

The continual search for new antitumor and antiviral agents from marine sponge extracts has resulted in the identification of a number of structurally complex and diverse natural products with intriguing biological activity. In 1996 and 1997 Minale and co-workers reported the isolation and structural assignment of callipeltosides A–C (Figure 1), the first three members of a new class of marine macrolides from the lithistid sponge Callipelta sp. indigenous to the shallow waters off New Caledonia.[1]
Figure 1.

The callipeltoside class of natural products.
Biological activity, in particular cytotoxicity, varies among the callipeltosides, and their IC50 values range from 11.3 to 30.0 μg mL−1 against human bronchopulmonary NSCLC-N6 cell lines.[1] These natural isolates were shown to block cell proliferation in the G1 phase, but unfortunately the exact mode of action cannot as yet be determined owing to the prohibitively small quantities of callipeltosides A–C that have been isolated to date (≤3.5 mg).[1] A more detailed annotation of the biological mechanism will await the availability of substantial synthetic quantities of the callipeltosides and their analogues as the natural isolates have been consumed.[1]
In terms of structural complexity, the callipeltosides share a common 12-membered macrocycle comprising seven stereocenters and a unique dienyne chlorocyclopropane side chain; this class is differentiated only by the composition of the saccharide moiety. In particular, callipeltosides A and B are characterized by the presence of two unique deoxyamino sugars, while callipeltoside C incorporates the novel deoxy sugar 2-O-methylevalose. Perhaps more notable, the carbohydrate moieties of callipeltosides B and C were reported to exist in the opposite enantiomeric series to that found in callipeltoside A (callipeltosides B and C=d sugar, callipeltoside A=l sugar).
Their stereochemical and structural complexity coupled with their unique biological activity make the callipeltoside family an attractive target for total synthesis. Indeed, to date, the groups of Trost,[2] Evans,[3] Paterson,[4] and Panek[5] have completed the asymmetric construction of callipeltoside A, yet surprisingly callipeltosides B and C have remained unchallenged by chemical synthesis. In this communication we describe the first total synthesis of callipeltoside C and present a structural revision with respect to the enantioseries of the pendent 2-O-methylevalose carbohydrate. This synthetic sequence, which is founded upon the use of organocatalytic and organometallic technologies, provides access to useful quantities of callipeltoside C in 18 chemical steps and 12% overall yield.
Our disconnection of callipeltoside C into four components 2–5 of similar complexity (Figure 2) revealed the possibility of a convergent synthesis with broad latitude in the sequence of fragment coupling. Inspection of fragments 2–4 brought to mind the possibility of exploiting three asymmetric organocatalytic transformations that have recently been developed in our laboratories.[6-9] In particular, we proposed the use of a direct aldehyde–aldehyde aldol coupling[6] in combination with a Semmelhack alkoxycarbonylation for the rapid construction of tetrahydropyran 2. Furthermore, we envisioned that the stereogenicity of the protected iodoalcohol 3 could be furnished by means of an enantioselective formyl α-oxyamination.[7] Last, we recognized the opportunity to further evaluate the versatility of our enamine-catalyzed two-step carbohydrate synthesis[8,9] to rapidly assemble the desired deoxy sugar 4 from simple achiral starting materials. Moreover, we hoped that the amino acid proline would function as a suitable organocatalyst for all of these asymmetric processes.
Figure 2.

Retrosynthetic analysis of callipeltoside C. PMB=para-methoxybenzyl, TBS=tert-butyldimethylsilyl, TIPS=triisopropylsilyl.
Assembly of the tetrahydropyran 2 and iodoalcohol fragment 3 to give the callipeltoside aglycone was envisioned to result from a two-point coupling strategy involving a Yamaguchi esterification and a diastereoselective aldehyde vinylation, both of which could be sequentially interchanged to serve as the macrocyclization event. Based on the studies of Evans et al., we anticipated that attachment of the dienyne side chain would readily be accomplished with high levels of stereocontrol using a Horner–Wadsworth–Emmons (HWE) olefination.[3] Finally, we recognized the importance of forming the glycosidic linkage at a late stage to 1) establish rapidly the absolute configuration of the callipeltose C sugar moiety and 2) enable convenient access to a range of callipeltoside analogues from the aglycone.
