

Abstract
Hutchinson–Gilford progeria syndrome (HGPS) is caused by point mutations that increase utilization of an alternate splice donor site in exon 11 of LMNA (the gene encoding lamin C and prelamin A). The alternate splicing reduces transcripts for wild-type prelamin A and increases transcripts for a truncated prelamin A (progerin). Here, we show that antisense oligonucleotides (ASOs) against exon 11 sequences downstream from the exon 11 splice donor site promote alternate splicing in both wild-type and HGPS fibroblasts, increasing the synthesis of progerin. Indeed, wild-type fibroblasts transfected with these ASOs exhibit progerin levels similar to (or greater than) those in fibroblasts from HGPS patients. This progerin was farnesylated, as judged by metabolic labeling studies. The synthesis of progerin in wild-type fibroblasts was accompanied by the same nuclear shape and gene-expression perturbations observed in HGPS fibroblasts. An ASO corresponding to the 5′ portion of intron 11 also promoted alternate splicing. In contrast, an ASO against exon 11 sequences 5′ to the alternate splice site reduced alternate splicing in HGPS cells and modestly lowered progerin levels. Thus, different ASOs can be used to increase or decrease ‘HGPS splicing’. ASOs represent a new and powerful tool for recreating HGPS pathophysiology in wild-type cells.
INTRODUCTION
Hutchinson–Gilford progeria syndrome (HGPS), a rare pediatric progeroid syndrome, is caused by single-nucleotide substitutions in exon 11 of LMNA. These point mutations promote usage of an alternate splice donor site located within exon 11 (1–3). The alternate splicing simultaneously reduces wild-type prelamin A transcripts and increases the production of a mutant prelamin A transcript lacking the last 150 nucleotides of exon 11. This mutant transcript encodes a mutant prelamin A protein, commonly called progerin, containing an internal deletion of 50 amino acids. Progerin retains a C-terminal CaaX motif (encoded by exon 12) that triggers protein prenylation (4–7). The production of progerin interferes with the integrity of the nuclear lamina, causing misshapen nuclei, and also adversely affects other important cellular processes, such as mitosis and cell proliferation (4–7). A gene-targeted HGPS mouse model has clearly established that the hallmark phenotypes of progeria are caused by the production of progerin (8,9).
The most common point mutations causing HGPS optimize a cryptic splice donor site in exon 11 (10). This splice donor site, CAG↓GTGGGC, is used at trace levels in wild-type cells (10). However, the levels of alternatively spliced transcripts in wild-type cells are so low that they are generally undetectable on ethidium bromide-stained agarose gels of RT–PCR products (3) [although one group did report finding trace amounts of progerin transcripts when large amounts of the RT–PCR product was loaded (10)]. In any case, the levels of alternatively spliced transcripts are so low that progerin in wild-type cells is not normally detected by western blotting, even on heavily loaded gels (11,12). The most common point mutation causing HGPS [G608G(GGC>GGT)] increases usage of the alternate splice site so that ∼40% of all prelamin A transcripts are alternatively spliced (12). Rare point mutations associated with very high levels of alternate splicing and very high levels of progerin production lead to particularly severe forms of progeria (12).
Many studies investigating the effects of progerin have used fibroblasts from HGPS patients (2,3,11,13–15). These experiments have been informative, but their interpretation can be difficult because of prominent phenotypic differences between different HGPS fibroblast cell lines—presumably differences related to passage number and the age, sex and genetic background of the donor patient. Other studies have relied on expressing progerin in cell lines that already express a full complement of wild-type lamin proteins (6,7,13,14,16). Although these studies have also been useful, for example in defining the impact of progerin on nuclear shape and cell growth (6,7,16), they also have distinct limitations, given that overexpression of even wild-type lamins can cause abnormalities in nuclear shape and gene expression (17).
Because of the problems associated with progerin overexpression and the phenotypic variability of human HGPS fibroblasts, we reasoned that it would be useful to develop techniques to increase ‘HGPS alternate splicing’ and progerin production in wild-type cells. If it was possible to vastly increase the alternate splicing in wild-type cells, it would provide an important new approach for assessing the impact of progerin in cells—and one that would faithfully mirror the molecular pathophysiology of HGPS (i.e. increased progerin expression with reduced expression of wild-type prelamin A).
Several recent studies have shown that antisense oligonucleotides (ASOs) have the potential to modulate splice site utilization (18–21). For example, an ASO directed against exon 7 of SMN2 promotes the inclusion of exon 7 in the mature transcript, whereas other ASOs promote skipping of that same exon (18). In the current study, we examined multiple ASOs on the utilization of the splice donor site in exon 11 of LMNA. We identified multiple ASOs that increased levels of progerin transcripts. Remarkably, these ASOs converted wild-type fibroblasts into cells that synthesize high levels of progerin and manifest cellular and molecular hallmarks of HGPS fibroblasts. In addition, one of the ASOs, located upstream from the splice donor site, reduced alternate splicing and modestly lowered levels of progerin in HGPS fibroblasts.
RESULTS
To determine if ASOs could affect the utilization of the splice donor site in exon 11 of LMNA, we transfected fibroblasts with ASOs directed against exon 11 sequences. All of the ASOs contained a 2′-O-methoxy-ethyl (2′-MOE) ribose modification and a phosphodiester backbone. Testing of more than 25 ASOs, located within 50 bp of the alternative splice site, identified multiple ASOs that modulated utilization of the splice donor site, as judged by RT–PCR (Supplementary Material, Figs S1 and S2). Nine ASOs, all located ∼47 bp downstream from the alternative splice site (c.1849–1882) promoted alternative splicing in HGPS fibroblasts (e.g. ASO 324), resulting in more progerin transcripts and fewer wild-type prelamin A transcripts (Supplementary Material, Figs S1B and S2B, Fig. 1B). In contrast, only one ASO (ASO 365), located 51 bp upstream from the splice donor site, consistently reduced progerin transcripts (Fig. 1B). The ratio of progerin to prelamin A transcripts, as judged by image analysis, increased from 1.4 to 3.9 in HGPS cells transfected with ASO 324, relative to cells transfected with a scrambled ASO, ASO 333. In contrast, the ratio decreased from 1.4 to 0.92 in HGPS cells transfected with ASO 365. Interestingly, ASOs directed against the exon 11 splice donor site (e.g. ASOs 076 and 790) had minimal, if any, effects on splicing. To measure the changes in splicing more precisely, we assessed transcript levels by quantitative RT–PCR in two HGPS cell lines (AG03513 and GM01972). These studies revealed that ASO 324 increased progerin transcripts 2.6-fold relative to the control ASO, whereas ASO 365 lowered progerin levels 1.8-fold (relative to the control ASO) (Fig. 1C).
