

Abstract
Natural killer (NK) cells recognize and destroy cancer cells through a variety of mechanisms. They may also modulate the adaptive immune response to cancer by interacting with DCs and T cells. Although NK cells play an important role in tumor suppression, little is known about the mechanisms of their recruitment to tumors. Previously it has been shown that subcutaneous tumor growth is enhanced in mice lacking selectins, a family of cell adhesion molecules that mediate the first step of immune cell entry into tissue from the blood. Here we demonstrate that NK cell recruitment to tumors is defective in selectin-deficient mice. In vivo NK cell depletion, either pharmacologic or genetic, leads to enhanced subcutaneous tumor growth, similar to the phenotype observed in the selectin-deficient animals. We also show that, although NK cells from selectin-deficient mice appear developmentally normal, and are functional in in vitro assays, their in vivo function is impaired. This study reveals a role for selectins in NK cell recruitment to tumors and in regulation of effective tumor immunity.
Keywords: Cancer, tumor surveillance, natural killer cells, selectins, leukocyte migratio
Introduction
Primary tumor growth is a complex process, involving many interactions between the tumor and surrounding tissue. A developing tumor influences and is influenced by its stroma, initiates angiogenesis, and interacts with both the adaptive and innate immune systems. The concept of the immune system surveying the body for nascent tumors and suppressing tumor growth dates to the beginning of the 20th century. While the immune surveillance hypothesis has had a turbulent history (1, 2), it is now generally accepted that the immune system plays an important role in controlling cancer progression (1, 3-5).
Innate immune cells clearly participate in immune surveillance. There is clinical and experimental evidence to implicate macrophages (6, 7), mast cells (8, 9), eosinophils (10), dendritic cells (DCs) (11) and natural killer (NK) cells (12, 13) in tumor interactions. While many innate immune cells have both tumor-promoting and tumor-suppressive functions (14, 15), NK cells are generally considered tumor-suppressive (1, 16, 17). Through a variety of positive and negative receptors, NK cells can recognize (2, 16) and eliminate tumor cells by several mechanisms (16, 18). In addition to their well-known cytotoxic functions, NK cells are also gaining recognition as key regulators of the adaptive immune response. Several recent studies indicate that NK cells can activate DCs (19, 20) and influence T cell differentiation (20, 21). NK cell-related cancer immunotherapies are being actively investigated (22, 23).
Although much progress has been made in understanding the processes of NK cell target recognition, target killing, and cross-talk with other immune cells, the cell adhesion mechanisms that mediate NK cell traffic to the tumor are still unclear (24, 25). NK cells are not tissue-resident cells in most organs. However, NK cells need contact with tumor cells in order to recognize and eliminate them (16). Therefore cell adhesion mechanisms that would allow NK cells to exit the circulation and enter into tissues must play an important role in NK-mediated tumor surveillance.
Classical immune cell recruitment requires chemokines, integrins and selectins. Selectins are members of the C-type lectin family of cell adhesion molecules. E-selectin is expressed on activated endothelium, P-selectin is rapidly translocated to the cell surface from intracellular storage granules in activated endothelium and in platelets, and L-selectin is constitutively expressed on leukocytes. Selectins mediate initial rolling of leukocytes along activated endothelium, as has been demonstrated for neutrophils, monocytes and T cells (26-28). The rolling step allows for activation of leukocyte integrins, leading to firm leukocyte adhesion to endothelium, and subsequent extravasation into tissue (27, 28). Selectins are critical for immune cell trafficking, as revealed by defective leukocyte rolling and recruitment to lymphoid organs and to sites of inflammation in selectin-deficient mice (29-32). It is known that NK cells express L-selectin and selectin ligands, and can use them for rolling in in vitro studies (24, 25). However, there is little prior work to indicate whether selectin-dependent recruitment mechanisms are important for NK cell traffic in vivo.
Previous experiments from our laboratory demonstrated that human tumors injected subcutaneously into Rag2-/- mice grow significantly larger in the absence than in the presence of selectins (33). This phenotype is largely transferable with bone marrow transplant. These results implicated selectins and the innate immune system in tumor immune surveillance. Since selectins are known to play a role in immune cell traffic, we hypothesized enhanced tumor growth in the absence of selectins may be due to defective migration of tumor-suppressing immune cells.