Our first step towards the construction of callipeltoside C involved an enamine-catalyzed double diastereo-differentiating aldol reaction between propionaldehyde and the Roche ester-derived aldehyde 6 (Scheme 1). This matched aldol union provides the desired product[10] with high levels of stereocontrol (12:1 anti/syn, > 19:1 Felkin/anti-Felkin),[11] while affording a product carbonyl compound that can be used directly in subsequent alkylation reactions without the need for oxidation state adjustment.[6] Indeed, Felkin-selective chelation-controlled addition of propargyl zinc to aldehyde 7 afforded alkynyl diol 8 in nearly quantitative yield with 6:1 selectivity for the desired 4,5-syn diastereomer. With the four stereogenic centers of fragment 2 in place, we sought to employ the Semmelhack reaction to build the central heterocyclic ring of callipeltoside C by a palladium-catalyzed alkoxycarbonylation.[12] Using the conditions developed by Marshall and co-workers,[12b] exposure of alkynyl alcohol 8 to the PdII catalyst in the presence of CO and MeOH did indeed furnish the highly functionalized tetrahydropyran 9 in 75% yield and with > 95:5 anomeric diastereocontrol. At this stage, protection of the remaining secondary hydroxy group as a tert-butyldimethylsilyl ether followed by cleavage of the PMB protecting group and then Parikh–Doering oxidation[13] provided the key tetrahydropyran coupling fragment 2 in six steps and 44% overall yield.
Scheme 1.

Synthesis of the tetrahydropyran fragment: a) l-proline (10 mol%), DMSO, +4°C; b) HCCCH2Br, Zn, THF; c) 5% [PdCl2-(CH3CN)2], p-benzoquinone, CO, MeOH, 0°C; d) TBSCl, imidazole, DMF; e) DDQ, CH2Cl2; f) SO3·pyridine, Et3N, CH2Cl2, DMSO. brsm=based on recovered starting material, DDQ=2,3-dichloro-5,6-dicyano-1,4-benzoquinone.
Synthesis of fragment 3 began with Negishi carbometalation–iodination of 4-pentynol[14] followed by Swern oxidation to provide the trisubstituted vinyl iodide 10 with excellent reaction efficiency and control of olefin geometry (Scheme 2). In 2003 the Zhong group, along with our own laboratory, published a new organocatalytic transformation for the direct and enantioselective α-oxyamination of aldehydes.[7] We proposed that implementation of this technology with iodoaldehyde 10 might furnish the desired α-oxidized product with high levels of enantioselectivity, while we hoped the mild reaction conditions would leave the vinyl iodide moiety intact. Indeed, deployment of iodoaldehyde 10 in a one-pot, three-step sequence consisting of proline-catalyzed α-oxyamination,[7] borohydride reduction, and reductive O–N bond cleavage, successfully provided the diol 12 in 99% ee and 77% yield (over three steps). Selective protection of the resulting 1,2-diol then provided the requisite vinyl iodide 3 in 57% overall yield for the seven-step sequence (Scheme 2).
Scheme 2.

Synthesis of the iodoalcohol fragment: a) AlMe3, [Cp2ZrCl2], I2, THF, −30°C; b) (COCl)2, Et3N, DMSO, CH2Cl2, −50°C; c) l-proline (20 mol%), PhNO, DMSO; d) NaBH4, EtOH; e) Zn, AcOH, EtOH; f) Bu2Sn(OMe)2, PMBCl, Bu4NI, PhCH3; g) TBSCl, imidazole, DMF.