Figure 1.

Modulating the utilization of the exon 11 splice donor site with antisense oligonucleotides corresponding to sequences within exon 11 of human LMNA. All of the antisense oligonucleotides (ASOs) contained a 2′-O-methoxy-ethyl ribose backbone. (A) Exons 11 and 12 of LMNA, depicting both ‘wild-type splicing’ and ‘HGPS splicing’ (which uses the alternate splice donor site in exon 11). Also shown are the locations of ASOs located upstream of the alternate splice donor site (i.e. 365), ASOs downstream from the splice donor site (i.e. 324) and ASOs that straddle the site (i.e. 076 and 790). (B) SYTO 60-stained agarose gel showing RT–PCR products of RNA prepared from an HGPS cell line. The PCR products correspond to wild-type prelamin A and progerin transcripts. Transfection with ASO 324 increases progerin transcripts, whereas ASO 365 lowers progerin transcripts, as judged by image analysis on a LI-COR infrared scanner. (C) Quantification of progerin transcripts, by quantitative RT–PCR, in HGPS fibroblasts (AG03513 and GM01972) transfected with different ASOs. Results are plotted relative to progerin transcript levels in fibroblasts transfected with ASO 333. (D) SYTO 60-stained gel showing relative levels of wild-type prelamin A and progerin transcripts in wild-type fibroblasts, HGPS fibroblasts and wild-type fibroblasts transfected with either ASO 324 or control ASO 333. Also shown are DNA sequencing chromatograms of RT–PCR products corresponding to wild-type prelamin A transcripts in ASO 324-treated wild-type cells (upper), progerin transcripts in ASO 324-treated wild-type cells (middle) and progerin transcripts in HGPS fibroblasts (lower).
These studies involving HGPS fibroblasts showed that ASOs can change progerin transcript levels, but the mechanism was not entirely clear. One possibility, of course, is that the ASOs affected splice site utilization, but alternatively, the results could be explained by an effect of the ASO on the relative stability of the two transcripts. We reasoned that if the ASOs changed progerin transcript levels by altering splicing, then the ASOs might induce high levels of progerin transcripts in wild-type cells, where progerin transcripts are normally present in trace amounts (undetectable on ethidium bromide-stained agarose gels of RT–PCR products, as shown in Supplementary Material, Figs S1B and S2B). To explore this idea, wild-type fibroblasts were transfected with ASO 324 and progerin transcript levels were evaluated by RT–PCR. Remarkably, the predominant RT–PCR product in ASO 324-transfected wild-type cells was shorter and had the same mobility as the progerin product obtained with HGPS fibroblasts; in addition, the levels of wild-type prelamin A transcripts were decreased (Fig. 1D and Supplementary Material, Fig. S2). Wild-type cells that had been transfected with the control ASO (ASO 333) expressed only the full-length prelamin A transcript.
Both of the RT–PCR products from ASO 324-transfected wild-type cells were cloned and sequenced; the longer RT–PCR product was derived from the full-length prelamin A transcript, whereas the shorter product was derived from the alternatively splice progerin transcript (Fig. 1D).
We identified multiple ASOs that promoted the production of progerin transcripts in HGPS fibroblasts (Supplementary Material, Fig. S2). Of these, ASOs 074 and 324 (located 43 and 47 bp downstream from the HGPS splice-donor site, respectively) were the most potent. Each ASO that stimulated synthesis of progerin transcripts in HGPS fibroblasts had similar effects in wild-type fibroblasts (Supplementary Material, Fig. S2).
We were concerned that ASOs that promoted alternate splicing, such as ASOs 074 and 324, might bind to and promote the degradation of progerin transcripts, thereby preventing the synthesis of progerin protein. However, this was not the case, as large amounts of progerin protein were observed in ASO-transfected wild-type cells (Fig. 2A). Transfection of ASO 074 into either wild-type or HGPS fibroblasts resulted in a concentration- and time-dependent increase in the amount of progerin, as judged by western blotting with a lamin A/C antibody (Fig. 2A and B). Progerin persisted for at least 5 days after transfection, and the amounts of progerin in wild-type cells were similar to, or even greater, than the amount in non-transfected HGPS fibroblasts (Fig. 2A and B). RT–PCR studies on ASO 074-transfected cells showed that wild-type prelamin A transcripts fell at the same time that progerin transcripts increased. Consistent with these RT–PCR results, quantitative image analyses of western blots revealed that mature lamin A levels (relative to lamin C) in ASO 074-transfected cells decreased by 68.3 ± 8.5%, compared with cells transfected with the control ASO (average of results with three different wild-type fibroblast cell lines). In contrast, transfection of ASO 365 into HGPS fibroblasts reduced progerin levels in cells (Fig. 2C), a result that was predictable from the RT–PCR studies (Fig. 1B).
Figure 2.

