

Abstract
A significant consequence of protein phosphorylation is to alter protein-protein interactions, leading to dynamic regulation of the components of protein complexes that direct many core biological processes. Recent proteomic studies have populated databases with extensive compilations of cellular phosphoproteins and phosphorylation sites and a similarly deep coverage of the subunit compositions and interactions in multi-protein complexes. However, considerably less data are available on the dynamics of phosphorylation, composition of multiprotein complexes or that define their interdependence. We describe a method to identify candidate phosphoprotein complexes by combining phosphoprotein affinity chromatography, separation by size, denaturing gel electrophoresis, protein identification by tandem mass spectrometry, and informatics analysis. Toward developing phosphoproteome profiling, we have isolated native phosphoproteins using a phosphoprotein affinity matrix, Pro-Q Diamond resin (Molecular Probes-Invitrogen). This resin quantitatively retains phosphoproteins and associated proteins from cell extracts. Pro-Q Diamond purification of a yeast whole cell extract followed by 1-D PAGE separation, proteolysis and ESI LC-MS/MS, a method we term PA-GeLC-MS/MS, yielded 131 proteins, the majority of which were known phosphoproteins. To identify proteins that were purified as parts of phosphoprotein complexes, the Pro-Q eluate was separated into two fractions by size, < 100 kDa and > 100 kDa, before analysis by PAGE and ESI LC-MS/MS and the component proteins queried against databases to identify protein-protein interactions. The < 100 kDa fraction was enriched in phosphoproteins indicating the presence of monomeric phosphoproteins. The > 100 kDa fraction contained 171 proteins of 20 to 80 kDa, nearly all of which participate in known protein-protein interactions. Of these 171, few are known phosphoproteins, consistent with their purification by participation in protein complexes. By mining our phosphoprotein purification with the informational databases on phosphoproteomics, protein-protein interactions and protein complexes, we have developed an approach to examining the correlation between protein interactions and protein phosphorylation.
Keywords: Protein complexes, phosphorylation, mass spectrometry, ESI LC MS/MS, phosphoprotein affinity enrichment, databases
Introduction
Systems biology is the study of the interactions between components of a biological system and how these interactions bring about the function and behavior of that system (reviewed in 1). In contrast to the classical enzyme-substrate paradigm that underlies the biosynthesis of most metabolites, other basic biological processes such as replication, transcription, splicing, translation, or secretion require dynamic assembly of multiprotein complexes whose subunits function in concert to perform highly regulated reactions that synthesize or modify macromolecules (discussed in 2). Protein complexes bring proteins in close proximity and can facilitate sequential reactions on the same substrate. A relatively simple example is the proteasome, a large protein complex that breaks down polyubiquitinylated proteins via ATP-dependent proteolysis (reviewed in 3). As a major mechanism by which cells regulate the concentration of proteins and degrade misfolded proteins, the proteasome must recognize properly tagged substrates, unfold the polypeptide and thread it into the proteolytic chamber and degrade the protein to short peptides. The proteasome itself is comprised of over 20 individual stably associated protein subunits that provide structural support or are involved in recognition, unfolding and/or proteolytic roles.
The limiting factor for identifying protein complexes is the method for their isolation or enrichment. Large protein complexes and organelles such as nucleasome and centrosomes can be enriched on a sucrose gradient and analyzed by mass spectrometry 4, 5. Only a subset of protein complexes is large enough in size and sufficiently dissimilar from other complexes to be suited for this type of analysis. Blue native polyacrylamide gel electrophoresis (BN-PAGE) has also been used to separate native protein complexes in the first dimension followed by denaturing SDS-PAGE electrophoresis in the second dimension creating a 2-D gel where the spots can be analyzed by mass spectrometry 6–8. This method was recently applied to complex protein samples 7. As with conventional denaturing 2-D gel electrophoresis limitations include reproducibility, recovery of protein and visualization and selection of proteins spots in gel for mass spectrometry analysis. Alternatively, sedimentation of protein complexes in a rate zonal gradient allows for estimation of the relative size of protein complexes as recently described for Arabidopsis thaliana 9.
Methods for mass spectrometry analysis of protein complexes are reviewed in 10. A comprehensive genome wide study of protein complexes was completed in yeast where the authors affinity purified individual tandem affinity purification (TAP) tagged proteins from whole-cell lysates 11, 12. Each TAP-tagged protein and any copurifying polypeptides were analyzed by mass spectrometry from SDS-PAGE gel bands. This approach identified over 500 distinct protein complexes containing two to dozens of proteins and showed that many proteins serve as subunits of a number of different complexes. The interactions identified in this experiment were combined in a protein complex database (http://yeast-complexes.embl.de). This rich database provides a blueprint of protein-protein interactions in yeast, however, as these types of databases are constantly being updated and reevaluated, it is incomplete. For example the http://yeast-complexes.embl.de dataset was generated using exponentially growing cells, and thus did not capture cell cycle dynamics of protein abundance and interactions. Similar concerns affect the interpretation of databases compiling interaction information including affinity based mass spectrometry results, two-hybrid analysis and other approaches as found in the BOND (http://bond.unleashedinformatics.com/, Thomson Scientific) and BioGRID (http://www.thebiogrid.org/database) 13 databases.
The composition of protein complexes is determined both by the availability of subunits and their post-translational modifications 12, 14. A common motif is regulation by protein phosphorylation 14, 15. A third of eukaryotic proteins may be subject to phosphorylation 16, but low stoichiometry contributes to a low abundance of most phosphorylated species. Phosphorylation is readily reversible and highly dynamic, which further complicates its detection. Proteomic analysis of unfractionated cellular proteins typically yields only a tiny fraction of the expected phosphorylated species. Beyond their low abundance, factors such as increased hydrophilicity, lower pK and inefficient fragmentation conspire to limit detection of phosphopeptides by conventional ESI LC-MS/MS approaches.
One successful approach to increasing the coverage of the phosphoproteome has been to enrich phosphorylated species prior to proteomic analysis (reviewed in 17, 18). The best characterized methodologies are based on isolation of phosphopeptides after proteolysis. Anti-phosphotyrosine 19, 20 and other phosphoepitope antibodies 21, 22 are effective reagents, but as yet, a robust antibody-based approach to capturing serine or threonine phosphorylated peptides remains to be described. A chemical approach exploits beta-elimination of the phosphate moieties of phosphoserine and phosphothreonine to permit chemical activation and tagging 23, 24, 25 facilitating tethering to a solid support. The phosphate moiety itself can be used for affinity purification as by immobilized metal-affinity chromatography (IMAC)26, 27 or on titanium dioxide (TiO2) 28, 29. A recent comparison of three common phosphopeptide isolation methods, beta elimination with covalent tethering, IMAC and TiO2, showed that while each method recovered a large and reproducible population of phosphopeptides from a complex mixture, only a small fraction of these could be isolated by two or more of the methods 25. These results argue that we remain far from achieving comprehensive analysis of a phosphoproteome in a single experiment, even with abundant samples.
A major limitation in this approach to phosphoproteomics is the reliance on a single phosphopeptide as a tag for identification of the phosphorylated protein. Even though methods and equipment vary widely among laboratories, only a small fraction of the expected peptides are reliably and reproducibly detected and from a complex mixture and these are termed proteotypic peptides. Determinants of detectability include the length, hydrophobicity, charge and amino acid composition of these peptides 30, 31. Importantly, phosphopeptides fail to satisfy many of these criteria.
An alternative to enrichment of phosphopeptides is to purify the phosphoproteins, exploiting one or another phospho-affinity approaches 19, 32–38. Phosphoprotein profiling, has the potential practical advantage of improving the statistical significance of protein identification. Intact proteins maintain characteristic properties such as their native molecular weight, allowing use of polyacrylamide gel separations. After proteolysis, the proteins can be recognized by multiple peptides, including any proteotypic peptides (30) they may contain. However, phosphoprotein enrichment suffers from the drawback that phosphorylation sites are unlikely to be identified, raising the possibility of misidentification. In turn, performing phosphoprotein enrichment under native conditions is likely to co-purify any associated, nonphosphorylated proteins, further complicating analysis. Nonetheless, phosphorylation site prediction and the emergence of large databases of phosphopeptides as well as databases of known protein-protein interactions and protein complexes permit another method of validation, through data mining approaches.
Several phosphopeptide enrichment strategies have been applied with limited success to enrichment of native or denatured phosphoproteins. Chemical methods are likely to offer only limited utility while IMAC has demonstrated some value 32. Enrichment of phosphotyrosine containing proteins via anti-phosphotyrosine antibodies has been successful 19, 33–35 but, as with peptides, anti-phosphoserine and/or -threonine antibodies have proven to be less successful 36. Recently, phosphotyrosine immunoprecipitation was used to identify proteins involved in erythropoietin receptor (EPOR) signaling 39. The phosphoprotein enriched sample was split and analyzed by two different proteomic strategies, 1-D electrophoresis with LC-MS/MS (1-D LC-MS/MS) or 2-D gel electrophoresis, silver staining and identification of proteins by MALDI-TOF (2-D MALDI). The majority of proteins identified using the 2-D MALDI technique were highly abundant housekeeping proteins and no proteins were identified from the EPOR-dependent pathways. The 1-D LC-MS/MS method, however, allowed for identification of multiple lower abundance proteins known to be part of the EPOR-dependent pathways, but also a number of new candidates 39.