At this stage in our synthesis we recognized the possibility to forge the 12-membered macrocycle by using two different sequences that would combine fragments 2 and 3. Specifically, we proposed that intermolecular esterification followed by intramolecular Nozaki–Hiyama–Kishi coupling,[15] or alternatively, intermolecular addition of a vinyl anion to the aldehyde prior to Yamaguchi lactonization[16] would result in formation of the macrocycle. Initial attempts focused upon the former route, and while macrocycle formation was possible, all attempts at the intramolecular Nozaki–Hiyama–Kishi reaction resulted exclusively in the production of the undesired R alcohol stereoisomer at C9. Turning to our latter route, we were optimistic that the correct stereochemistry at C9 could be delivered by diastereoselective intermolecular aldehyde alkylation. Indeed, after some optimization we found that treatment of the pyranyl aldehyde 2 with MgBr2·Et2O prior to addition of vinyl Grignard 13 (derived in situ from iodoalcohol 3) resulted in formation of the desired anti-Felkin addition product 14 with excellent yield and diastereoselectivity (98% yield, 16:1 d.r.) (Scheme 3). We presume that the observed anti-Felkin selectivity arises from substrate chelation to MgBr2 (through the aldehyde and pyranyl oxygen moieties), prior to vinyl addition across the least hindered carbonyl π face. The resulting C9 hydroxy group was then converted to a methyl ether followed by cleavage of the PMB protecting group and oxidation of the resultant primary hydroxyl function to furnish aldehyde 15. Installation of the cyclopropyl dienyne side chain 5[17] using the HWE protocol developed by Evans et al.[3a] resulted in optimal yields in addition to excellent levels of stereoselectivity (19:1 E/Z).[18] Selective removal of the C13 silyloxy group and saponification of the methyl ester then afforded the seco-acid 16, which is poised for macrolactonization. Yamaguchi cyclization[16] proceeded in 83% yield, albeit with elimination of methanol to give the dihydropyran 17. Fortunately, treatment of the resulting cyclic enol with triphenylphosphine hydrogen bromide introduced the requisite hemiacetal at C3, while subsequent exposure to acid removed the remaining TBS group to complete the synthesis of the callipeltoside aglycone 18 in 16 steps and 19% overall yield. Spectral and optical rotation data for the aglycone were in complete accord with the previous reports of Paterson et al.[4a] and Trost et al.[2a]
Scheme 3.

Coupling the iodoalcohol and tetrahydropyran: a) MgBr2·Et2O, CH2Cl2, −78°C; b) MeOTf, 2,6-DTBP, CH2Cl2; c) DDQ, CH2Cl2, pH 7 buffer; d) SO3·pyridine, Et3N, CH2Cl2, DMSO; e) LiHMDS, then 5, THF, −78°C; f) TBAF, THF, 0°C; g) Ba(OH)2·8 H2O, MeOH; h) Yamaguchi: 2,4,6-Cl3C6H2COCl, iPr2EtN, THF, DMAP, toluene, 60°C; i) PPh3·HBr, H2O, CH2Cl2. j) TFA, THF, H2O. DMAP=4-dimethylaminopyridine, 2,6-DTBP=2,6-di-tert-butylpyridine, HMDS=hexamethyldisilazane, TBAF=tetrabutylammonium fluoride, TFA=trifluoroacetic acid.
In 2004 our group reported a two-step protocol for the de novo synthesis of carbohydrates, in which we found that Lewis acid mediated Mukaiyama aldol reactions could be used to selectively access a variety of aldohexose sugars.[9] Most relevant to this current report, we observed that the MgBr2-mediated variant leads to the differentially protected mannose architecture, a structural and stereochemical topology that closely related to the callipeltoside C hexose. From the outset we recognized the possibility that the carbohydrate moieties of callipeltosides A–C might, in fact, be of the same enantiomeric series (in contrast to the findings of the isolation paper). Indeed, the recent isolation of phorbaside A,[19] which contains l-2-O-methylevalose, provides additional support for such a possibility. With this in mind, we thought that a de novo carbohydrate synthesis seemed logical as it would provide access to both enantiomers of the callipeltose C sugar (a convenience not likely available by synthetic manipulation of a naturally abundant carbohydrate).