Western blots with an antibody against lamin A/C showing that transfection of antisense oligonucleotide 074 yields higher levels of progerin expression. (A) Western blot of wild-type fibroblasts (GM01651) and HGPS fibroblasts (AG11513) 2 days after transfection with ASO 074, revealing progerin in the wild-type fibroblasts and increased levels of progerin in the HGPS fibroblasts. (B) Western blot of a wild-type fibroblast cell line (GM01651) 1–5 days after transfection with ASO 074, revealing a time-dependent increase in progerin expression (similar results were observed with cell line AG07095; data not shown). In both experiments, the levels of progerin in wild-type fibroblasts exceeded those in HGPS fibroblasts. (C) Western blot showing that transfection of ASO 365 into HGPS fibroblasts (AG11513) results in slightly reduced levels of progerin. (D) Western blot of ASO 074-transfected restrictive dermopathy (RD) fibroblasts and HeLa cells. Progerin was found in cell extracts prepared 2 days after the transfection.
Most studies on progerin have relied on fibroblast cell lines harvested from patients with HGPS. However, the ASO-based approach provides the opportunity to studying progerin in cell lines derived from other tissues. For example, transfection of HeLa cells with ASO 074 resulted in the production of significant amounts of progerin (Fig. 2D). This technique also makes it possible to induce progerin expression in fibroblasts with other defects in prelamin A processing. Fibroblasts from restrictive dermopathy (RD) patients normally accumulate farnesyl-prelamin A as a result of a deficiency in ZMPSTE24 (the prelamin A protease) (22). However, when RD fibroblasts were transfected with ASO 324, they also produced progerin (Fig. 2D).
In a recent study, Moulson et al. (12) showed that a particularly severe form of progeria was caused by a LMNA point mutation involving the first nucleotide of intron 11 (c.1968 + 1G > A). That mutation promoted utilization of the alternate splice donor site in exon 11, resulting in large amounts of progerin synthesis. We therefore speculated that ASOs directed against the beginning of intron 11 might also promote alternate splicing and progerin synthesis. Indeed, when human fibroblasts were transfected with an ASO directed against the first 20 nucleotides of intron 11 (ASO 893), the fibroblasts produced progerin (Fig. 3A and B). ASOs directed against sequences immediately upstream or downstream from the intron 11 splice donor site (ASOs 890, 891, 892 and 894) had no effect (Fig. 3A and B).
Figure 3.

Western blot with an antibody against lamin A/C showing that transfection of an antisense oligonucleotide directed against the splice donor sequence at the beginning of intron 11 results in progerin expression. (A) Schematic showing the location of five ASOs near the splice donor sequence at the beginning of intron 11. (B) Western blot of extracts from wild-type fibroblasts (AG07095) showing that transfection of ASO 893 (corresponding to the first 20 nucleotides of intron 11) results in the production of progerin.
Progerin is predicted to be farnesylated, and this modification has been proposed to be important in the pathogenesis of disease (23,24). To determine if the progerin produced by ASO-transfected cells is farnesylated, ASO 074-transfected fibroblasts were metabolically labeled with 8-anilinogeraniol (AG) in the presence or absence of a farnesyltransferase inhibitor (FTI). AG is incorporated into anilinogeranyl diphosphate (AGPP) within cells and used as a substrate for protein farnesyltransferase (FTase). The incorporation of the AGPP into proteins can be monitored by western blotting with a monoclonal antibody against AG. This approach was recently used to show that a GFP-tagged progerin fusion protein was farnesylated in HeLa cells (7). We used AG metabolic labeling to examine progerin farnesylation in HGPS fibroblasts. These studies showed that the progerin in HGPS fibroblasts is farnesylated and that this farnesylation could be blocked with an FTI (Fig. 4A). Interestingly, the AG experimental approach is quite sensitive; we detected farnesylation of the full-length prelamin A, even though only small amounts of farnesyl-prelamin A are present in cells (Fig. 4A). AG labeling studies also revealed that the progerin in ASO 074-transfected fibroblasts was farnesylated (Fig. 4B). When the ASO 074-transfected fibroblasts were treated with an FTI, the farnesylation of progerin was eliminated (Fig. 4B).
Figure 4.

Metabolic labeling experiments showing that the progerin that accumulates in antisense oligonucleotide-transfected wild-type fibroblasts is farnesylated. Fibroblasts were metabolically labeled with 8-anilinogeraniol (AG) in the presence or absence of a protein farnesyltransferase inhibitor (FTI) (ABT-100). After entering cells, AG is incorporated into anilinogeranyl diphosphate (AGPP), which is used as a substrate by protein farnesyltransferase. The incorporation of the AGPP into proteins can be monitored by western blotting with an antibody against AG. (A) Western blot of HGPS fibroblast extracts, performed with an Odyssey infrared imaging system, showing that progerin is farnesylated. Shown is the merged image of western blots with an antibody specific for lamin A/C (red) and an antibody against AG (green) before (left panel) and after (right panel) immune precipitation with a lamin A/C antibody. Incorporation of AG into progerin was blocked by the FTI. (B) Western blot of ASO-transfected wild-type fibroblasts showing that the progerin is farnesylated. Wild-type fibroblasts were transfected with ASO 074 to induce progerin synthesis and incubated with the AG substrate. After 2 days, extracts were prepared and the A-type lamins isolated by immune precipitation with an antibody against lamin A/C. The progerin in the wild-type cells was farnesylated (green), and this farnesylation could be blocked with an FTI.
We predicted that the increased production of progerin in ASO 074-transfected wild-type and HGPS fibroblasts would be associated with a higher frequency of misshapen cell nuclei. Indeed, in two independent experiments, the frequency of cells with misshapen nuclei was higher in ASO 074-transfected cells (P < 0.0001, χ2-test) (Fig. 5A and B). We found a somewhat lower level of misshapen nuclei in ASO 074-transfected fibroblasts than in HGPS fibroblasts, but this was not unexpected. The HGPS fibroblasts used in these experiments were late-passage cells, which are known to exhibit more severe nuclear shape abnormalities (16).
Figure 5.
