron microscopy
Control and mutant animals were deeply anesthetized by intraperitoneal injection of Avertin (250 mg/kg). A 20 gauge needle was inserted into the left cardiac ventricle and animals were perfused with Karnovsky solution (1.25% formaldehyde, 2.5% glutaraldehyde and 0.03% picric acid in 100 mM cacodylate buffer; Polysciences, Inc.). Brains were then removed and postfixed overnight. Samples from the SVZ were fixed with a solution containing 3% glutaraldehyde plus 2% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.3 for I hour. After fixation, the samples were washed and treated with 0.1% Millipore-filtered cacodylate-buffered tannic acid, post-fixed with 1% buffered osmium tetroxide for 30 minutes, and stained en bloc with 1% Millipore-filtered uranyl acetate. The samples were dehydrated in increasing concentrations of ethanol, infiltrated, and embedded in Polybed812 medium. The samples were polymerized in a 60°C oven for 2 days. Ultrathin sections were cut in a Leica Ultracut microtome (Leica), stained with uranyl acetate and lead citrate in a Leica EM Stainer, and examined in a JEM 1010 transmission electron microscope (JEOL, Inc.) at an accelerating voltage of 80 kV. Digital images were obtained using AMT Imaging System (Advanced Microscopy Techniques Corp.).
Statistical analyses
Analysis of variance (ANOVA) model was used to determine differences in NPC self-renewal and proliferation between wild-type and mutant groups. Student's t-test was used to determine if there are any statistically significant differences, which are shown on the graphs as standard deviations.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/11/1842/DC1
We thank Adam Lacy-Hulbert (Massachusetts General Hospital) and Howard Colman (M. D. Anderson Cancer Center) for insightful comments on the manuscript. We are also grateful to Roderick Bronson (Harvard Medical School) for helpful discussions regarding mouse brain pathophysiology. Gordon Mills (M. D. Anderson Cancer Center) provided the PAI1-luciferase plasmid and Kenneth J. Dunner (M. D. Anderson Cancer Center) provided electron microscopy technical assistance. This research was supported by grants awarded to J.H.M. from the Ellison Medical Foundation (AG-NS-0324-06), the National Institute of Neurological Disease and Stroke (R01NS059876-01A2) and the University Cancer Foundation at the University of Texas M.D. Anderson Cancer Center. Deposited in PMC for release after 12 months.
References
- Abbott, N. J., Ronnback, L. and Hansson, E. (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41-53. [DOI] [PubMed] [Google Scholar]
- Abe, M., Harpel, J. G., Metz, C. N., Nunes, I., Loskutoff, D. J. and Rifkin, D. B. (1994). An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 216, 276-284. [DOI] [PubMed] [Google Scholar]
- Aluwihare, P., Mu, Z., Zhao, Z., Yu, D., Weinreb, P. H., Horan, G. S., Violette, S. M. and Munger, J. S. (2009). Mice that lack activity of {alpha}v{beta}6- and {alpha}v{beta}8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J. Cell Sci. 122, 227-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes, J. P., Munger, J. S. and Rifkin, D. B. (2003). Making sense of latent TGFbeta activation. J. Cell Sci. 116, 217-224. [DOI] [PubMed] [Google Scholar]
- Bader, B. L., Rayburn, H., Crowley, D. and Hynes, R. O. (1998). Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell 95, 507-519. [DOI] [PubMed] [Google Scholar]
- Battista, D., Ferrari, C. C., Gage, F. H. and Pitossi, F. J. (2006). Neurogenic niche modulation by activated microglia: transforming growth factor beta increases neurogenesis in the adult dentate gyrus. Eur. J. Neurosci. 23, 83-93. [DOI] [PubMed] [Google Scholar]
- Belvindrah, R., Hankel, S., Walker, J., Patton, B. L. and Muller, U. (2007). Beta1 integrins control the formation of cell chains in the adult rostral migratory stream. J. Neurosci. 27, 2704-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier, S., Gline, S., Mu, D., Collins, R., Araya, J., Dolganov, G., Einheber, S., Boudreau, N. and Nishimura, S. L. (2005). Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am. J. Pathol. 166, 1883-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, C. S., Weeber, E. J., Kurup, S., Sweatt, J. D. and Davis, R. L. (2003). Integrin requirement for hippocampal synaptic plasticity and spatial memory. J. Neurosci. 23, 7107-7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazal, G., Durbec, P., Jankovski, A., Rougon, G. and Cremer, H. (2000). Consequences of neural cell adhesion molecule deficiency on cell migration in the rostral migratory stream of the mouse. J. Neurosci. 20, 1446-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak, D., Mori, T., Brill, M. S., Pfeifer, A., Falk, S., Deng, C., Monteiro, R., Mummery, C., Sommer, L. and Gotz, M. (2008). Adult neurogenesis requires Smad4-mediated bone morphogenic protein signaling in stem cells. J. Neurosci. 28, 434-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasch, J. R., Pace, D. R., Waegell, W., Inenaga, D. and Ellingsworth, L. (1989). Monoclonal antibodies recognizing transforming growth factor-beta. Bioactivity neutralization and transforming growth factor beta 2 affinity purification. J. Immunol. 142, 1536-1541. [PubMed] [Google Scholar]