Several commercial kits for native or denatured phosphoprotein enrichment are available including PhosphoProtein Purification Kit (Qiagen), Pro-Q Diamond Phosphoprotein enrichment kit (Invitrogen/Molecular Probes) and BD Phosphoprotein enrichment kit (BD Biosciences). Phosphoproteins can be visualized on SDS-PAGE gels using the Pro-Q Diamond fluorescent stain from Invitrogen. Pro-Q Diamond detects phosphate groups attached to tyrosine, serine or threonine residues, with a sensitivity limit between 1–16 ng/protein spot, depending on the phosphorylation state of the protein 40–42. Makrantoni et al 38 combined the PhosphoProtein Purification Kit from Qiagen with 2-D gel electrophoresis and Pro-Q Diamond fluorescent phosphoprotein staining to show specific enrichment of phosphoproteins. Furthermore, the authors excised 13 protein spots from the gel and used matrix assisted laser desorption ionization (MALDI) mass spectrometry to identify the proteins; 11 of which were known phosphoproteins 38. Metodiev et al 37 used the PhosphoProtein Purification Kit from Qiagen and MALDI to identify several proteins from human and yeast sources that were known phosphoproteins. Another study showed that phoshpoproteins from undifferentiated and early differentiated mouse embryonic stem cells could be enriched using the Qiagen Phosphoprotein purification kit and identified over 30 proteins using 2-D PAGE and either MALDI-MS/MS or LC-MS/MS that exhibited differential recovery from the column, indicating a change in phosphorylation status 43.
To test the potential for integrating phosphoprotein chemistry, proteomics and bioinformatics to identify phosphoproteins and phosphoprotein complexes on a systems level, we utilized the Pro-Q Diamond phosphoprotein enrichment kit and phosphoprotein stain from Invitrogen/Molecular Probes. We subjected a G2/M phase whole cell yeast extract to phosphoaffinity purification followed by gel electrophoresis and reverse phase HPLC combined with tandem electrospray mass spectrometry (PA-GeLC-MS/MS). By querying several freely available, large scale databases we found that most proteins had previously been identified as phosphoproteins and/or as components of protein complexes containing phosphoproteins. In summary, we have made progress toward a phosphoprotein profiling method that permits phosphorylation to be studied on a global scale and offers insight into the relationship between phosphorylation of proteins and their association with other proteins in multisubunit complexes.
Materials and Methods
Reagents
The Pro-Q Diamond Phosphoenrichment kits used for this study were kindly provided by Invitrogen/Molecular Probes. Pro-Q Diamond stain and NuPAGE 2–12% gradient gels were obtained from Invitrogen/Molecular Probes, HALT phosphatase inhibitor cocktail from Pierce, Vivaspin centrifugal concentrators from Vivascience, sequencing grade modified trypsin from Promega, Lys-C protease from Princeton Separations, and Zorbax 300SB-C18 reversed phase HPLC columns (dimensions: 3.5 μm packing, 150 mm × 75 μm) from Agilent. Other reagents were purchased from Sigma-Aldrich.
Phosphoprotein enrichment
The lysate/sample was diluted and loaded onto a column pre-equilibrated with Pro-Q Diamond resin. Fresh Pro-Q Diamond resin was used for each experiment. The column was washed and phosphoproteins eluted in buffers supplied with the Pro-Q Diamond phosphoenrichment kit, with all steps performed at 4° C. The protein sample or lysate, flow-through from wash step and eluate were concentrated by centrifugation in 10 kDa MWCO Vivaspin concentrators at 4° C and washed with 50 mM Tris pH 7.5. The proteins were precipitated using methanol/chloroform/water as described in the Pro-Q Diamond phosphoenrichment kit, resuspended in 4X Laemmli buffer and boiled for 10 min before loading on NuPAGE 2–12% gradient gels. The gel was stained with Coomassie for proteins and/or with Pro-Q Diamond stain for phosphoproteins, following manufacturer’s instructions. Coomassie stained gels were scanned with a Microtek Scan-Maker 6800. Pro-Q Diamond stained gels were visualized with a Bio-Rad Molecular Imager FX.
Phosphorylated GST-SH3n-Abltide
Purification of Abl tyrosine kinase substrate, GST-SH3n-Abltide, and in vitro phosphorylation by c-Abl kinase was as described 44, except that 100 μCi of [γ-32P]-ATP was added and the reaction was scaled up 7-fold. The reaction was loaded onto a Vivaspin centrifugal concentrator with a molecular weight cutoff (MWCO) of 10 kDa. The retentate was collected and subjected to phosphoprotein enrichment as above. Fractions from the lysate, flow-through, wash and eluate of the Pro-Q Diamond column were scanned for radioactivity by Geiger counter, pooled, precipitated and subjected to SDS-PAGE on NuPAGE 2–12% gradient gels. Protein was imaged using Pierce Imperial Coomassie Stain and incorporated radioactivity was imaged with a GE Storm 860 phosphorimager.
K562 lysate preparation
K562 cells were grown in suspension at 37° C and 5% CO2 in RPMI 1640 medium (Sigma) containing 10% heat-inactivated fetal bovine serum (Gemini), 1% penicillin/streptomycin and 0.3 mg/ml L-glutamine. Whole cell lysates were prepared from 5 × 106 cells in 0.5 ml Pro-Q Diamond phosphoenrichment kit lysis buffer with 1:1000 dilution of Pierce HALT phosphatase inhibitor cocktail; a proprietary mixture of sodium fluoride, sodium orthovanadate, sodium pyrophosphate and sodium glycerophosphate. The supernatant was collected, and protein yields were determined by Bradford analysis using Bio-Rad protein assay reagent. Phosphoprotein enrichment was performed as described above.
Yeast lysate preparation
Yeast cells were treated with 30 μg/ml nocodazole for 3 hours at 30° C to arrest cells in G2/M phase and then harvested by centrifugation at 3,000 × g for 5 minutes. The pellet was resuspended in ice cold Pro-Q Diamond phosphoenrichment kit lysis buffer or a buffer composed of 50 mM HEPES pH 7.5, 10% glycerol, 150 mM NaCl, 0.1% NP-40 supplemented with 1 μM okadaic acid. The sample was subjected to bead beating (3 × 30 seconds with 60 seconds on ice) on a BioSpec Products Mini-Bead-Beater-8. After centrifugation at 14,000 × g for 30 min at 4° C, the supernatant was collected, yield was determined with Bio-Rad protein assay reagent and phosphoprotein enrichment was performed as above.
Pro-Q eluate from nocodazole arrested yeast extract (1.2 mg) was further fractionated on a 100 kDa MWCO Vivaspin centrifugal concentrator. The retentate was collected as the >100 kDa sample. The flow-through was concentrated on a 10 kDa MWCO Vivaspin centrifugal concentrator and the retentate was collected as the <100 kDa sample. Both the >100 kDa and <100 kDa samples were precipitated and loaded onto a NuPAGE 2–12% gradient gel.
Western blotting
Samples were separated on 12% NuPAGE gels and transferred to nitrocellulose membranes according to standard methods. Uniform sample loading and transfer were confirmed using Pierce Memcode reversible protein stain kit. Membranes were blocked in 10% bovine serum albumin (BSA) for 1 hour at 25° C. The membrane containing K562 cell extract was probed with 1:1000 4G10 anti-phosphotyrosine primary antibody (Upstate Cell Signaling Solutions) in 5% BSA at 25° C for 1 hour and detected with 1:5000 horseradish peroxidase (HRP)-conjugated anti-mouse IgG secondary antibody (GE-Amersham) in 5% BSA for 30 min. The membrane from yeast cell extract was probed with 1:1000 anti-phosphothreonine primary antibody (Upstate Cell Signaling Solutions) in 5% Carnation dried milk at 25° C overnight and 1:3000 HRP conjugated secondary antibody in 5% milk for 1 hour. Blots were developed using Supersignal WestPico chemiluminescent substrate (Pierce), recorded on autoradiography film and scanned with a Microtek Scan-Maker 6800 at 600ppi resolution.