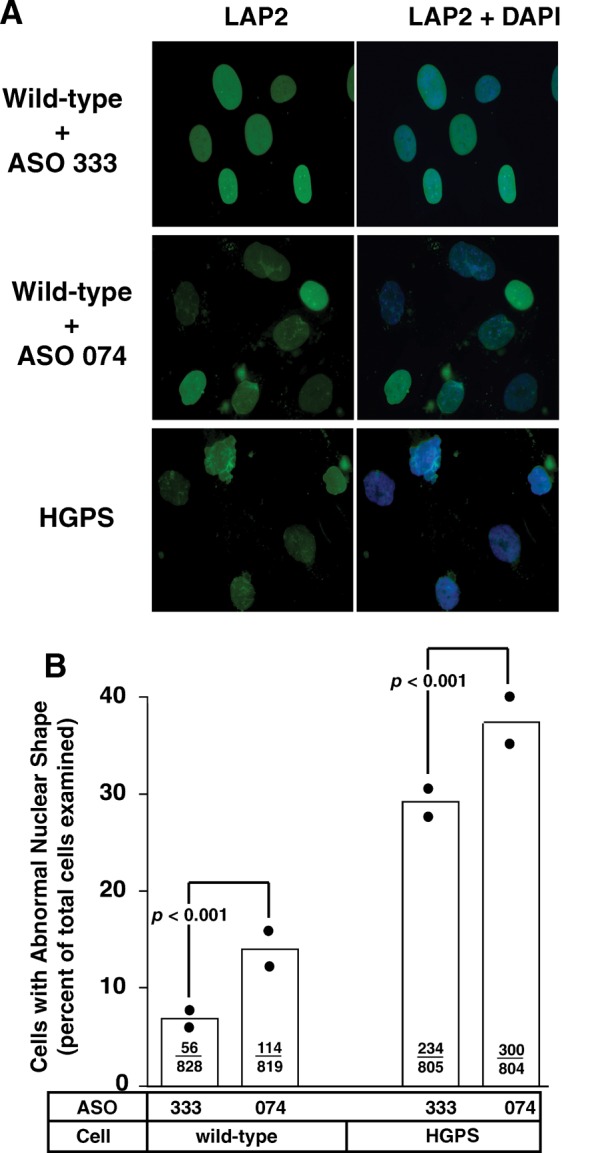
Increased percentage of misshapen nuclei in wild-type and HGPS fibroblasts after transfection with antisense oligonucleotide 074. (A) Epifluorescence microscopy showing increased numbers of misshapen nuclei in wild-type fibroblasts transfected with ASO 074, which promotes alternate splicing and progerin synthesis. ASO 333, which has no effect on splicing, was used as a control. (B) Bar graph comparing the frequency of misshapen nuclei in wild-type and HGPS fibroblasts transfected with ASOs 074 and 333. The ratios inside the bars reflect the number of misshapen nuclei divided by the total number of nuclei that were scored. The solid black circles show the percentage of misshapen nuclei in two independent experiments. Differences in the frequency of misshapen nuclei were assessed with a χ2-test.
To study consequences of progerin expression in cells, we examined gene and protein expression patterns in ASO-transfected wild-type fibroblasts. Previous studies had suggested that the expression of five genes (i.e. HAS3, CCL8, MMP14, MMP3 and TIMP3) was perturbed in HGPS fibroblasts when STAT3 was unchanged (4). In cells that had been transfected with ASO 324, we found that, in addition to progerin, the expression of HAS3 and CCL8 was significantly increased (P = 0.030 and 0.005, respectively, in three independent cell lines); however, there were no consistent changes in the other three genes (Supplementary Material, Fig. S3). In follow-up experiments, we were unable to confirm consistent perturbations of the other three genes (i.e. MMP14, MMP3 and TIMP3) in six different HGPS fibroblast cell lines (data not shown). More recently, increased expression of Notch signaling pathway effectors was observed in fibroblasts expressing an inducible GFP–progerin fusion protein (25). Consistent with those findings, HES1 was increased by 1.6-fold (P = 0.003) and HEY1 was increased by 2.0-fold (P = 0.044) in wild-type fibroblasts 3 days after transfection with ASO 074 (relative to levels in wild-type fibroblasts transfected with ASO 333).
The proteome of fibroblasts expressing progerin was analyzed by two-dimensional difference gel electrophoresis (2D-DIGE). Fibroblasts were transfected with ASO 324 or ASO 333 (the control ASO), and after 3 days, proteins from cell extracts were prepared and labeled with cyanine dyes. The extracts were mixed together, separated by 2D-gel electrophoresis, and differences in expression were identified by fluorescence image analysis (after normalization to the internal standard). In two independent experiments, 24 proteins with perturbed expression patterns were identified (P < 0.05, ANOVA) (Fig. 6); 15 proteins had decreased expression and 9 were increased. Proteins with a change in expression more than 1.5-fold were submitted for mass spectrometry, and 12 of the 15 that were submitted were identified (Supplementary Material, Table S3). It was reassuring that one of the proteins with increased expression in the ASO 324-transfected cells was progerin, and one of the proteins with reduced expression was lamin A. Several mitochondria-associated proteins were increased in the progerin-expressing cells, including Mn-superoxide dismutase (Mn-SOD), Mn-SOD precursor, voltage-dependent anion channel 2 and phosphoenolpyruvate carboxykinase 2 (Supplementary Material, Table S3).
Figure 6.

Two-dimensional gel electrophoresis experiments revealing distinct patterns of protein expression in wild-type human fibroblasts after transfection with ASO 324, an antisense oligonucleotide that promotes alternate splicing and progerin synthesis, or ASO 333, a control oligonucleotide that has no effect on splicing. Wild-type fibroblasts (CRL-1474) were transfected with ASO 324 or ASO 333, and soluble extracts were labeled with cyanine dyes (red and green). Equal amounts of the extracts were mixed together and fractionated by 2D-gel electrophoresis. The gel was imaged on a Typhoon TRIO scanner and individual protein spots were quantified with DeCyder. In the merged image (far right), red spots represent proteins that increase in ASO 324-transfected cells, whereas green spots represent proteins that decrease (relative to cells transfected with ASO 333). Two independent experiments are shown. The locations of eight proteins (identified by mass spectrometry) are shown in one of the experiments.