In this paper, we have extended the previous xenograft tumor model to immunocompetent C57BL/6 mice. In concordance with previous work on human cell lines, we found that several syngeneic murine tumor cell lines grow significantly better in selectin-deficient mice. We explored leukocyte traffic in selectin-deficient mice, and discovered that NK cell recruitment to tumors is impaired in the absence of selectins. NK cells isolated from selectin-deficient mice appear otherwise normal and functional. In mice depleted of NK cells, either pharmacologically by antibody injection, or genetically in NK-deficient GrzA-Ly49A transgenic mice (34), tumor growth is significantly enhanced. Increased tumor growth in the absence of NK cells is not augmented further by the absence of both NK cells and selectins, arguing that selectins and NK cells act in the same pathway to suppress tumor growth. Furthermore, the ability of NK cells to clear tumors in selectin-deficient mice is defective. These results suggest that NK cells act to suppress tumor growth in our system and depend on selectins to do so.
Materials and Methods
Mice
Mice missing all combinations of selectins were created in our laboratory (29, 31, 32) and back-crossed for 8-9 generations onto C57BL/6, BALB/c, or 129S4 backgrounds. Mice are referred to by the selectins they are missing: L-/- for L-selectin knockouts, ELP-/- for triple selectin knockouts. Immune-deficient mice missing the recombination activating gene 2 (Rag2-/-) on C57BL/6 background were obtained from the laboratory of Dr. Chen (MIT) (35). Mice where NK cell development is disrupted by the insertion of the Ly49A NK receptor under the control of the Granzyme A promoter (GrzA-Ly49Atg) backcrossed to the C57BL/6 background were obtained from the laboratory of Dr. Yokoyama (Washington University St. Louis) (34).
Subcutaneous tumors
For tumor growth assays, B16F0 melanoma, LL/2 Lewis lung carcinoma, and CT26 adenocarcinoma cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). MC38GFP colon carcinoma cells were obtained from the laboratory of Dr. Varki (UCSD) (36). B16-puro and B16-puroRae1ε variants were generated by infecting B16F0 cells with lentiviral vectors containing appropriate constructs (empty vector or vector containing Rae1ε, respectively). The cells were infected and selected in parallel; Rae1ε expression was validated by FACS. The Rae1ε construct was a kind gift from Dr. Raulet (UC Berkeley). All tumor cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), 10% heat-inactivated fetal bovine serum (FBS), glutamine and penicillin/streptomycin (100X stock from Gibco-Invitrogen). For in vivo experiments, anesthetized 8- to 12-week-old mice were injected subcutaneously in the flank with 1×104 tumor cells/mouse in Hank’s Balanced Salt Solution (HBSS, Gibco-Invitrogen), or in 50% HBSS/50% growth factor-reduced Matrigel (BD Biosciences) for Matrigel experiments. For time-course experiments, mice were monitored every 2-3 days by palpation for tumor presence at the injection site. Tumor surface area was calculated by multiplying the length of the tumor by its width. For endpoint experiments, tumors were removed and weighed 21 days after tumor cell injection.
Immune cell purification from Matrigel
Matrigel “plugs” were removed on days 1, 3, 5, 7, and 10 after tumor injection; 4-7 mice of each genotype (C57BL/6, L-/-, ELP-/-) were analyzed at every time point. The “plug” was mechanically dissociated and incubated with agitation at 37°C for 2 hours in 0.2% auto-digested collagenase I (Worthington Biochemical Corporation) in PBS. Digested samples were strained through a 70μm-mesh sieve. Larger pieces were mashed through the sieve with the rubber-capped end of a syringe plunger. A 100μL sample was counted by hemocytometer to establish total cell number in the Matrigel plug. Recovered cells were pelleted by centrifugation and analyzed by FACS.