In-gel tryptic digestion and mass spectrometry
Gel lanes to be analyzed were excised from SDS-PAGE gels by razor blade and divided into twelve ~ 1 cm slices. Each slice was then further divided into ~ 1 mm3 pieces. Each section was washed in water and completely destained using 100 mM ammonium bicarbonate in 50% acetonitrile. A reduction step was performed by addition of 100 μl 50 mM ammonium bicarbonate pH 8.9 and 10 μl of 10 μM TCEP (Tris(2-carboxyethyl)phosphine HCl) at 37 °C for 30 min. The proteins were alkylated by adding 100 μl 50 mM iodoacetamide and allowed to react in the dark at 20° C for 40 min. Gel sections were first washed in water, then acetonitrile, and finally dried by SpeedVac for 30 min.
Digestion was carried out using 20 μg/ml sequencing grade modified trypsin in 50 mM ammonium bicarbonate. Sufficient trypsin solution was added to swell the gel pieces, which were kept at 4° C for 45 min and then incubated at 37° C overnight. Sections containing proteins larger than 150 kDa were pre-digested with 0.25 mg/ml Lys-C in 6–8 M urea overnight at 25° C, diluted to final concentration of < 2 M urea then digested with trypsin as described above. Peptides were extracted from the gel pieces with 5% formic acid.
The peptides were loaded onto the Zorbax 300SB-C18 reversed phase column on an online Dionex Ultimate multidimensional nanoHPLC system and separated with a gradient of 5% to 60% acetonitrile in 0.1% trifluoroacetic acid over 110 minutes and then analyzed by electrospray tandem mass spectrometry (LC-MS/MS) on either an Agilent XCT ion trap or a Thermo Instruments LTQ FT hybrid ion-trap/FTICR mass spectrometer.
The Agilent XCT ion trap was operated in Ultrascan Mode at a speed of 26000 m/z/sec with the following settings: Skimmer at 40V, Cap exit at 65V, Capillary at 1700 V, and End Plate Offset at −500V. After a 150 ms MS scan over 200–2200m/z with 3 averages, the 5 most intense ions above an intensity of 6000 were subjected to MS/MS. The MS/MS was set to exclude single charged ions and prefer double charged ions. Active exclusion was used excluding ions after 2 scans for 1 min.
The LTQ FT was operated in positive ion mode, and parent ions were selected for fragmentation by data-dependent analysis (five most abundant ions in each cycle): 1 scan LTQ FT-MS (m/z 400–2000) and maximum 5 scan LTQ-MS/MS (m/z 50–2000), 60-second dynamic exclusion. A normalized collision energy of 35 was used for low energy CID MS/MS of peptide ions.
Database searching and criteria for protein identification
Mascot (version 2.1.01, Matrix Science) and X! Tandem (version 2007.01.01.1, www.thegpm.org) were used to identify proteins based on MS/MS spectra. Mascot was set up to search the NCBInr_20060910 database (selected for S. cerevisiae, 11101 entries) assuming the digestion enzyme trypsin. A fragment ion mass tolerance of 1.0 Da and a parent ion tolerance of 0.6 Da was specified for the Agilent XCT data. A fragment ion mass tolerance of 1.0 Da and a parent ion tolerance of 0.2 Da was specified for the LTQ FT data. Oxidation of methionine, N-formylation of the amino terminus and iodoacetic acid derivative of cysteine were specified as variable modifications.
X! Tandem was set up to search the scd.fasta.pro database (selected for S. cerevisiae, 6794 entries) also assuming trypsin. X! Tandem was searched with a fragment ion mass tolerance of 0.60 Da and a parent ion tolerance of 10.0 PPM. Iodoacetamide derivative of cysteine was specified as a fixed modification. Deamidation of asparagine and glutamine, oxidation of methionine and tryptophan, sulphone of methionine, tryptophan oxidation to formylkynurenin of tryptophan and acetylation of lysine and the amino terminus were specified as variable modifications.
Scaffold (version Scaffold-01_06_00, Proteome Software) was used to visualize and validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 90% probability as specified by the Peptide Prophet algorithm 45. Protein identifications were accepted if they could be established at greater than 95% probability and contained at least one identified peptide. Proteins identified with only one peptide were manually confirmed. Protein probabilities were assigned by the Protein Prophet algorithm 46. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Results
Phosphorylated proteins are bound to and eluted from Pro-Q Diamond resin
To evaluate the Pro-Q Diamond affinity media (Invitrogen/Molecular Probes) as a phosphoprotein enrichment tool, we first tested the resin with a single model protein, GST-SH3n-Abltide 44. We have previously shown that GST-SH3n-Abltide 44, a synthetic 35 kDa protein affinity purified from bacteria consisting of glutathione S transferase, the 56 residue SH3n domain of CrkL and the substrate peptide Abltide (EAIYAAPFAKKK) can be phosphorylated quantitatively by the tyrosine kinase c-Abl in vitro. Our prior work suggests that this protein can only be phosphorylated by c-Abl on a single site, the Tyr residue in the Abltide peptide. By including [γ-32P]-ATP in the reaction as a tracer, the phosphorylated protein could be followed through Pro-Q Diamond fractionation. Aliquots of the kinase reaction, column flow-through, wash, and eluate were subjected to SDS-PAGE and protein detection by Coomassie stain and phosphorimaging (Figure 1). 32P-labelled GST-SH3n-Abltide formed in the kinase reaction was applied to the resin. No radioactivity was detected in the flow-through, indicating that the Pro-Q Diamond resin bound most if not all of the phosphorylated protein. Nearly all the radioactivity was recovered in the eluate and very little remained on the beads after elution. These results show that the Pro-Q Diamond resin selectively and quantitatively binds GST-SH3n-Abltide, a model singly phosphorylated protein.
Figure 1. Pro-Q Diamond phosphoenrichment resin selectively binds phosphorylated protein.

Bcr-Abl kinase was incubated with GST- SH3n-Abltide and [γ-32P]-ATP to allow for phosphorylation of GST- SH3n-Abltide. The solution was passed over a column containing Pro-Q Diamond resin and samples were taken from the loaded sample (L), flow-through (FT), elution (E) and remaining beads after elution (Beads). (a) Each fraction was subjected to SDS-PAGE and the phosphoprotein detected using a phosphorimager. The two E lanes represent the same sample loaded twice, first a 5uL aliquot of the sample (1X) and then with a 4-fold higher amount of sample or 20 uL (4X). (b) Total phosphorylation in each sample after accounting for sample dilution and loading. The star (*) represents that only a small sample (3%) of the loaded sample (L) was loaded on the gel. The remainder of sample (L) was used for the Pro-Q phosphoenrichment column and is represented in lanes FT, E, E2 and Beads.
Enrichment of phosphotyrosine and phosphothreonine containing proteins from complex samples
To determine if the Pro-Q Diamond resin enriches for phosphoserine/threonine proteins, we tested whole cell extracts of yeast, which has little or no tyrosine phosphorylation. The extract was passed over the Pro-Q Diamond resin and tested for the presence of phosphothreonine (Figure 2A) by Western blotting. The yeast extract shows enrichment of phosphoproteins in the eluate and little if any phosphoproteins in the flow-through fraction. To determine if Pro-Q Diamond also enriches for phosphotyrosine containing proteins we tested whole cell extract from K562 leukemia cells. Whole cell extract was passed over the Pro-Q Diamond resin and tested for the presence of phosphotyrosine (Figure 2B) by Western blotting. Like the yeast extract, the K562 extract shows enrichment of phosphoproteins in the eluate and little if any phosphoproteins in the flow-through fraction. These experiments were repeated twice resulting in identical staining patterns (data not shown). Our data suggest that Pro-Q Diamond resin provides a reasonably efficient enrichment of both phosphothreonine and phosphotyrosine containing proteins from complex samples.
Figure 2. Enrichment of phosphothreonine and phosphotyrosine containing proteins from complex samples using Pro-Q Diamond resin.
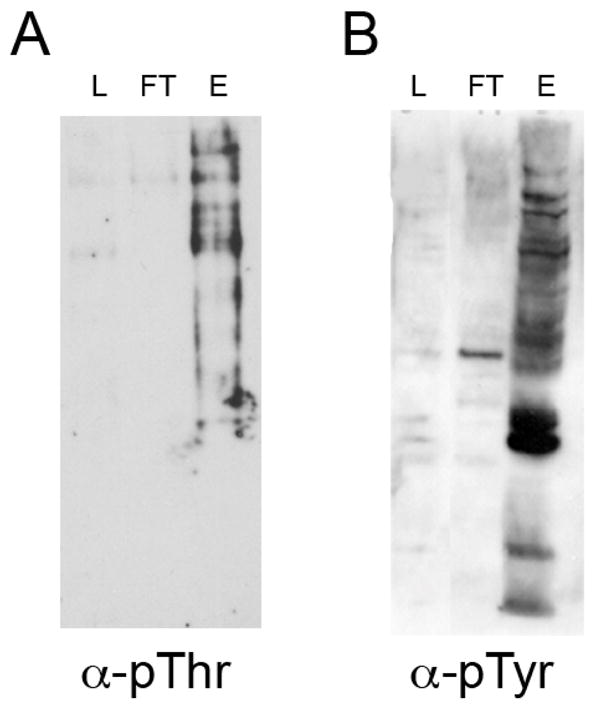
Cell lysates (L) were passed over a column containing Pro-Q Diamond resin, the flow-through (FT) collected and phosphoproteins eluted (E). Each sample was subjected to SDS-PAGE and Western blotting with (A) anti-phosphothreonine (α-pThr) for the yeast sample or (B) anti-phosphotyrosine (α-pTyr) for the K562 human cell line. In each case, phosphorylated species are bound to and eluted from the Pro-Q resin with minimal amounts of the phosphorylated species in the flow-through sample.