- Del Bigio, M. R. and Zhang, Y. W. (1998). Cell death, axonal damage, and cell birth in the immature rat brain following induction of hydrocephalus. Exp. Neurol. 154, 157-169. [DOI] [PubMed] [Google Scholar]
- Falk, S., Wurdak, H., Ittner, L. M., Ille, F., Sumara, G., Schmid, M. T., Draganova, K., Lang, K. S., Paratore, C., Leveen, P. et al. (2008). Brain area-specific effect of TGF-beta signaling on Wnt-dependent neural stem cell expansion. Cell Stem Cell 2, 472-483. [DOI] [PubMed] [Google Scholar]
- Garcia, C. M., Darland, D. C., Massingham, L. J. and D'Amore, P. A. (2004). Endothelial cell-astrocyte interactions and TGF beta are required for induction of blood-neural barrier properties. Brain Res. Dev. Brain Res. 152, 25-38. [DOI] [PubMed] [Google Scholar]
- Hall, P. E., Lathia, J. D., Miller, N. G., Caldwell, M. A. and Ffrench-Constant, C. (2006). Integrins are markers of human neural stem cells. Stem Cells 24, 2078-2084. [DOI] [PubMed] [Google Scholar]
- Hynes, R. O. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673-687. [DOI] [PubMed] [Google Scholar]
- Hynes, R. O., Lively, J. C., McCarty, J. H., Taverna, D., Francis, S. E., Hodivala-Dilke, K. and Xiao, Q. (2002). The diverse roles of integrins and their ligands in angiogenesis. Cold Spring Harb. Symp. Quant. Biol. 67, 143-153. [DOI] [PubMed] [Google Scholar]
- Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat. Rev. Neurosci. 5, 347-360. [DOI] [PubMed] [Google Scholar]
- Jacques, T. S., Relvas, J. B., Nishimura, S., Pytela, R., Edwards, G. M., Streuli, C. H. and ffrench-Constant, C. (1998). Neural precursor cell chain migration and division are regulated through different beta1 integrins. Development 125, 3167-3177. [DOI] [PubMed] [Google Scholar]
- Kerever, A., Schnack, J., Vellinga, D., Ichikawa, N., Moon, C., Arikawa-Hirasawa, E., Efird, J. T. and Mercier, F. (2007). Novel extracellular matrix structures in the neural stem cell niche capture the neurogenic factor fibroblast growth factor 2 from the extracellular milieu. Stem Cells 25, 2146-2157. [DOI] [PubMed] [Google Scholar]
- Lacy-Hulbert, A., Smith, A. M., Tissire, H., Barry, M., Crowley, D., Bronson, R. T., Roes, J. T., Savill, J. S. and Hynes, R. O. (2007). Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc. Natl. Acad. Sci. USA 104, 15823-15828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty, J. H. (2005). Cell biology of the neurovascular unit: implications for drug delivery across the blood-brain barrier. Assay Drug Dev. Technol. 3, 89-95. [DOI] [PubMed] [Google Scholar]
- McCarty, J. H., Monahan-Earley, R. A., Brown, L. F., Keller, M., Gerhardt, H., Rubin, K., Shani, M., Dvorak, H. F., Wolburg, H., Bader, B. L. et al. (2002). Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Mol. Cell. Biol. 22, 7667-7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty, J. H., Cook, A. A. and Hynes, R. O. (2005a). An interaction between {alpha}v{beta}8 integrin and Band 4.1B via a highly conserved region of the Band 4.1 C-terminal domain. Proc. Natl. Acad. Sci. USA 102, 13479-13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty, J. H., Lacy-Hulbert, A., Charest, A., Bronson, R. T., Crowley, D., Housman, D., Savill, J., Roes, J. and Hynes, R. O. (2005b). Selective ablation of {alpha}v integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development 132, 165-176. [DOI] [PubMed] [Google Scholar]
- Menn, B., Garcia-Verdugo, J. M., Yaschine, C., Gonzalez-Perez, O., Rowitch, D. and Alvarez-Buylla, A. (2006). Origin of oligodendrocytes in the subventricular zone of the adult brain. J. Neurosci. 26, 7907-7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier, F., Kitasako, J. T. and Hatton, G. I. (2002). Anatomy of the brain neurogenic zones revisited: fractones and the fibroblast/macrophage network. J. Comp. Neurol. 451, 170-188. [DOI] [PubMed] [Google Scholar]