Flow cytometry
Cells harvested from Matrigel “plugs” or from mouse tissues were analyzed on a four-color FACSCalibur (BD Biosciences) flow cytometer supported by FlowJo software. Antibodies used were against CD3, CD4, CD8, NK1.1, Gr1, CD107a, NKG2D, NKG2A/C/E, Ly49C/I/F/H, Ly49A/D, MEL14 (L-selectin, CD62L), PSGL-1 (CD162), DX5 (α2 integrin, CD49b), LFA-1 (αL integrin, CD11a), Mac-1 (αM integrin, CD11b), CD11c (αX integrin), αV integrin, α5 integrin, and isotype controls (BD Biosciences). Pan-specific anti-mRae1 antibody was obtained from R&D Systems.
In vitro cytotoxicity assay
Yac-1 lymphoma cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in RPMI medium/10% FBS, L-glutamine and penicillin/streptomycin (100X stock from Gibco-Invitrogen). NK cells were isolated by positive selection with anti-DX5 beads (Miltenyi Biotech), plated at ∼1×106 NK cells/well, and activated overnight in complete RPMI medium supplemented with 125U/mL IL-2 (BD Biosciences) and 75ng/mL IL-15 (Sigma). Activated NK cells were co-incubated at varying effector:target ratios with target cells (1×104 Yac-1 cells/well; 2×103 RMA or LL/2 tumor cells/well) for 4 hours. Target cell killing was assayed using the CytoTox Non-Radioactive Cytotoxicity assay (Promega). In L-selectin blocking experiments, NK cells were pre-treated with the L-selectin blocking antibody MEL14 or an isotype control antibody (BD Biosciences), which were present throughout the assay.
NK cell interferon γ production and degranulation
Purified splenic NK cells were either left in culture medium (RPMI+5%FBS) alone for 4 or 30 hours, stimulated with LL/2 tumor cells, or stimulated with NK1.1 activating antibody or an isotype control antibody for 6 hours as described previously (37). Interferon γ (IFNγ) secretion was assayed using the DuoSet ELISA Development System for IFNγ (R&D Systems). In some cases, NK cells were recovered after 6-hour stimulation and analyzed by FACS for surface CD107a expression to assess degranulation in response to activating stimuli.
NK cell antibody depletion
TM-β1 NK-depleting antibody (against mouse IL-2 receptor β) (38) and RR4-7 control antibody (against human T cell receptor variant) were purified in the laboratory from hybridoma supernatants. Hybridoma cells were provided by, respectively, Dr. Tanaka (Osaka University, Japan), and Dr. Eisen (MIT). For NK cell depletion experiments, antibodies were injected intraperitoneally at 1mg/mouse in 500μL of sterile PBS. Antibody injection was performed once, 3 days prior to tumor cell injection. The efficiency of NK cell depletion was monitored throughout the experiment by FACS analysis of small blood samples collected from the tail vein of antibody-treated mice.
Statistical analysis
All data have been subjected to statistical analysis. For graphic representations, data are presented as mean values for at least 3 individual experiments with error bars representing standard error of the mean (SEM). For determining the statistical significance of the differences in the data, two-tailed Student’s t test analysis with the assumption of unequal variance was used. In cases where tumor incidence varied, statistical significance was additionally assayed by a chi2 test. All statistically significant (p<0.05) differences in the data are indicated by asterisks (*) in the figures.
Results
LL/2 tumor growth is enhanced in selectin-null mice
To investigate the mechanism of selectin-dependent tumor suppression in the context of an intact immune system, we extended our previous studies to fully murine models, using inbred mouse strains and subcutaneous injection of syngeneic tumor cell lines. Growth of MC38GFP, B16F0, and LL/2 cells syngeneic to C57BL/6, as well as CT26 cells syngeneic to BALB/c, was assayed in selectin-deficient and in control selectin-replete mice (Fig. 1). Tumors were analyzed at 3 weeks after injection. In all cases, tumors arose more frequently and grew larger in selectin-deficient mice than in matched selectin-replete controls (Fig. 1). In both C57BL/6 and in BALB/c backgrounds, the tumors were largest and most frequent in ELP-/- mice. Using LL/2 cells, which showed the clearest difference in tumor growth between selectin-deficient and control mice, we further explored tumor growth in mice missing single selectins and in mice missing both of the endothelial selectins (PE-/-). Of the single selectin knockouts, L-/- mice were most similar in tumor growth to the triple knockouts, P-/- mice and PE-/- double knockouts showed a mild tumor enhancement, and E-/- animals showed no tumor growth (Fig. 1D). These findings are consistent with previous data using human cell lines (33), and indicate that selectin-dependent tumor suppression is an important phenomenon for a variety of tumor cell lines in multiple mouse strains.