To evaluate the degree of nonspecific binding, we loaded yeast extract onto the beads used to make Pro-Q Diamond resin, CL-4B, and then eluted with the Pro-Q elution buffer. Pro-Q Diamond and Coomassie staining revealed abundant phosphoproteins in the flow-through fraction and no discernable protein bands in the eluate (data not shown), indicating that the Pro-Q purification is specific for phosphoproteins and their binding partners.
Phosphatase inhibitors
Many phosphatase inhibitors are competitive with phospho-amino acids for binding to the catalytic site. One concern is that the Pro-Q affinity column may also be subject to saturation by such phospho-amino acid mimics. Indeed, we found that excess phosphatase inhibitors apparently bound to the column competitively with phosphoproteins. For example, adding the recommended amount of Halt Inhibitor cocktail (Pierce), a proprietary mixture of sodium fluoride, sodium orthovanadate, sodium pyrophosphate and sodium glycerophosphate, to K562 extracts resulted in significant detection of phosphotyrosine proteins in the flow-through and decreased amounts in the eluate (data not shown). Nonetheless, omission of phosphatase inhibitors could result in loss of binding due to dephosphorylation, suggesting the need for balancing phosphatase inhibition against loss of resin capacity.
We analyzed the binding of K562 extract to Pro-Q Diamond resin in the presence of Halt Inhibitor cocktail diluted 10-fold. A strong phosphotyrosine signal was detected in the eluate of ProQ resin with a very faint signal in the flow-through (data not shown, and Figure 2B). We next analyzed the binding of yeast extract to Pro-Q Diamond resin in the presence of several specific inhibitors of the serine/threonine phosphatases Protein Phosphatase 1 (PP1) and 2 (PP2) 47–49. The inhibitors were added at values 5–100-fold above the experimentally determined IC50 values. Calyculin at 10 nM, Microcystin at 20 nM, and Cantharadin at 2 μM each individually resulted in reduced binding and elution of phosphoproteins from the column. Okadaic acid at 1 μM, however, did not impair binding to the column (data not shown, and Figure 2A). Okadaic acid inhibits PP2A at concentrations as low as 1–2 nM but PP1 type phosphatases are much less sensitive; complete inhibition is observed only at 1 μM Okadaic acid 50. Thus, Okadaic acid at 1 μM was used in yeast whole cell extract preparations and 10-fold diluted Halt Inhibitor Cocktail was used for phosphotyrosine containing extracts such as K562.
Identification of phosphoproteins and associated proteins enriched from G2-arrested yeast
To visualize phosphoproteins enriched from the G2-arrested yeast, the eluate was subjected to 1-D SDS-PAGE and stained for phosphoproteins using Pro-Q Diamond stain and then for total protein with Coomassie stain. Pro-Q staining revealed a scarceness of phosphoproteins in the flow-through fraction versus abundance in the eluate while Coomassie stained proteins were present in both flow-through and eluate (Figure 3). To identify proteins, the gel was sliced into 12 even sections that were subjected to tryptic digestion and extraction of peptides. The peptides were separated and detected on an Ion Trap XCT LC-MS/MS (Agilent) and data analysis was performed with Mascot (Matrix Science) and X!Tandem (The Global Proteome Machine Organization) software. Figure 4 shows a mass spectrum of a peptide originating from protein Cor1 (inset) and the tandem mass spectrum of this peptide. Four other peptides were identified from Cor1, confirming the identification. The runs from each section were compiled and analyzed with Scaffold (Proteome Software) software, yielding identification of 131 proteins with 90% protein confidence levels based upon 95% peptide confidence with a minimum of 1 peptide per identification (Supplemental Table 1). More stringent criteria of 99% protein confidence levels with a minimum of 2 peptides per identification resulted in identification of 108 proteins. The identification software (Mascot, X!Tandem) suggested the presence of several phosphopeptides. However, manual inspection revealed low quality spectra and/or poor fragmentation in all of the proposed phosphopeptide MS/MS spectra (data not shown). Instead, mining proteomic databases and literature revealed that over half of the 131 identified proteins have previously been described as phosphoproteins based on identification of the site of phosphorylation (Supplemental Table 1 and 51–55). The remaining proteins could contain novel phosphoproteins since phosphoenrichment protocols are not comprehensive as evidenced by the non-overlapping results of published phosphopeptide studies.
Figure 3. PA-GeLC-MS/MS results in apparent enrichment of phosphoproteins and identification of several known phosphoproteins.

Cell lysates (L) were passed over a Pro-Q Diamond column, the flow-through (FT) collected and phosphoproteins eluted (E) and subjected to SDS-PAGE. The gel was stained with Coomassie for proteins and Pro-Q Diamond stain for phosphoproteins. The peptides were eluted from the gel with trypsin and subjected to LC-MS/MS. Phosphoproteins are selectively bound and eluted from the phosphoaffinity column. A sampling of proteins is listed to the right, grouped by which fraction they were identified in and the range of molecular weight of proteins identified within that fraction.
Figure 4. An example of peptide mass spectra used for protein identification.

The insert (top right) shows the mass spectrum of the precursor ion. The remainder of the figure depicts the tandem mass spectrum of the same peptide, LAAQIFGSYNAFEPASR. The identified y- and b-fragments are indicated in the sequence above the spectrum. Four additional peptides identified the Cor1 protein.
Enrichment of phosphoprotein complexes
Phosphoprotein enrichment was performed under native conditions to retain proteins that are components of phosphoprotein complexes. Thus, it is likely that several of the proteins identified are isolated as non-phosphorylated components of phosphoprotein complexes. To investigate whether phosphoprotein complexes remained intact under the experimental conditions we first performed a gene annotation analysis using the BioGRID protein interaction database. Osprey software was used for visualization of protein-protein interaction networks 56. The analysis reveals several functional clusters and many potential protein-protein interactions within a subset of our 131 identified proteins (Figure 5A). The functional subsets include ribosomal proteins, metabolic enzymes, proteins involved in translation, RNA polymerase components, heat shock proteins, and proteasomal proteins.
Figure 5. Gene annotation analysis of enriched proteins reveals clusters of functionalities.

Protein interactions within a subset of the Pro-Q Diamond enriched proteins were obtained from the BioGRID database 13 and are indicated by solid lines (http://www.thebiogrid.org/). Osprey software was used to generate the figures, where each node represents a single protein 56. (A) Functionalities identified include the proteasome (lower left), ribosome (upper left), heat shock proteins (lower right), RNA polymerase (upper right) and proteins involved in translation (middle). The nodes are in (A) are color coded (grayscale) according to gene annotation. (B) Protein protein interactions between a subset of the proteins identified from the 20S subunit of the proteasome.
Utilizing protein-protein interactions databases, we then queried our data against protein-protein interactions within the proteasome. One of the components, Pre2, has 39 known binding partners in BioGRID, encompassing several protein complexes. Our dataset contains 11 of these binding partners, including Pre1, Pre3, Pre5, Pre7, Pre8, Pre9, Pre10, Pup2, Scl1 and Rpn5 (Supplemental Table 1, Figure 5B). When studying individual affinity purifications of a Pre proteins complex we obtained even better coverage. Our dataset contained 10 of 13 identified binding partners for Pre2 in the BOND database and 9 out of 13 interactions in the http://yeast-complexes.embl.de database for Pre10 57. Examining the components of the 20S core particle of the proteasome on the Saccharomyces Genome Database (http://www.yeastgenome.org) reveals 15 proteins of which we found 14 in this study. The core particle can be divided into the alpha and beta subunit complexes. All of the components of these subunit complexes are identified in our study with the alpha subunit containing Pre10, Pre5, Pre6, Pre8, Pre9, Pup2 and Scl1 and the beta complex containing Pre1, Pre2, Pre3, Pre4, Pre7, Pup1 and Pup2. Thus our affinity purification is highly enriched in proteasome components even though only three of these proteins have previously been described as phosphoproteins: Pre5, Pre7 and Pup2. It is likely that phosphorylated Pre5, Pre7 and/or Pup2 proteins bound to the Pro-Q Diamond resin and other components of the protein complex were co-purified in the phosphoprotein enrichment.