DISCUSSION
The discovery of mutations causing HGPS (1–3) was quite important, as it provided an entry point for investigating the molecular mechanisms underlying progeria. During the past few years, the investigation of HGPS pathogenesis has relied heavily on studies involving either HGPS fibroblasts or tumor cell lines overexpressing progerin (26). Although these approaches have been useful, they have clear limitations. Different HGPS fibroblast cell lines from cell repositories are phenotypically different (16), and entirely appropriate control cell lines are rarely available. With the overexpression approach, it is often difficult to match expression levels for progerin and control lamin constructs, and it is sobering that wild-type lamins as well as commonly used N-terminal protein tags can alter nuclear morphology (17) and gene expression (25). In the current study, we describe an entirely new approach for expressing progerin in wild-type cells—by inducing ‘HGPS alternative splicing’ within exon 11 of LMNA. We demonstrate that ASOs directed against sequences downstream from the alternate splice donor site promote ‘HGPS splicing’ in wild-type cells, leading to significant progerin levels in cells and morphological and gene-expression hallmarks of HGPS. This ASO approach faithfully reproduces the proximal event in the pathogenesis of HGPS—increased alternate splicing at the expense of reduced levels of normal splicing. We found no evidence that two of the most potent ASOs in promoting alternate splicing (ASOs 074 and 324) did not lower the overall expression of lamin proteins. This is not particularly surprising, as the 2′-MOE ribose oligonucleotides that we used do not support elimination of LMNA transcripts by a short interference RNA activity (27).
These studies provide a robust and reproducible approach for defining the impact of progerin on cells. The amount of progerin in the ASO-transfected wild-type fibroblasts exceeded those in HGPS fibroblasts, and the progerin in ASO-transfected cells was farnesylated (like the progerin in HGPS fibroblasts). When the ASOs were transfected into HGPS fibroblasts, the levels of progerin increased markedly. The ASO method for expressing progerin is versatile, in that it is possible to achieve different levels of progerin synthesis simply by titrating ASO concentrations.
Alternative splicing within exon 11 was promoted most effectively by ASOs directed against sequences centered 47 bp downstream from the alternate splice donor site (e.g. ASOs 324 and 074). ASOs centered > 9 bp upstream or downstream from ASO 324 were less effective. What is the mechanism by which ASOs 324 and 074 promote the alternate spice site utilization? We suspect that these ASOs likely interfere with exonic splicing enhancers (ESEs). Proper mRNA splicing clearly depends on intronic cis-elements—the 5′ splice site, the 3′ splice site and the branch site (28). However, proper splicing also depends on ESEs, which are specific sequences within exons that promote proper exon definition. ESEs are thought to serve as binding sites for specific serine/arginine-rich (SR) proteins; the SR proteins bind to specific RNA sequences and promote proper exon identification by the splicing machinery (28). Of note, a strong SRp40 binding site (TCACTCG) is located within the exon 11 sequences that are targeted by ASO 074 and 324 (http://rulai.cshl.edu/tools/ESE2/index.html). Thus, it seems likely that ASOs 074 and 324 mask an ESE required for exon definition, reducing normal splicing and increasing abnormal splicing.
Although our studies showed that specific ASOs directed against exon 11 sequences promoted alternate splicing, it would be a mistake to conclude that exon 11 sequences were the only sequences that affect splicing. Indeed, an ASO corresponding to sequences at the beginning of intron 11 also induced significant amounts of progerin expression. The latter findings were in keeping with genetic observations by Moulson et al. (12), who showed that a single-nucleotide mutation at the beginning of intron 11 dramatically increased levels of progerin transcripts.
We identified one ASO (ASO 365), located upstream from the alternate splice donor site, that suppressed alternative splicing, lowered levels of progerin transcripts and lowered progerin levels in cells. However, the magnitude of the effects was fairly modest and certainly was less impressive, at both the RNA and protein levels, than the effects of ASO 324 or ASO 074 in promoting progerin synthesis. An earlier study found that repeated transfections of a morpholino oligonucleotide directed against the exon 11 splice donor site inhibited alternate splicing (4). In the current study, we tested multiple ASOs with sequences spanning the exon 11 splice donor site, and the effects of these oligonucleotides were small, and clearly were less impressive than the modest effects of ASO 365. We do not why our results were different from the earlier study (4), but several factors could be relevant. The ASOs in our studies were 2′-MOE ribose oligonucleotides rather than morpholino oligonucleotides. Also, the concentrations of ASOs that we used (2.5–100 nm) were lower than those used in the earlier study (15–40 µm).
Although the reduction in progerin synthesis with ASO 365 was modest, it is conceivable that this ASO, or other ASOs with similar properties, could be therapeutically useful. Progerin transcript and protein levels were reduced by only ∼30% with ASO 365, but even this small effect might still be relevant from a therapeutic perspective. Relatively small changes in progerin production have been shown to have a major impact on the severity of progeria (12). Also, reducing the levels of farnesyl-prelamin A in Zmpste24-deficient mice by 50% completely eliminated disease phenotypes (23). Another reason for optimism about the possibility of ASO therapy is that 2′-MOE ribose oligonucleotides are extremely stable and have already been tested extensively in humans (29–31).
Our ability to modulate alternative splicing within exon 11 ASOs raises an extremely intriguing possibility for future studies—that it might be possible to use ASOs to modulate the physiologically normal alternative splicing event that occurs within exon 10. Alternative splicing within exon 10 controls the relative rates of synthesis of lamin C and prelamin A transcripts. Recent studies by our group (32) showed that mice lacking prelamin A (‘lamin C-only’ mice) are healthy, fertile and robust, strongly suggesting that prelamin A and lamin A are dispensable, at least in the laboratory mouse. Identifying an ASO that promotes the production of lamin C transcripts at the expense of prelamin A transcripts might pave the way to an entirely new therapeutic approach for HGPS (32).