Synthesis of the d-callipeltose sugar commenced with the well-established d-proline-catalyzed aldol dimerization of 2-triisopropylsilanoxyacetaldehyde[8] to afford the erythrose equivalent 19, which we hoped would participate in a Mukaiyama aldol reaction with a nascent aldehyde enolate according to our group!s precedent (Scheme 4).[9] To our satisfaction, reaction of the aldol dimer 19 with the triethylsilyl enol ether derived from 2-methoxy acetaldehyde in the presence of MgBr2·OEt2 did indeed provide the desired polyol-differentiated mannose 20 with excellent selectivity, and in moderate yield (> 20:1 d.r., 47% yield). Acid-catalyzed benzyl protection of the anomeric hydroxy group and concomitant removal of the primary silyloxy protecting group was accomplished prior to selective formation of the corresponding primary phenyl thiocarbonate. Deoxygenation following the Barton–McCombie protocol[20] and then Dess–Martin oxidation of the secondary alcohol provided the desired tetrahydropyranyl ketone 21 in good yield over four steps (Scheme 4). At this juncture, selective π-facial addition of a methyl group to the carbonyl of 21 would install the requisite tertiary oxy stereocenter of the evalose saccharide ring. Semiempirical calculations suggested that carbanion nucelophiles would approach the low-energy pseudo-chair conformation of 21 by means of the desired axial trajectory to avoid steric interactions with the neighboring methoxy group, while alleviating torsional strain in the bond-forming event. Indeed, addition of MeMgBr to a solution of 21 and MgBr2·OEt2 in CH2Cl2 produced the desired stereoisomer in 90% yield and 20:1 d.r. The relative stereochemistry of 21 was confirmed by NOE analysis and by X-ray analysis of a substance that was chemically correlated to the minor diastereomer. Palladium-catalyzed hydrogenolysis cleanly removed the benzyl group prior to activation of the sugar as a trichloroacetimidate furnished the d-2-O-methylevalose coupling partner 22 in eight steps and 14.5% overall yield (Scheme 4). Central to our design plan, production of the antipodal l-callipeltose sugar (ent-22) was also accomplished using this eight-step protocol (using catalytic l-proline in lieu of d-proline in the initial asymmetric aldol event).
Scheme 4.

De novo synthesis of a differentially protected mannose: a) d-proline, DMF; b) MgBr2·Et2O, CH2Cl2; c) AcCl, BnOH, 110°C; d) PhOCSCl, pyridine, CH2Cl2; e) Bu3SnH, AIBN, toluene, 120°C; f) Dess–Martin periodinane, CH2Cl2, 0°C; g) MgBr2·Et2O, CH3MgBr, CH2Cl2; h) H2, Pd/C, EtOAc; Cl3CCN, Cs2CO3, CH2Cl2; i) l-proline, DMF. AIBN=2,2′-azobisisobutyronitrile.
The coupling of the d-2-O-methylevalose hexose 22 and the aglycone 18 was accomplished according to the glycosylation procedure of Tietze et al (Scheme 5).[21] Silyl deprotection of the resulting material using tris(dimethylamino)-sulfonium difluorotrimethylsilicate (TASF) in DMF at 40°C then provided a compound having the reported structure of callipeltoside C (1) in 90% yield (Scheme 5). Unfortunately, spectroscopic data for the macrolide 1 did not match the characterization data reported for the natural isolate.[1,22] With this knowledge in hand, we next focused upon linking the l antipode of the callipeltose C sugar (ent-22) to the macrocyclic core 18. Using again the Tietze glycosylation/TASF desilylation sequence to combine fragments 18 and ent-22, we were delighted to find that the resulting macrolide 23 (formed as a single diastereomer) was identical to callipelto-side C with respect to the spectroscopic data obtained for the natural isolate.[23] On this basis we report that the structure of callipeltoside C should be revised to that shown in Scheme 5.[24,25]
Scheme 5.