We repeated the phosphoprotein enrichment using a lysis buffer (50 mM HEPES pH 7.5, 10% glycerol, 150 mM NaCl, 0.1% NP-40) similar in composition to the buffers used in Tandem Affinity Purifications (TAP) for identification of protein complexes in yeast 58. This lysis buffer is not as optimized for the phosphoprotein resin, and did result in an increase of phosphoproteins in the flow-through fraction (data not shown). However, since this lysis buffer is optimized to retain protein-protein interactions at neutral pH and a sizeable number of phosphoproteins were still bound and eluted from the resin we decided to accept this decrease in binding. To identify which proteins are components of protein complexes we added a fractionation step based on size. Protein complexes composed of multiple proteins are typically large and would be expected to be retained by a 100 kDa molecular weight cut-off (MWCO) centrifugal concentrator, while monomeric proteins and protein complexes smaller than 100 kDa should filter through. The retentate and flow-through samples were subjected to 1-D SDS PAGE and analyzed by Coomassie staining (Figure 6). Preparing the samples for SDS-PAGE by boiling in SDS denatured the proteins and resulted in dissolution of all protein complexes allowing each protein to migrate on the gel according to the monomeric molecular weight. Most proteins were found in the retentate fraction despite that fact that the vast majority of the proteins have a monomeric MW of less than 100kDa (Figure 6).
Figure 6. Separation of proteins and protein complexes by size.
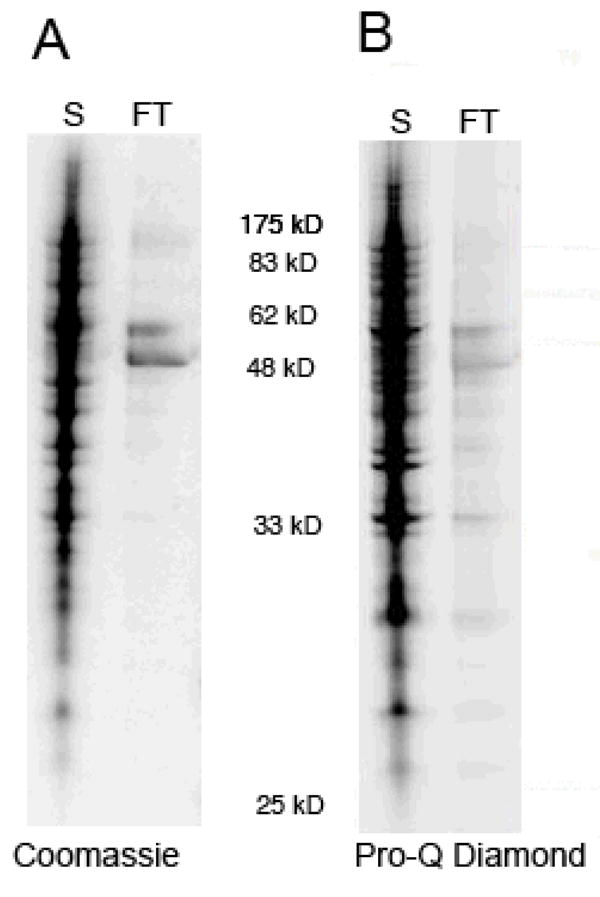
Yeast extract was enriched for phosphoproteins and associated proteins using Pro-Q Diamond resin (see Figure 3) and then separated by size using a Vivaspin centrifugal concentrator with a 100kDa MW cutoff. The supernatant (S) or retentate of the concentrator was collected and contains protein complexes and proteins larger than 100 kDa. The flow-through (FT) contains the proteins and protein complexes that are smaller than 100 kDa. The samples were denatured and subjected to SDS-PAGE and the gel stained with Pro-Q Diamond stain (A) and Coomassie stain (B). Molecular weight markers are indicated between gels.
Proteins were identified by mass spectrometry as described above except a newer instrument was used, the FT LTQ (Thermo Finnigan). Using these methods, 237 proteins were identified in the retentate (>100 kDa fraction) using 90% protein confidence levels based upon 95% peptide confidence with a minimum of two peptides per identification (Figure 6, Supplemental Table 2). To avoid the need for manual interpretation and increase confidence levels at least two peptides were required for each protein identification. Applying the same conditions to the flow-through sample (<100 kDa fraction) resulted in identification of 45 proteins (Supplemental Table 3). The total amount of proteins identified was 250 with some proteins identified in both sections. This number is nearly double the amount of proteins identified in the original enriched sample (131 proteins, Figure 3, Supplementary Tables 1). Several factors could have contributed to the increase in proteins identified. First, we moved the mass spectrometry analysis to a newer, higher performance instrument (from an Agilent XCT Ion Trap to a Thermo LTQ FT). Second, we used a greater amount of starting material and finally, we increased the fractionation of the sample (additional separation into >100kDa and <100kDa fractions). Comparing the two experiments reveals that over 80% of the proteins are identified in both experiments for two peptide identifications and 89% when identifications with one peptide were allowed. Thus our PA-GeLC-MS/MS method is highly reproducible.
As expected, the retentate (>100 kDa) contained several large proteins, including 23 proteins ranging between 100–250 kDa (Supplemental Table 2). Only three proteins in the flow-through fraction were larger than 100 kDa: Kap123, Yef3 and Hsp104 at 123, 116 and 102 kDa, respectively (Supplemental Table 3). It is not surprising that proteins close in size to 100 kDa are able to travel through the membrane given that the molecular weight cut-off (MWCO) is assigned based on 90% retention of a standard with that molecular weight. Indeed, these three proteins are detected in both the flow-through and the retentate fractions.
Proteins in the retentate fraction are highly connected, with several known binding partners (Figure 7). Non-complexed proteins in the retentate would include monomeric and homodimeric proteins with molecular weights around 100 kDa including Arg5,6, Vtc2, Vtc3, Ala1 and Cdc3. The heterodimeric proteins Pdi1-Ero1, Eno2-YIL091C, Cor1-Qcr2 and Ses1-Dre2 are around 100 kDa in size. Individual nodes with no lines connecting to them indicate that the protein has not been described to bind to any of the other proteins identified in this study. Several individual nodes, however, including Rps18B, Rpl27B, Bfr2 and Qcr7 are small proteins and thus should not have been retained as monomers in the retentate. This could be due to the fact that the proteins are a part of a protein complex that has not been previously reported. Alternatively, they might be part of a known complex but the interacting partner(s) was not identified in our sample. One caveat to our experimentation is that the sample is concentrated on a 10 kDa MWCO centrifugal concentrator, resulting in loss of any proteins smaller than 10 kDa. Several ribosomal proteins are smaller than 10 kDa including Rpl29, Rpl39, Rpl41A/B, Rps29A/B and RPS30A/B. Indeed, one of the individual nodes mentioned above has been found to bind to both Rpl29 and RPS30A.
Figure 7. Protein interactions within the >100 kDa sample reveal a high density of protein-protein interactions.

Phosphoproteins and phosphoprotein complexes were obtained by passing yeast extract over the Pro-Q Diamond resin. The sample was then separated by size using a 100 kDa centrifugal concentrator. The retentate contained proteins and protein complexes larger than 100 kDa. Figure 8 shows the proteins found in the flow-through indicating a size smaller than 100 kDa. Osprey software was used to generate a figure depicting all possible interactions within the sample, where each node represents a single protein 56. Proteins are represented by grey and black nodes where black nodes represent proteins previously reported to be phosphorylated (see Supplemental Table 2). Protein interactions from the entire retentate of the 100 kDa centrifugal concentrators were obtained from the BioGRID database 13 and are indicated by solid lines (http://www.thebiogrid.org/).
Since monomeric proteins should be purified based on their phosphorylation state we would expect the <100 kDa fraction to contain a high percentage of phosphoproteins. This can be seen visually in Figure 8, where black nodes represent proteins previously reported to be phosphorylated and grey nodes represent nonphosphorylated proteins. Indeed over 78% of the proteins identified in the <100 kDa fraction have previously been identified as phosphoproteins (Supplementary Table 3). On the other hand, the >100 kDa fraction should contain phosphoprotein complexes and as such should have a higher percentage of non-phosphorylated proteins and thus a lower overall percentage of phosphorylated proteins. Indeed, the percentage of phosphoproteins in the >100 kDa fraction is much lower at 43% (Figure 7, Supplemental Table 2).
Figure 8. The majority of proteins in the <100 kDa sample are known phosphoproteins.