Figure 1. Syngeneic subcutaneous tumor growth is increased in the absence of selectins.
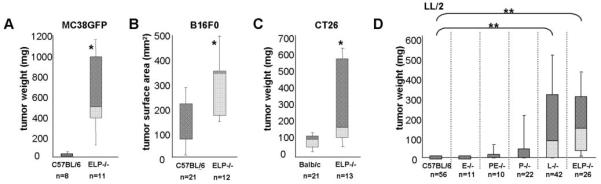
5×104 MC38GFP colon carcinoma (A) or 1×104 B16F0 melanoma (B) syngeneic to C57BL/6, 1×104 CT26 colon carcinoma syngeneic to Balb/c (C), or 1×104 Lewis lung carcinoma (LL/2) syngeneic to C57BL/6 (D), cells were injected subcutaneously into the flank of either control mice or mice lacking selectins. Three weeks after injection, tumors were dissected out and weighed. Where dissection was difficult, tumor growth was assayed by measuring tumor surface area instead of weight (B). Tumor incidence is reported as % mice with tumors out of total mice of the given genotype that were assayed (n). The weights or sizes of the tumors are displayed as box-whisker plots, the central line denoting the mean, the shaded gray boxes and the lines denoting the 90th, 75th, 25th, and 10th percentiles.
*, p<0.05 by two-tailed Student’s t test
**, p<0.0005 two-tailed Student’s t test and p<0.005 by chi2 analysis
Selectin-deficient mice are defective in NK cell recruitment to tumors
Having established an immune-competent fully murine model, we proceeded to investigate the causes of the differences in tumor growth. Since selectins are known to be involved in immune cell recruitment, we hypothesized that selectin-deficient mice fail to reject tumors because tumoricidal immune cells are unable to reach the tumor. To obtain samples which could be readily harvested and assayed for immune cell recruitment, tumor cells were injected as a suspension in Matrigel, a commercial preparation of mouse tumor matrix (39, 40). LL/2 cells embedded in growth-factor-reduced Matrigel were injected into the right flank of selectin-replete or selectin-deficient mice, while Matrigel alone was injected into the left flank of the same mice. Matrigel plugs were removed from the mice over a course of 10 days following injection and analyzed by FACS for immune cell content.
Matrigel enhanced tumor growth in all mice, but differences in tumor size between selectin-replete and selectin-deficient mice were preserved (Fig. 2A). Analysis of total immune cell infiltration into Matrigel plugs demonstrated that the overall immune response to tumor-containing Matrigel was significantly more robust than the immune response to Matrigel alone (Fig. 2B). Matrigel plugs were assayed for the presence of neutrophils, CD4+ T cells, CD8+ T cells, macrophages, DCs and NK cells. Neutrophil recruitment proved neither tumor-specific nor significantly impaired by the absence of selectins. No significant differences in T cell recruitment were detected between selectin-deficient and control mice. We observed some reduction in tumor-specific recruitment of macrophages and DCs in selectin-deficient mice (Supp. Fig. 1). However, since defects in macrophage and DC recruitment were only present either in ELP-/- or in L-/- mice but not in both, while the tumor growth increase was observed both in ELP-/- and in L-/- mice, these differences were not pursued further.
Figure 2. Selectin-null mice are defective in NK cell recruitment to tumors in Matrigel.