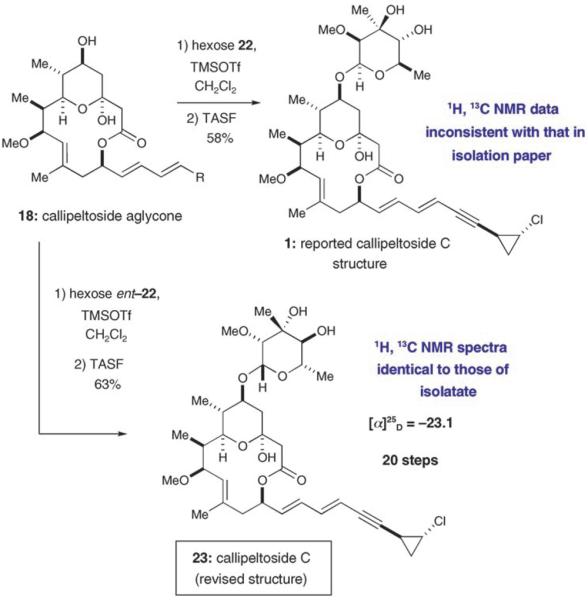
Completion of the synthesis of callipeltoside C with structural revision.
In conclusion, a highly efficient enantioselective synthesis of callipeltoside C has been accomplished with a longest linear sequence of 20 steps in 11% overall yield from the commercially available Roche ester.[26] As a result of our synthetic efforts we also present a structural revision of the originally reported carbohydrate stereochemistry. Highlights of our approach include the proline-catalyzed direct aldol reaction, enantioselective α-oxyamination reaction, and rapid access to the carbohydrate framework using a de novo synthesis protocol.[27]
Acknowledgments
We thank Professor L. Minale and co-workers for the Herculean task of isolating and characterizing callipeltosides A–C. We thank Professor M. Valeria D'Auria for providing comparison spectra of natural 23 to D.W.C.M.
Footnotes
Dedicated to Professor Luigi Minale and co-workers
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Zampella A, D'Auria MV, Minale L, Debitus C, Roussakis C. J. Am. Chem. Soc. 1996;118:11085. [Google Scholar]; b) Zampella A, D'Auria MV, Minale L. Tetrahedron. 1997;53:3243. [Google Scholar]
- 2.a) Trost BM, Gunzner JL, Dirat O, Rhee YH. J. Am. Chem. Soc. 2002;124:10396. doi: 10.1021/ja0205232. [DOI] [PubMed] [Google Scholar]; b) Trost BM, Dirat O, Gunzner JL. Angew. Chem. 2002;114:869. doi: 10.1002/1521-3773(20020301)41:5<841::aid-anie841>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2002;41:841. [Google Scholar]; c) Trost BM, Gunzner JL. J. Am. Chem. Soc. 2001;123:9449. doi: 10.1021/ja011424b. [DOI] [PubMed] [Google Scholar]
- 3.a) Evans DA, Hu E, Burch JD, Jaeschke G. J. Am. Chem. Soc. 2002;124:5654. doi: 10.1021/ja026235n. [DOI] [PubMed] [Google Scholar]; b) Evans DA, Hu E, Tedrow JS. Org. Lett. 2001;3:3133. doi: 10.1021/ol016416e. [DOI] [PubMed] [Google Scholar]; c) Evans DA, Burch JD. Org. Lett. 2001;3:503. doi: 10.1021/ol0155182. [DOI] [PubMed] [Google Scholar]
- 4.a) Paterson I, Davies RDM, Heimann AC, Marquez R, Meyer A. Org. Lett. 2003;5:4477. doi: 10.1021/ol0357853. [DOI] [PubMed] [Google Scholar]; b) Paterson I, Davies RDM, Marquez R. Angew. Chem. 2001;113:623. doi: 10.1002/1521-3773(20010202)40:3<603::AID-ANIE603>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2001;40:603. [Google Scholar]
- 5.a) Huang H, Panek JS. Org. Lett. 2004;6:4383. doi: 10.1021/ol0480325. [DOI] [PubMed] [Google Scholar]; b) Huang H, Panek JS. Org. Lett. 2003;5:1991. doi: 10.1021/ol034582b. [DOI] [PubMed] [Google Scholar]
- 6.Northrup AB, MacMillan DWC. J. Am. Chem. Soc. 2002;124:6798. doi: 10.1021/ja0262378. [DOI] [PubMed] [Google Scholar]
- 7.a) Brown SP, Brochu MP, Sinz CJ, MacMillan DWC. J. Am. Chem. Soc. 2003;125:10808. doi: 10.1021/ja037096s. [DOI] [PubMed] [Google Scholar]; b) Zhong G. Angew. Chem. 2003;115:4379. [Google Scholar]; Angew. Chem. Int. Ed. 2003;42:4247. [Google Scholar]
- 8.a) Northrup AB, Mangion IK, Hettche F, MacMillan DWC. Angew. Chem. 2004;116:2204. doi: 10.1002/anie.200453716. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:2152. [Google Scholar]
- 9.Northrup AB, MacMillan DWC. Science. 2004;303:1752. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]
- 10.Stereochemistry has been previously established: Horita K, Inoue T, Tanaka K, Yonemitsu O. Tetrahedron. 1996;52:531.