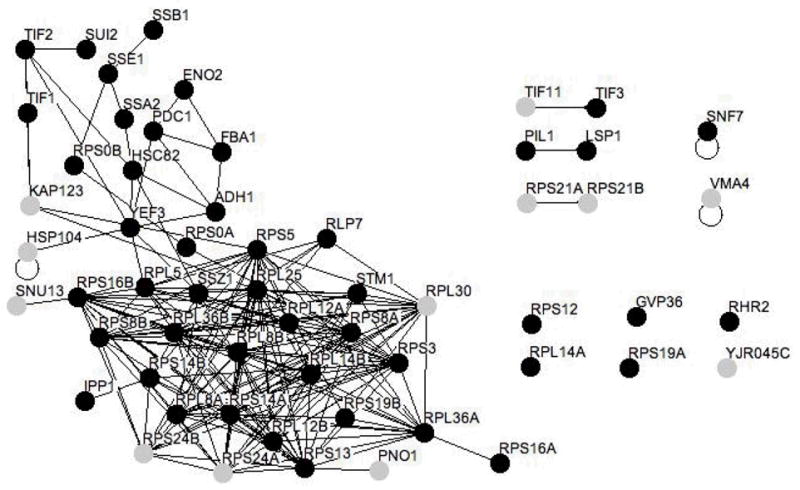
Phosphoproteins and phosphoprotein complexes were obtained by passing yeast extract over the Pro-Q Diamond resin. The sample was then separated by size using a 100 kDa centrifugal concentrator. The figure shows proteins found in the flow-through of the concentrator indicating a size smaller than 100 kDa. Figure 7 depicts the retentate, which should contain proteins and protein complexes larger than 100 kDa. Osprey software was used to generate a figure depicting all possible interactions within the sample, where each node represents a single protein 56. Proteins are represented by grey and black nodes where black nodes represent proteins previously reported to be phosphorylated (see Supplemental Table 3). Protein interactions within the entire retentate of the 100 kDa centrifugal concentrator were obtained from the BioGRID database (http://www.thebiogrid.org/) 13 and are indicated by solid lines.
A global analysis of protein expression levels in yeast has been performed in which the authors TAP- and/or GFP-tagged every protein and calculated the copy number of proteins in a cell 59. Using these measurements on the proteins identified from Figure 3 reveals an average abundance of 123,000 copies per cell (Supplemental Table 1). Separating the enriched phosphoproteins into two fractions (Figure 6) and analyzing each fraction separately resulted in a decrease of average abundance to 73,000 copies per cell (Supplemental Table 2). The lowest abundant protein was Rpl3 at 450copies per cell (Supplemental Table 1) and from the fractionated sample, YIL091C at 172 copies per cell (Supplemental Tables 2–3). Comparing the abundance of proteins within the two sections revealed a similar distribution of abundances with the exception of an absence of low abundance proteins (<1000 copies) in the <100 kDa fraction (Lane FT from Figure 6).
Discussion
Recent proteomic studies have populated databases with extensive compilations of cellular phosphoproteins and phosphorylation sites and similarly deep coverage of the subunit compositions and interactions in multi-protein complexes. We describe a method, PA-GeLC-MS/MS, to identify candidate phosphoprotein complexes by combining phosphoaffinity chromatography, size separation, 1-D SDS-PAGE, mass spectrometry and informatics analysis. On a global scale, we show successful enrichment of 250 phosphoproteins and associated proteins from G2-phase yeast using the commercially available Pro-Q Diamond phosphoprotein affinity resin. Comparing the identified proteins to several large-scale phosphoproteomic studies reveals that about half were previously identified as phosphoproteins, many in several different studies. To identify components of phosphoprotein complexes we fractionated the enriched sample under non-denaturing conditions using a centrifugal concentrator into components larger (retentate) or smaller (flow-through) than 100 kDa. The results show that the concentrator provided good separation with only three proteins larger than 100 kDa found in the flow-through. Furthermore, these proteins were also found in the retentate suggesting that their size was too close to the molecular weight exclusion size of the membrane for efficient separation. As expected all larger proteins were found in the retentate, including Acc1 and Ura2 at around 250 kDa. In accordance with isolation of protein complexes, more than 200 of the proteins identified in the retentate are smaller than 100 kDa. It appears that under the experimental conditions used in this study, the majority of proteins are not single, monomeric proteins but components of protein complexes in vivo. Indeed, the percentage of known phosphoproteins was higher in the flow-through (78%) than in the retentate (43%), which correlates with an abundance of monomeric phosphoproteins in the flow-through. We took advantage of the extensive protein interaction databases (BioGRID, BOND, http://yeast-complexes.embl.de) to identify proteins previously identified as components of protein complexes. We noticed enrichment of several functional groups including chaperones. Chaperones are abundant proteins that bind to unfolded proteins and help them fold correctly. Chaperones thus participate in protein-protein interactions and each chaperone has several potential binding partners. Indeed we find several chaperones in our retentate including Hsc82, Hsp104, Hsp60, Hsp82, Ssa1, Sse1, Sti1, and Ydj1. We also find several chaperones in the flow-through fraction representing either small chaperones bound to small proteins or monomeric chaperones. To further analyze the oligomeric status of these proteins a more comprehensive size separation experiment is in order. Several methodologies exist to study the size of protein complexes. Centrifugal concentrators with different MWCO or gel filtration/size exclusion columns can be used to obtain a gradual series of molecular weights. Blue native polyacrylamide gel electrophoresis (BN-PAGE) has also been used to separate native protein complexes in the first dimension followed by denaturing SDS-PAGE electrophoresis in the second dimension creating a 2-D gel where the spots can be analyzed by mass spectrometry 6, 7. Alternatively, sedimentation of protein complexes in a rate zonal gradient allows for estimation of the relative size of protein complexes as recently described for Arabidopsis thaliana 9. Any of these methods could be combined with PA-GeLC-MS/MS for a more comprehensive analysis of protein complexes.
Reproducibility was evaluated both on the phosphoaffinity level and the mass spectrometry level. On the phosphoaffinity level, Pro-Q staining revealed a paucity of phosphoproteins in the flow-through fraction versus abundance in the eluate while Coomassie stained proteins were present in both flow-through and eluate fractions. This staining pattern was consistently produced in replicate experiments (data not shown). Two independent experiments (Figure 3, Table 1 vs. Figure 6, Tables 2 and 3) using different lysis conditions and with or without an exclusion step still have 89% overlap of proteins identified when 1 peptide was required for protein identification showing the reproducibility of the PA-GeLC-MS/MS method.
Phosphoprotein enrichment results in identification of several peptides from each protein and a corresponding high confidence of protein identification. However, the enriched fraction is much more complex than phosphopeptide enriched fractions because of the surplus of non-phosphorylated peptides both in number and abundance. Thus our method does a poor job of identifying phosphorylation sites within the proteins identified. To improve the identification of phosphorylation sites, the sample complexity needs to be reduced. First, the experiment could be done under denaturing conditions to eliminate the presence of protein complexes containing non-phosphorylated components. Secondly, the sample could be further fractionated. A promising approach was described by Hung et al 60 in which tryptic peptides are subjected to isoelectric focusing (IEF), which is a gel based method that separates species based on overall charge or pI value. Phosphopeptides and peptides composed of more acidic than basic amino acid residues have low pI values (2.1–6.5). Peptides containing a higher or equal number of basic compared to acidic residues have high pI values (5.8–9.8). Acidic amino acids were neutralized by esterification increasing the pI value of the corresponding peptides resulting in their separation from phosphopeptides (low pI). This method allows for isolation of a highly enriched phosphopeptide fraction in addition to allowing for isolation of non-phosphorylated proteins. It would likely result in a large increase in identification of phosphorylation sites.
This proteomic analysis of phosphoprotein complexes is not meant to be comprehensive, however, comparing our results to the global proteome reveals interesting over- and underrepresented subpopulations. We analyzed the gene ontology (GO) of the proteins identified in this study and compared them to the yeast proteome. Analysis of protein localization reveals a large overrepresentation of cytosolic proteins (5.5-fold) and to a lesser extent cytoplasmic (1.7-fold), mitochondrial (1.4-fold) and endoplasmic proteins (1.4-fold). On the other hand, our protein set is slightly under populated by nuclear proteins (1.1-fold). A quarter of the proteome are defined as having “cellular component unknown”. We do not identify any of these proteins, which might reflect that they are either low abundance or only expressed under special conditions. Additionally, a large portion of the proteins with “cellular compartment unknown” are classified as dubious open reading frames and might thus not be expressed as proteins.
Analysis of GO terms for proteins associated with different processes revealed that the trend remained the same since the most populated category in the proteome, cellular processes, was also the most populated in our sample and so forth. However, metabolic processes and the general category of cellular processes were overrepresented in our results by about 35% probably due to these proteins being relatively high abundance. Transport, transcription, signal transduction and amino acid metabolic processes were between 55 and 75% underrepresented. Cell cycle related proteins were only 6% underrepresented.