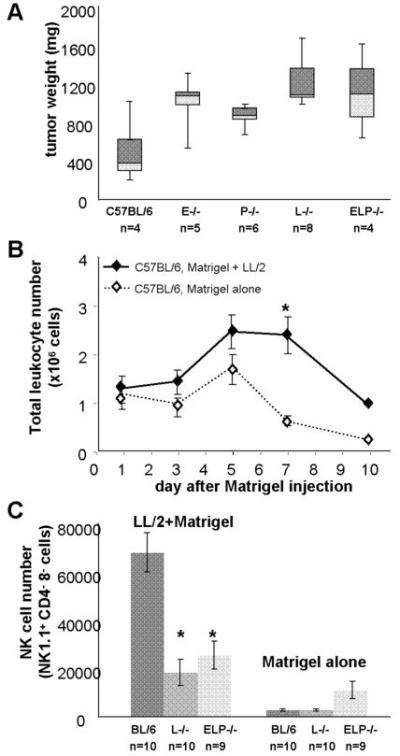
1×104 LL/2 cells in 200μL of Matrigel were injected subcutaneously into the right flank of either C57BL/6 w.t. mice or C57BL/6 selectin-deficient mice. A. Tumors were dissected and weighed three weeks after injection. Results are expressed as box-whisker plots, the central line denoting the mean, the shaded gray boxes and the lines denoting the 90th, 75th, 25th, and 10th percentiles. B. Tumors were dissected over a 10-day period after injection, dissociated into a single-cell suspension, and analyzed by FACS for immune cell content. A plug of 200μL of Matrigel alone injected into the left flank of the same mice at the time of tumor injection was used as an internal control. Total leukocyte counts in the plugs of C57BL/6 control animals established that the tumor-specific immune response was particularly prominent on day 7 after tumor injection. C. At day 7 after tumor injection, NK cell recruitment to Matrigel containing LL/2 tumor cells, but not to Matrigel alone, was significantly reduced in L-/- and ELP-/- mice compared with C57BL/6 w.t. controls. NK cells were defined as NK1.1+, CD4 and CD8- live leukocytes. Results are expressed as mean leukocyte counts obtained from 5-10 individual animals of each genotype, error bars = SEM.
*, p<0.005 by two-tailed Student’s t test.
Analysis of immune cell content of tumors in Matrigel showed a clear and consistent defect in tumor-specific NK cell recruitment both in ELP-/- and in L-/- mice. Early NK cell recruitment to tumors was similar in all mice and independent of tumor cell presence in Matrigel plugs. In Matrigel plugs without tumor cells, NK cell numbers slowly declined over time in all mice. However, NK cell recruitment to tumor cell-containing Matrigel plugs increased steadily in selectin-replete mice, peaking on day 7 after tumor injection. In contrast, NK cell recruitment to tumors in ELP-/- and L-/- mice was markedly reduced, particularly at the peak of NK cell infiltration (Fig. 2C).
These data demonstrate, first, that NK cells are recruited to Matrigel plugs containing tumor cells, but not to Matrigel alone, confirming that NK cell recruitment in our system is not a general reaction to Matrigel nor to tissue injury caused by the injection, but a tumor cell-specific response. Second, the results show that NK cell recruitment to tumors is defective in ELP-/- and L-/- mice. These data establish NK cells as good candidates for mediating selectin-dependent tumor suppression.
NK cell development is unimpaired in the absence of selectins
To ascertain that the defect in NK cell recruitment to tumors in selectin-deficient mice is not due to an overall NK cell deficit in these animals, we performed a comparative analysis of NK cells from selectin-replete and selectin-deficient mice.
First, we confirmed that NK cells isolated from spleens of C57BL/6 mice express L-selectin and the main ligand for all three selectins, PSGL-1, as has been reported (24, 41-43). We found that L-selectin is highly expressed on splenic NK cells, while PSGL-1 is expressed at low levels, and that expression levels of L-selectin and PSGL-1 are not altered by overnight NK cell activation with cytokines (Fig. 3A). To investigate NK cell development in selectin-deficient mice, we performed FACS analysis of blood, bone marrow, and lymphoid organs. NK cells were present in the blood and bone marrow of the ELP-/- and L-/- mice in normal numbers (Fig. 3B). NK cell numbers in the lymph nodes of L-/- mice were reduced, while NK cell numbers in the spleens of ELP-/- mice were increased, in keeping with known disturbances of leukocyte recirculation in these mouse strains (30, 32). NK cell receptor repertoire remained largely unaltered in the absence of selectins (Supp. Fig. 2). We next assayed selectin-deficient and selectin-replete mice for changes in NK cell integrin expression that are known to correlate to NK cell developmental stages (44). As with NK cells from selectin-replete animals, integrin αV was downregulated on NK cells from selectin-deficient mice as the cells exited the bone marrow, while the expression of integrin αMβ2, which is a marker of mature activated NK cells, was upregulated (Fig. 3C).