- 11.With d-proline catalysis: 12:1 anti/syn, 1:2 Felkin/anti-Felkin.
- 12.a) Semmelhack MF, Kim C, Zhang N, Bodurow C, Sanner M, Dobler W, Meier M. Pure Appl. Chem. 1990;62:2035. [Google Scholar]; b) Marshall JA, Yanik MM. Tetrahedron Lett. 2000;41:4717. [Google Scholar]
- 13.Parikh JR, Doering W. v. E. J. Am. Chem. Soc. 1967;89:5505. [Google Scholar]
- 14.Negishi E, Van Horn DE, Yoshida T. J. Am. Chem. Soc. 1985;107:6639. [Google Scholar]
- 15.a) Okude Y, Hirano S, Hiyama T, Nozaki H. J. Am. Chem. Soc. 1977;99:3179. [Google Scholar]; b) Jin H, Uenishi J, Christ WJ, Kishi Y. J. Am. Chem. Soc. 1986;108:5644. [Google Scholar]
- 16.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989. [Google Scholar]
- 17.The side chain was synthesized according to the procedure of Evans: Evans DA, Burch JD. Org. Lett. 2001;3:503. doi: 10.1021/ol0155182.
- 18.The HWE coupling was less effective when performed after the macrocyclization step (84% vs. 54% yield).
- 19.Skepper KC, MacMillan JB, Zhou G, Masuno MM, Molinski TF. J. Am. Chem. Soc. 2007;129:4150. doi: 10.1021/ja0703978. [DOI] [PubMed] [Google Scholar]
- 20.Barrett AGM, Prokopiou PA, Barton DHR. J. Chem. Soc. Chem. Commun. 1979:1175. [Google Scholar]
- 21.a) Tietze LF, Fischer R, Guder HJ, Goerlach A, Meumann M, Krach T. Carbohydr. Res. 1987;164:177. [Google Scholar]; b) Tietze LF, Fischer R, Remberg G. Liebigs Ann. Chem. 1987:971. [Google Scholar]
- 22.The macrolide 1 was formed as a 4:1 mixture of diastereomers at the anomeric position. Spectral data for each anomer was inconsistent with the characterization data for the natural isolate.
- 24.At the present time we cannot state definitively the stereo-chemistry of the C1′ anomeric position of callipeltoside C. Based on the isolation studies for callipeltoside B, we tentatively suggest that it exists as the β-equatorial anomer as shown in Scheme 5.
- 25.We anticipate that the structure of callipeltoside B will likely be revised with respect to the enantiomer series of the pendent deoxyamino carbohydrate.
- 26.The longest linear sequence is 18 steps and 12% overall yield from the Roche ester-derived aldehyde 6.
- 27.For a review on the organocatalytic synthesis of bioactive molecules: Figueirido RMF, Christmann M. Eur. J. Org. Chem. 2007:2575.