MATERIALS AND METHODS
Fibroblast cell lines
Human HGPS fibroblasts (AG11498, AG06917, AG11513, AG03513, AG06297 and GM01972; all with the G608G mutation), control human fibroblasts (CRL-1474, AG07095, AG08470, AG16409, CRL-7469 and GM01651) and HeLa cells were from the American Type Culture Collection (Manassas, VA, USA) or Coriell Cell Repository (http://locus.umdnj.edu/ccr/) (3). Skin fibroblasts from a RD patient (22) were provided by Drs Casey Moulson and Jeffrey Miner (Washington University, St Louis, MO, USA). Cells were cultured with conditions recommended by the cell repositories. In some experiments, fibroblasts were incubated with an FTase inhibitor, ABT-100 (33). Untreated fibroblasts were incubated with the vehicle alone (dimethylsulfoxide).
ASOs and cell transfection experiments
Uniform 2′-MOE ASOs with a phosphorothioate backbone were synthesized with an Applied Biosystems 380B automated DNA synthesizer (Applied Biosystems, Foster City, CA, USA) as described previously (29). ASOs used in this study are listed in Supplementary Material, Table S1.
The ASOs were transfected into fibroblasts as described (32,34). Fibroblasts were plated in 6- or 24-well dishes and allowed to adhere overnight. The fibroblasts were washed with reduced serum Opti-MEM I medium (Invitrogen, Carlsbad, CA, USA) and transfected with 2′-MOE-modified oligonucleotides (2.5–100 nm) with 4 µl/ml Lipofectamine 2000 (Invitrogen) at 37°C for 5–7 h. The medium was then removed and replaced with fresh culture medium, and the fibroblasts were cultured for an additional 1–5 days.
RT–PCR studies
RNA was prepared with Trizol reagent (Gibco, Rockville, MD, USA), and RT–PCR reactions were performed with AmpliTaq (ABI, Foster City, CA, USA) and 50 ng of cDNA. A portion of the prelamin A transcript (corresponding to exon 8–12 sequences) was amplified with oligonucleotides 5′-GATGATCCCTTGCTGACTTAC-3′ and 5′-GATTACATGATGCTG CAGTTCTG-3′. The thermal cycling conditions were as follows: 95°C for 3 min, 95°C for 15 s, 60°C for 30 s and 72°C for 15 s (for a total of 26 cycles). PCR products were size-fractionated on 1.3% agarose gels, and DNA was visualized by ethidium bromide staining and UV photography or by SYTO 60 staining (Invitrogen) (1:16 000 dilution) and imaging with an Odyssey infrared scanner (LI-COR Biosciences, Lincoln, NE, USA).
Levels of progerin transcripts were quantified by SYBR Green-based quantitative RT–PCR with previously described primers and reaction conditions (4). Levels of other transcripts were also measured by quantitative PCR with the oligonucleotide primers listed in Supplementary Material, Table S2.
Western blot analysis
Fibroblasts were washed with phosphate-buffered saline, and urea-soluble extracts were prepared as described (35). Protein extracts were size-fractionated on 4–12% gradient SDS–polyacrylamide Bis-Tris gels (Invitrogen), and the separated proteins were electrophoretically transferred to a sheet of nitrocellulose. Western blots were performed with a 1:400 dilution of anti-lamin A/C goat IgG (sc-6215, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and a 1:1000 dilution of goat anti-actin goat IgG (sc-1615, Santa Cruz Biotechnology). Antibody binding was detected with a 1:6000 dilution of horseradish peroxidase-labeled anti-goat IgG (sc-2020, Santa Cruz Biotechnology) and the ECL Plus chemiluminescence system (GE Healthcare, Buckinghamshire, UK) or with a 1:6000 dilution of IRDye 700 anti-goat IgG antibody (Rockland Immunochemicals, Gilbertsville, PA, USA) and scanning on the Odyssey infrared image scanner.
In some studies, lamins A, lamin C and progerin were isolated by immune precipitation. For these experiments, cell pellets collected from six-well plates were resuspended in 0.3 ml ice-cold lysis buffer (50 mm Tris, 0.15 m NaCl, 1 mm EDTA, 1% NP-40, 2.5 mg/ml deoxycholate, 0.1% SDS, 1 mm PMSF) containing Complete Mini protease inhibitors (Roche, Indianapolis, IN, USA) and incubated on ice for 10 min. After removing insoluble material by centrifugation, cell extracts were pre-cleared by incubating with 25 µl of Protein G–agarose (50% suspension in lysis buffer) (Roche) at 4°C for 30 min. Next, goat anti-lamin A/C antibody (15 µl) (Santa Cruz Biotechnology) was added and incubated overnight at 4°C. Immune complexes were isolated with Protein G–agarose (4°C for 2 h), followed by centrifugation. The bound proteins were released by boiling for 3 min in SDS–PAGE loading buffer containing 2-ME.
Metabolic labeling studies
Fibroblasts were incubated for 36 h with an analog of farnesol, AG (30 µm) (36) in the presence and absence of a protein FTI ABT-100. Cell extracts were prepared and the proteins analyzed by western blotting with an AG-specific monoclonal antibody (36). Binding of the antibody was detected with a 1:5000 dilution of IRDye 700 anti-mouse IgG antibody (Rockland Immunochemicals) and infrared scanning. In some studies, lamin proteins were isolated by immune precipitation prior to analysis.