GO analysis of protein complexes reveals an overrepresentation of components of 69 complexes including ribonucleoprotein complex, ribosome, proteasome, translation initiation factor complexes, vacuolar transporter chaperone complex and mitochondrial respiratory chain complex. Six complexes identified in our study are underrepresented compared to the proteome: DNA-directed RNA polymerase II complex, transcription factor complex small nuclear ribonucleoprotein complex, kinetochore, nuclear pore, and histone acetyltransferase complex. The majority of these complexes are localized in the nucleus. This is not surprising since the sample preparation was not targeted towards nuclear proteins and based on the GO analysis of protein localization mentioned previously. A large number of complexes (220) were not identified in our sample. This could be due to several factors such as the absence of a phosphorylated protein in the complex and would thus not be isolated via the PA-GeLC-MS/MS method, formation of complexes under certain conditions (for example cell cycle stage and stress conditions) and/or low abundance of the protein complex.
Most of the proteins identified in this study, including the chaperones, are highly abundant proteins. This is a recognized problem in proteomics, mostly stemming from the dynamic range of protein concentrations being much larger than the dynamic range of the mass spectrometer. The limit of detection for Pro-Q Diamond phosphostain in an SDS-PAGE gel is around 1 ng, which is similar to silver stain, however, the limit for Coomassie stain is much higher at about 50 ng 40–42, 61. Pro-Q staining is proportional to the number of phosphates on the protein thus more highly phosphorylated proteins will be detected at low levels. The method of subjecting samples to 1-D PAGE, trypsin digestion, and identification using reverse phase chromatography on an LC-MS/MS has been shown to be more sensitive than, for example 2-D SDS PAGE coupled to MALDI 39. The level of sensitivity highly depends on the type and model of mass spectrometer. The FT-LTQ, which is used for the size separation experiment in Figure 6 has been shown to identify proteins at levels as low as 20 femtomoles (about 0.5 ng for a 40 kDa protein) 62. Thus, we should be identifying all proteins that are detected by Coomassie stain and the vast majority of Pro-Q stained proteins depending on the phosphorylation status and peptide fragmentation pattern by trypsin.
We were able to detect some low abundance proteins, as low as two hundred copies per cell and as mentioned previously, we identified several signal transduction and cell cycle regulated proteins. However, there is significant room for improvement and for purposes of studying cell cycle regulated events, the number of low abundance proteins detected needs to be increased. To reach this goal we are scaling up the experiment, performing more extensive fractionation and improving identification software. Another approach that could be taken is to combine our PA-GeLC-MS/MS method with targeting of certain cellular compartments and/or organelles, which has proven to be very successful and is reviewed in 63.
We have demonstrated that PA-GeLC-MS/MS can be used to catalog potential phosphoproteins and phosphoprotein complexes at a given time and/or condition. The challenge remains to create software to analyze and predict the presence of protein complexes based on experimental criteria and protein interaction databases. Our method relies greatly on the quality and density of protein-protein interaction databases. We anticipate much improvement in density, sensitivity and accuracy of the protein-protein interaction networks as more and more data and databases are combined and re-evaluated.
Supplementary Material
Acknowledgments
Proteomics on the Thermo LTQ FT instrument were performed in the CBC-UIC Research Resources Center Proteomics and Informatics Services Facility, which was established by a grant from The Searle Funds at the Chicago Community Trust to the Chicago Biomedical Consortium. Proteomics on the Agilent XCT instrument were performed at the University of Chicago Proteomics Core. We thank Tamara Nyberg and Brian Agnew at Molecular Probes-Invitrogen for generously providing Pro-Q Diamond phosphoprotein enrichment media, other reagents and helpful suggestions for their use. We thank Donald L. Helseth Jr. for technical support and helpful discussions. We thank Samuel L. Volchenboum for critical reading of the manuscript. This work was supported by NIH R01s GM60443 and HG003864. KK was supported by University of Chicago Cancer Biology NRSA Training Grant CA09594 and SJK was a Leukemia & Lymphoma Society Scholar.
Footnotes
Supporting Information Available: Supporting Tables 1-3 list the proteins identified in each proteomic experiment. This material is available free at http://pubs.acs.org
References
- 1.Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–72. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- 2.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402(6761 Suppl):C47–52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 3.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–68. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 4.Andersen JS, Lam YW, Leung AK, Ong SE, Lyon CE, Lamond AI, Mann M. Nucleolar proteome dynamics. Nature. 2005;433(7021):77–83. doi: 10.1038/nature03207. [DOI] [PubMed] [Google Scholar]
- 5.Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426(6966):570–4. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 6.Schagger H, Cramer WA, von Jagow G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem. 1994;217(2):220–30. doi: 10.1006/abio.1994.1112. [DOI] [PubMed] [Google Scholar]
- 7.Camacho-Carvajal MM, Wollscheid B, Aebersold R, Steimle V, Schamel WW. Two-dimensional Blue native/SDS gel electrophoresis of multi-protein complexes from whole cellular lysates: a proteomics approach. Mol Cell Proteomics. 2004;3(2):176–82. doi: 10.1074/mcp.T300010-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199(2):223–31. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 9.Hartman NT, Sicilia F, Lilley KS, Dupree P. Proteomic complex detection using sedimentation. Anal Chem. 2007;79(5):2078–83. doi: 10.1021/ac061959t. [DOI] [PubMed] [Google Scholar]
- 10.Kocher T, Superti-Furga G. Mass spectrometry-based functional proteomics: from molecular machines to protein networks. Nat Methods. 2007;4(10):807–15. doi: 10.1038/nmeth1093. [DOI] [PubMed] [Google Scholar]
- 11.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415(6868):141–7. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 12.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MH, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O’Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440(7084):637–43. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 13.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34(Database issue):D535–9. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300(5618):445–52. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 15.Slepnev VI, Ochoa GC, Butler MH, Grabs D, De Camilli P. Role of phosphorylation in regulation of the assembly of endocytic coat complexes. Science. 1998;281(5378):821–4. doi: 10.1126/science.281.5378.821. [DOI] [PubMed] [Google Scholar]
- 16.Zolnierowicz S, Bollen M. Protein phosphorylation and protein phosphatases. De Panne, Belgium, September 19–24, 1999. Embo J. 2000;19(4):483–8. doi: 10.1093/emboj/19.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLachlin DT, Chait BT. Analysis of phosphorylated proteins and peptides by mass spectrometry. Curr Opin Chem Biol. 2001;5(5):591–602. doi: 10.1016/s1367-5931(00)00250-7. [DOI] [PubMed] [Google Scholar]
- 18.Goshe MB. Characterizing phosphoproteins and phosphoproteomes using mass spectrometry. Brief Funct Genomic Proteomic. 2006;4(4):363–76. doi: 10.1093/bfgp/eli007. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Li R, Du D, Zhang C, Yuan H, Zeng R, Chen Z. Proteomic analysis reveals novel molecules involved in insulin signaling pathway. J Proteome Res. 2006;5(4):846–55. doi: 10.1021/pr050391m. [DOI] [PubMed] [Google Scholar]
- 20.Gold MR, Yungwirth T, Sutherland CL, Ingham RJ, Vianzon D, Chiu R, van Oostveen I, Morrison HD, Aebersold R. Purification and identification of tyrosine-phosphorylated proteins from B lymphocytes stimulated through the antigen receptor. Electrophoresis. 1994;15(3–4):441–53. doi: 10.1002/elps.1150150161. [DOI] [PubMed] [Google Scholar]
- 21.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 22.Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316(5828):1194–8. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda Y, Nagasu T, Chait BT. Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome. Nat Biotechnol. 2001;19(4):379–82. doi: 10.1038/86783. [DOI] [PubMed] [Google Scholar]
- 24.Meyer HE, Eisermann B, Heber M, Hoffmann-Posorske E, Korte H, Weigt C, Wegner A, Hutton T, Donella-Deana A, Perich JW. Strategies for nonradioactive methods in the localization of phosphorylated amino acids in proteins. Faseb J. 1993;7(9):776–82. doi: 10.1096/fasebj.7.9.7687226. [DOI] [PubMed] [Google Scholar]