- Mu, D., Cambier, S., Fjellbirkeland, L., Baron, J. L., Munger, J. S., Kawakatsu, H., Sheppard, D., Broaddus, V. C. and Nishimura, S. L. (2002). The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell Biol. 157, 493-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, Z., Yang, Z., Yu, D., Zhao, Z. and Munger, J. S. (2008). TGFbeta1 and TGFbeta3 are partially redundant effectors in brain vascular morphogenesis. Mech. Dev. 125, 508-516. [DOI] [PubMed] [Google Scholar]
- Munger, J. S., Huang, X., Kawakatsu, H., Griffiths, M. J., Dalton, S. L., Wu, J., Pittet, J. F., Kaminski, N., Garat, C., Matthay, M. A. et al. (1999). The integrin alpha v beta 6 binds and activates latent TGF beta 1, a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319-328. [DOI] [PubMed] [Google Scholar]
- Nishimura, S. L., Boylen, K. P., Einheber, S., Milner, T. A., Ramos, D. M. and Pytela, R. (1998). Synaptic and glial localization of the integrin alphavbeta8 in mouse and rat brain. Brain Res. 791, 271-282. [DOI] [PubMed] [Google Scholar]
- Nitta, T., Hata, M., Gotoh, S., Seo, Y., Sasaki, H., Hashimoto, N., Furuse, M. and Tsukita, S. (2003). Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 161, 653-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, T. D., Willhoite, A. R. and Gage, F. H. (2000). Vascular niche for adult hippocampal neurogenesis. J. Comp. Neurol. 425, 479-494. [DOI] [PubMed] [Google Scholar]
- Plate, K. H. (1999). Mechanisms of angiogenesis in the brain. J. Neuropathol. Exp. Neurol. 58, 313-320. [DOI] [PubMed] [Google Scholar]
- Proctor, J. M., Zang, K., Wang, D., Wang, R. and Reichardt, L. F. (2005). Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. J. Neurosci. 25, 9940-9948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietze, R. L. and Reynolds, B. A. (2006). Neural stem cell isolation and characterization. Methods Enzymol. 419, 3-23. [DOI] [PubMed] [Google Scholar]
- Roland, E. H. and Hill, A. (2003). Germinal matrix-intraventricular hemorrhage in the premature newborn: management and outcome. Neurol. Clin. 21, 833-851, vi-vii. [DOI] [PubMed] [Google Scholar]
- Roussa, E., Wiehle, M., Dunker, N., Becker-Katins, S., Oehlke, O. and Krieglstein, K. (2006). Transforming growth factor beta is required for differentiation of mouse mesencephalic progenitors into dopaminergic neurons in vitro and in vivo: ectopic induction in dorsal mesencephalon. Stem Cells 24, 2120-2129. [DOI] [PubMed] [Google Scholar]
- Sawamoto, K., Wichterle, H., Gonzalez-Perez, O., Cholfin, J. A., Yamada, M., Spassky, N., Murcia, N. S., Garcia-Verdugo, J. M., Marin, O., Rubenstein, J. L. et al. (2006). New neurons follow the flow of cerebrospinal fluid in the adult brain. Science 311, 629-632. [DOI] [PubMed] [Google Scholar]
- Sheppard, D. (2004). Roles of alphav integrins in vascular biology and pulmonary pathology. Curr. Opin. Cell Biol. 16, 552-557. [DOI] [PubMed] [Google Scholar]
- Tavazoie, M., Van der Veken, L., Silva-Vargas, V., Louissaint, M., Colonna, L., Zaidi, B., Garcia-Verdugo, J. M. and Doetsch, F. (2008). A specialized vascular niche for adult neural stem cells. Cell Stem Cell 3, 279-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita, S., Ueno, M., Sakamoto, M., Kitahama, Y., Ueki, M., Maekawa, N., Sakamoto, H., Gassmann, M., Kageyama, R., Ueda, N. et al. (2003). Defective brain development in mice lacking the Hif-1alpha gene in neural cells. Mol. Cell. Biol. 23, 6739-6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis, M. A., Reizis, B., Melton, A. C., Masteller, E., Tang, Q., Proctor, J. M., Wang, Y., Bernstein, X., Huang, X., Reichardt, L. F. et al. (2007). Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449, 361-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachs, F. P., Winner, B., Couillard-Despres, S., Schiller, T., Aigner, R., Winkler, J., Bogdahn, U. and Aigner, L. (2006). Transforming growth factor-beta1 is a negative modulator of adult neurogenesis. J. Neuropathol. Exp. Neurol. 65, 358-370. [DOI] [PubMed] [Google Scholar]
- Wolburg, H. and Lippoldt, A. (2002). Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul. Pharmacol. 38, 323-337. [DOI] [PubMed] [Google Scholar]
- Wurmser, A. E., Palmer, T. D. and Gage, F. H. (2004). Neuroscience: cellular interactions in the stem cell niche. Science 304, 1253-1255. [DOI] [PubMed] [Google Scholar]
- Yang, Z., Mu, Z., Dabovic, B., Jurukovski, V., Yu, D., Sung, J., Xiong, X. and Munger, J. S. (2007). Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J. Cell Biol. 176, 787-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, J., Motejlek, K., Wang, D., Zang, K., Schmidt, A. and Reichardt, L. F. (2002). beta8 integrins are required for vascular morphogenesis in mouse embryos. Development 129, 2891-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}