Microscopy and analysis of nuclear shape
Fibroblasts were grown on coverslips, fixed in 3% paraformaldehyde, permeabilized with 0.2% Triton X-100 and blocked with PBS containing 10% fetal bovine serum and 0.2% bovine serum albumin (23). Cells were incubated for 1 h with antibodies against LAP2β (1:200, BD Transduction Laboratories, San Jose, CA, USA) followed by a 1:600 Alexa Fluor 488-conjugated anti-mouse antibody (A21202, Molecular Probes). DNA was visualized with DAPI. Images were obtained on an Axiovert 200 MOT microscope (Carl Zeiss, Thornwood, NY, USA) with a 63 × 1.25 oil immersion objective and processed with AxoVision 4.2 software (Zeiss). Nuclear shape abnormalities were scored by two independent observers blinded to genotype (8,11,23). Differences in the numbers of misshapen nuclei were assessed with a χ2-test.
2D-DIGE
Proteomic analysis of cells was performed by 2D-DIGE. Fibroblasts in six-well plates were treated with ASO 324 (which promotes ‘HGPS splicing’) or ASO 333 (the control oligonucleotide), and after 2 days the cells were harvested. The cell pellets were frozen in liquid nitrogen and stored at −80°C until analysis. The samples were analyzed by Applied Biomics (Hayward, CA, USA). Equal amounts of protein were labeled with cyanine dyes (e.g. Cy2, Cy3), mixed together and separated by 2D-gel electrophoresis. The gels were imaged on a Typhoon TRIO scanner (American Biosciences) and individual protein spots were quantified with DeCyder software (version 6.5) after normalization to an internal standard generated by mixing equal portions of the two test samples and labeling with a third dye (e.g. Cy5). To identify protein spots of interest, preparative gels were run, stained with Deep Purple and the stained proteins imaged with DeCyder software. Protein spots were picked with an Ettan Spot Picker (Amersham Biosciences), digested with trypsin and analyzed by MALDI-TOF/TOF with an ABI 4700 mass spectrometer. The NCBI database was searched with MASCOT and candidates with either protein C.I.% or Ion C.I.% score > 95 were considered significant. All of the proteins identified had protein C.I.% scores > 97%.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by National Institutes of Health Grants AR050200, HL76839, HL86683, GM66152, a March of Dimes grant 6-FY2007-1012, and an Ellison Medical Foundation Senior Scholar Award. Funding to pay the Open Access charge was provided by the National Institutes of Health.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Casey Moulson and Jeffrey Miner (Washington University, St Louis, MO, USA) for ZMPSTE24-deficient human fibroblasts (from an infant with restrictive dermopathy).
Conflict of Interest statement. Two of the authors of this manuscript, T.A.V. and C.F.B., are employed by Isis Pharmaceuticals.
REFERENCES
- 1.Cao H., Hegele R.A. LMNA is mutated in Hutchinson–Gilford progeria (MIM 176670) but not in Wiedemann–Rautenstrauch progeroid syndrome (MIM 264090) J. Hum. Genet. 2003;48:271–274. doi: 10.1007/s10038-003-0025-3. [DOI] [PubMed] [Google Scholar]
- 2.de Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C.L., Munnich A., Le Merrer M., et al. Lamin A truncation in Hutchinson–Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 3.Eriksson M., Brown W.T., Gordon L.B., Glynn M.W., Singer J., Scott L., Erdos M.R., Robbins C.M., Moses T.Y., Berglund P., et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scaffidi P., Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson–Gilford progeria syndrome. Nat. Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang S., Chen L., Libina N., Janes J., Martin G.M., Campisi J., Oshima J. Correction of cellular phenotypes of Hutchinson–Gilford Progeria cells by RNA interference. Hum. Genet. 2005;118:444–450. doi: 10.1007/s00439-005-0051-7. [DOI] [PubMed] [Google Scholar]
- 6.Shumaker D.K., Dechat T., Kohlmaier A., Adam S.A., Bozovsky M.R., Erdos M.R., Eriksson M., Goldman A.E., Khuon S., Collins F.S., et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl Acad. Sci. USA. 2006;103:8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dechat T., Shimi T., Adam S.A., Rusinol A.E., Andres D.A., Spielmann H.P., Sinensky M.S., Goldman R.D. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc. Natl Acad. Sci. USA. 2007;104:4955–4960. doi: 10.1073/pnas.0700854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang S.H., Bergo M.O., Toth J.I., Qiao X., Hu Y., Sandoval S., Meta M., Bendale P., Gelb M.H., Young S.G., et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson–Gilford progeria syndrome mutation. Proc. Natl Acad. Sci. USA. 2005;102:10291–10296. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang S.H., Meta M., Qiao X., Frost D., Bauch J., Coffinier C., Majumdar S., Bergo M.O., Young S.G., Fong L.G. Treatment with a protein farnesyltransferase inhibitor improves disease phenotypes in mice with a targeted Hutchinson–Gilford progeria syndrome mutation. J. Clin. Invest. 2006;116:2115–2121. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scaffidi P., Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toth J.I., Yang S.H., Qiao X., Beigneux A.P., Gelb M.H., Moulson C.L., Miner J.H., Young S.G., Fong L.G. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl Acad. Sci. USA. 2005;102:12873–12878. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moulson C.L., Fong L.G., Gardner J.M., Farber E.A., Go G., Passariello A., Grange D.K., Young S.G., Miner J.H. Increased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromes. Hum. Mutat. 2007;28:882–889. doi: 10.1002/humu.20536. [DOI] [PubMed] [Google Scholar]