- 25.Bodenmiller B, Mueller LN, Mueller M, Domon B, Aebersold R. Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat Methods. 2007;4(3):231–7. doi: 10.1038/nmeth1005. [DOI] [PubMed] [Google Scholar]
- 26.Andersson L, Porath J. Isolation of phosphoproteins by immobilized metal (Fe3+) affinity chromatography. Anal Biochem. 1986;154(1):250–4. doi: 10.1016/0003-2697(86)90523-3. [DOI] [PubMed] [Google Scholar]
- 27.Posewitz MC, Tempst P. Immobilized gallium(III) affinity chromatography of phosphopeptides. Anal Chem. 1999;71(14):2883–92. doi: 10.1021/ac981409y. [DOI] [PubMed] [Google Scholar]
- 28.Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, Heck AJ. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal Chem. 2004;76(14):3935–43. doi: 10.1021/ac0498617. [DOI] [PubMed] [Google Scholar]
- 29.Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJ. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol Cell Proteomics. 2005;4(7):873–86. doi: 10.1074/mcp.T500007-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Craig R, Cortens JP, Beavis RC. The use of proteotypic peptide libraries for protein identification. Rapid Commun Mass Spectrom. 2005;19(13):1844–50. doi: 10.1002/rcm.1992. [DOI] [PubMed] [Google Scholar]
- 31.Craig R, Cortens JP, Beavis RC. Open source system for analyzing, validating, and storing protein identification data. J Proteome Res. 2004;3(6):1234–42. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- 32.Collins MO, Husi H, Yu L, Brandon JM, Anderson CN, Blackstock WP, Choudhary JS, Grant SG. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J Neurochem. 2005 doi: 10.1111/j.1471-4159.2005.03507.x. [DOI] [PubMed] [Google Scholar]
- 33.Imam-Sghiouar N, Laude-Lemaire I, Labas V, Pflieger D, Le Caer JP, Caron M, Nabias DK, Joubert-Caron R. Subproteomics analysis of phosphorylated proteins: application to the study of B-lymphoblasts from a patient with Scott syndrome. Proteomics. 2002;2(7):828–38. doi: 10.1002/1615-9861(200207)2:7<828::AID-PROT828>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 34.Ficarro S, Chertihin O, Westbrook VA, White F, Jayes F, Kalab P, Marto JA, Shabanowitz J, Herr JC, Hunt DF, Visconti PE. Phosphoproteome analysis of capacitated human sperm. Evidence of tyrosine phosphorylation of a kinase-anchoring protein 3 and valosin-containing protein/p97 during capacitation. J Biol Chem. 2003;278(13):11579–89. doi: 10.1074/jbc.M202325200. [DOI] [PubMed] [Google Scholar]
- 35.Zheng H, Hu P, Quinn DF, Wang YK. Phosphotyrosine proteomic study of interferon alpha signaling pathway using a combination of immunoprecipitation and immobilized metal affinity chromatography. Mol Cell Proteomics. 2005;4(6):721–30. doi: 10.1074/mcp.M400077-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Gronborg M, Kristiansen TZ, Stensballe A, Andersen JS, Ohara O, Mann M, Jensen ON, Pandey A. A mass spectrometry-based proteomic approach for identification of serine/threonine-phosphorylated proteins by enrichment with phospho-specific antibodies: identification of a novel protein, Frigg, as a protein kinase A substrate. Mol Cell Proteomics. 2002;1(7):517–27. doi: 10.1074/mcp.m200010-mcp200. [DOI] [PubMed] [Google Scholar]
- 37.Metodiev MV, Timanova A, Stone DE. Differential phosphoproteome profiling by affinity capture and tandem matrix-assisted laser desorption/ionization mass spectrometry. Proteomics. 2004;4(5):1433–8. doi: 10.1002/pmic.200300683. [DOI] [PubMed] [Google Scholar]
- 38.Makrantoni V, Antrobus R, Botting CH, Coote PJ. Rapid enrichment and analysis of yeast phosphoproteins using affinity chromatography, 2D-PAGE and peptide mass fingerprinting. Yeast. 2005;22(5):401–14. doi: 10.1002/yea.1220. [DOI] [PubMed] [Google Scholar]
- 39.Korbel S, Buchse T, Prietzsch H, Sasse T, Schumann M, Krause E, Brock J, Bittorf T. Phosphoprotein profiling of erythropoietin receptor- dependent pathways using different proteomic strategies. Proteomics. 2005;5(1):91–100. doi: 10.1002/pmic.200400883. [DOI] [PubMed] [Google Scholar]
- 40.Martin K, Steinberg TH, Cooley LA, Gee KR, Beechem JM, Patton WF. Quantitative analysis of protein phosphorylation status and protein kinase activity on microarrays using a novel fluorescent phosphorylation sensor dye. Proteomics. 2003;3(7):1244–55. doi: 10.1002/pmic.200300445. [DOI] [PubMed] [Google Scholar]
- 41.Schulenberg B, Goodman TN, Aggeler R, Capaldi RA, Patton WF. Characterization of dynamic and steady-state protein phosphorylation using a fluorescent phosphoprotein gel stain and mass spectrometry. Electrophoresis. 2004;25(15):2526–32. doi: 10.1002/elps.200406007. [DOI] [PubMed] [Google Scholar]
- 42.Steinberg TH, Agnew BJ, Gee KR, Leung WY, Goodman T, Schulenberg B, Hendrickson J, Beechem JM, Haugland RP, Patton WF. Global quantitative phosphoprotein analysis using Multiplexed Proteomics technology. Proteomics. 2003;3(7):1128–44. doi: 10.1002/pmic.200300434. [DOI] [PubMed] [Google Scholar]
- 43.Puente LG, Borris DJ, Carriere JF, Kelly JF, Megeney LA. Identification of candidate regulators of embryonic stem cell differentiation by comparative phosphoprotein affinity profiling. Mol Cell Proteomics. 2006;5(1):57–67. doi: 10.1074/mcp.M500166-MCP200. [DOI] [PubMed] [Google Scholar]
- 44.Wu D, Nair-Gill E, Sher DA, Parker LL, Campbell JM, Siddiqui M, Stock W, Kron SJ. Assaying Bcr-Abl kinase activity and inhibition in whole cell extracts by phosphorylation of substrates immobilized on agarose beads. Anal Biochem. 2005;347(1):67–76. doi: 10.1016/j.ab.2005.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74(20):5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 46.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75(17):4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 47.Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, et al. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun. 1989;159(3):871–7. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- 48.Honkanen RE, Zwiller J, Moore RE, Daily SL, Khatra BS, Dukelow M, Boynton AL. Characterization of microcystin-LR, a potent inhibitor of type 1 and type 2A protein phosphatases. J Biol Chem. 1990;265(32):19401–4. [PubMed] [Google Scholar]
- 49.Bialojan C, Takai A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J. 1988;256(1):283–90. doi: 10.1042/bj2560283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cohen P, Klumpp S, Schelling DL. An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 1989;250(2):596–600. doi: 10.1016/0014-5793(89)80803-8. [DOI] [PubMed] [Google Scholar]
- 51.Futcher B, Latter GI, Monardo P, McLaughlin CS, Garrels JI. A sampling of the yeast proteome. Mol Cell Biol. 1999;19(11):7357–68. doi: 10.1128/mcb.19.11.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20(3):301–5. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 53.Gruhler A, Olsen JV, Mohammed S, Mortensen P, Faergeman NJ, Mann M, Jensen ON. Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol Cell Proteomics. 2005;4(3):310–27. doi: 10.1074/mcp.M400219-MCP200. [DOI] [PubMed] [Google Scholar]
- 54.Li X, Gerber SA, Rudner AD, Beausoleil SA, Haas W, Villen J, Elias JE, Gygi SP. Large-scale phosphorylation analysis of alpha-factor-arrested Saccharomyces cerevisiae. J Proteome Res. 2007;6(3):1190–7. doi: 10.1021/pr060559j. [DOI] [PubMed] [Google Scholar]
- 55.Ignatovich O, Cooper M, Kulesza HM, Beggs JD. Cloning and characterisation of the gene encoding the ribosomal protein S5 (also known as rp14, S2, YS8) of Saccharomyces cerevisiae. Nucleic Acids Res. 1995;23(22):4616–9. doi: 10.1093/nar/23.22.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Breitkreutz BJ, Stark C, Tyers M. Osprey: a network visualization system. Genome Biol. 2003;4(3):R22. doi: 10.1186/gb-2003-4-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aloy P, Bottcher B, Ceulemans H, Leutwein C, Mellwig C, Fischer S, Gavin AC, Bork P, Superti-Furga G, Serrano L, Russell RB. Structure-based assembly of protein complexes in yeast. Science. 2004;303(5666):2026–9. doi: 10.1126/science.1092645. [DOI] [PubMed] [Google Scholar]
- 58.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17(10):1030–2. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 59.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425(6959):737–41. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 60.Hung CW, Kubler D, Lehmann WD. pI-based phosphopeptide enrichment combined with nanoESI-MS. Electrophoresis. 2007;28(12):2044–52. doi: 10.1002/elps.200600678. [DOI] [PubMed] [Google Scholar]
- 61.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68(5):850–8. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 62.de Godoy LM, Olsen JV, de Souza GA, Li G, Mortensen P, Mann M. Status of complete proteome analysis by mass spectrometry: SILAC labeled yeast as a model system. Genome Biol. 2006;7(6):R50. doi: 10.1186/gb-2006-7-6-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andersen JS, Mann M. Organellar proteomics: turning inventories into insights. EMBO Rep. 2006;7(9):874–9. doi: 10.1038/sj.embor.7400780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc Natl Acad Sci U S A. 2007;104(7):2193–8. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilson LK, Dhillon N, Thorner J, Martin GS. Casein kinase II catalyzes tyrosine phosphorylation of the yeast nucleolar immunophilin Fpr3. J Biol Chem. 1997;272(20):12961–7. doi: 10.1074/jbc.272.20.12961. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.