- 13.Capell B.C., Erdos M.R., Madigan J.P., Fiordalisi J.J., Varga R., Conneely K.N., Gordon L.B., Der C.J., Cox A.D., Collins F.S. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson–Gilford progeria syndrome. Proc. Natl Acad. Sci. USA. 2005;102:12879–12884. doi: 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glynn M.W., Glover T.W. Incomplete processing of mutant lamin A in Hutchinson–Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum. Mol. Genet. 2005;14:2959–2969. doi: 10.1093/hmg/ddi326. [DOI] [PubMed] [Google Scholar]
- 15.Csoka A.B., English S.B., Simkevich C.P., Ginzinger D.G., Butte A.J., Schatten G.P., Rothman F.G., Sedivy J.M. Genome-scale expression profiling of Hutchinson–Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell. 2004;3:235–243. doi: 10.1111/j.1474-9728.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- 16.Goldman R.D., Shumaker D.K., Erdos M.R., Eriksson M., Goldman A.E., Gordon L.B., Gruenbaum Y., Khuon S., Mendez M., Varga R., et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson–Gilford progeria syndrome. Proc. Natl Acad. Sci. USA. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Favreau C., Dubosclard E., Ostlund C., Vigoroux C., Capeau J., Wehnert M., Higuet D., Worman H., Courvalin J.-C., Buendia B. Expression of lamin A mutated in the carboxyl-terminal tail generates an aberrant nuclear phenotype similar to that observed in cells from patients with Dunnigan-type partial lipodystrophy and Emery-Dreifuss muscular dystrophy. Exp. Cell Res. 2003;282:14–23. doi: 10.1006/excr.2002.5669. [DOI] [PubMed] [Google Scholar]
- 18.Hua Y., Vickers T.A., Baker B.F., Bennett C.F., Krainer A.R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5:e73. doi: 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vickers T.A., Zhang H., Graham M.J., Lemonidis K.M., Zhao C., Dean N.M. Modification of MyD88 mRNA splicing and inhibition of IL-1beta signaling in cell culture and in mice with a 2′-O-methoxyethyl-modified oligonucleotide. J. Immunol. 2006;176:3652–3661. doi: 10.4049/jimmunol.176.6.3652. [DOI] [PubMed] [Google Scholar]
- 20.Hua Y., Vickers T.A., Okunola H.L., Bennett C.F., Krainer A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sazani P., Gemignani F., Kang S.H., Maier M.A., Manoharan M., Persmark M., Bortner D., Kole R. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat. Biotechnol. 2002;20:1228–1233. doi: 10.1038/nbt759. [DOI] [PubMed] [Google Scholar]
- 22.Moulson C.L., Go G., Gardner J.M., van der Wal A.C., Smitt J.H., van Hagen J.M., Miner J.H. Homozygous and compound heterozygous mutations in ZMPSTE24 cause the laminopathy restrictive dermopathy. J. Invest. Dermatol. 2005;125:913–919. doi: 10.1111/j.0022-202X.2005.23846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fong L.G., Ng J.K., Meta M., Cote N., Yang S.H., Stewart C.L., Sullivan T., Burghardt A., Majumdar S., Reue K., et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc. Natl Acad. Sci. USA. 2004;101:18111–18116. doi: 10.1073/pnas.0408558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young S.G., Fong L.G., Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid. Res. 2005;46:2531–2558. doi: 10.1194/jlr.R500011-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Scaffidi P., Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008;10:452–459. doi: 10.1038/ncb1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Worman H.J., Bonne G. ‘Laminopathies’: a wide spectrum of human diseases. Exp. Cell Res. 2007;313:2121–2133. doi: 10.1016/j.yexcr.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prakash T.P., Allerson C.R., Dande P., Vickers T.A., Sioufi N., Jarres R., Baker B.F., Swayze E.E., Griffey R.H., Bhat B. Positional effect of chemical modifications on short interference RNA activity in mammalian cells. J. Med. Chem. 2005;48:4247–4253. doi: 10.1021/jm050044o. [DOI] [PubMed] [Google Scholar]
- 28.Cartegni L., Chew S.L., Krainer A.R. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 29.Baker B.F., Lot S.S., Condon T.P., Cheng-Flournoy S., Lesnik E.A., Sasmor H.M., Bennett C.F. 2′-O-(2-methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J. Biol. Chem. 1997;272:11994–12000. doi: 10.1074/jbc.272.18.11994. [DOI] [PubMed] [Google Scholar]
- 30.Dean N.M., Bennett C.F. Antisense oligonucleotide-based therapeutics for cancer. Oncogene. 2003;22:9087–9096. doi: 10.1038/sj.onc.1207231. [DOI] [PubMed] [Google Scholar]
- 31.Crooke R.M., Graham M.J., Lemonidis K.M., Whipple C.P., Koo S., Perera R.J. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J. Lipid Res. 2005;46:872–884. doi: 10.1194/jlr.M400492-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Fong L.G., Ng J.K., Lammerding J., Vickers T.A., Meta M., Cote N., Gavino B., Qiao X., Chang S.Y., Young S.R., et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Invest. 2006;116:743–752. doi: 10.1172/JCI27125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferguson D., Rodriguez L.E., Palma J.P., Refici M., Jarvis K., O’Connor J., Sullivan G.M., Frost D., Marsh K., Bauch J., et al. Antitumor activity of orally bioavailable farnesyltransferase inhibitor, ABT-100, is mediated by antiproliferative, proapoptotic, and antiangiogenic effects in xenograft models. Clin. Cancer Res. 2005;11:3045–3054. doi: 10.1158/1078-0432.CCR-04-2041. [DOI] [PubMed] [Google Scholar]
- 34.Vickers T.A., Koo S., Bennett C.F., Crooke S.T., Dean N.M., Baker B.F. Efficient reduction of target RNAs by small interfering RNA and RNase H-dependent antisense agents. A comparative analysis. J. Biol. Chem. 2003;278:7108–7118. doi: 10.1074/jbc.M210326200. [DOI] [PubMed] [Google Scholar]
- 35.Dalton M., Sinensky M. Expression systems for nuclear lamin proteins: farnesylation in assembly of nuclear lamina. Methods Enzymol. 1995;250:134–148. doi: 10.1016/0076-6879(95)50068-5. [DOI] [PubMed] [Google Scholar]
- 36.Troutman J.M., Roberts M.J., Andres D.A., Spielmann H.P. Tools to analyze protein farnesylation in cells. Bioconjug. Chem. 2005;16:1209–1217. doi: 10.1021/bc050068+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.