Figure 3. NK cells are developmentally normal in the absence of selectins.
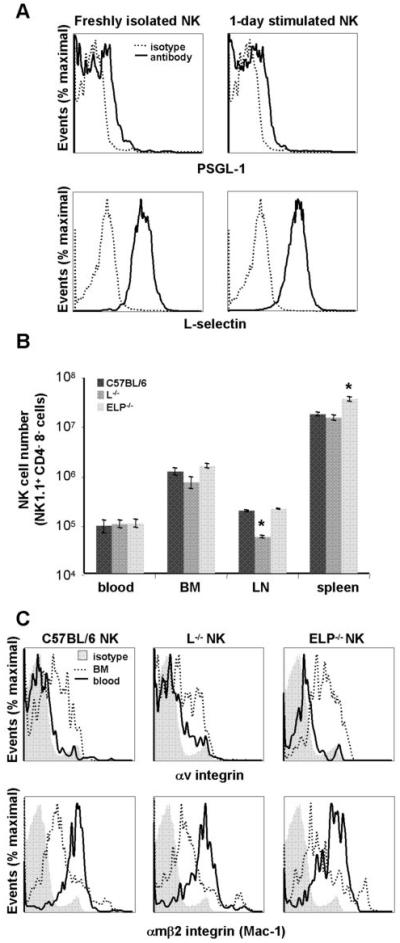
A. NK cells were isolated from spleens of C57BL/6 wt mice and either assayed directly or cultured overnight in activating medium containing IL-2 and IL-15. C57BL/6 NK cells were analyzed by FACS for expression of L-selectin and selectin ligand PSGL-1. B. NK cell numbers in blood (∼100μL), bone marrow (1 femur), lymph node (1 inguinal) and spleen of selectin-replete and selectin-deficient mice were determined from total cell number in the abovementioned tissues multiplied by percentage of NK1.1+, CD3- cells as determined by FACS. Data were obtained from at least 3 mice of each genotype and are presented as average cell numbers ± SEM; statistical significance for selectin-deficient mice as compared to selectin-replete mice within each organ has been calculated; *, p<0.05 by two-tailed Student’s t test. C. Freshly isolated blood and bone marrow collected from C57BL/6 selectin-replete and selectin-deficient mice were analyzed by FACS for expression of αV integrin and CD11b (Mac-1; αMβ2 integrin). As expected, αV integrin expression decreases on blood NK cells as compared with the immature NK cells in the bone marrow, while CD11b expression increases.
Overall, we did not find any large defects in NK cell number or development in the absence of selectins. It is therefore unlikely that such disturbances could account for the defect in NK cell recruitment to tumors in selectin-deficient mice. However, we did confirm that NK cells normally express L-selectin and a major selectin ligand PSGL-1, which could potentially contribute to NK cell recruitment to tumors.
Selectin-deficient NK cells retain tumoricidal and secretory functions
To explore whether the inability to reach the tumor was the only functional impairment of selectin-deficient NK cells, we assayed their ability to kill tumor targets and secrete cytokines in response to stimulation in vitro. NK cells from ELP-/- and L-/- mice were as efficient as control NK cells at destroying Yac-1 and RMA tumor targets (Fig. 4A, B). Similarly, when L-selectin was blocked acutely using the MEL14 antibody, no defects were observed in NK cell killing of LL/2 targets (Fig. 4C). NK cells are one of the major secretors of the tumor-suppressing cytokine IFNγ (16, 45). We tested NK cells isolated from spleens of selectin-deficient mice for their ability to produce IFNγ in response to stimulation. Selectin-deficient NK cells did not differ significantly from selectin-replete counterparts in IFNγ production in response to LL/2 tumor cell stimulation or to NK1.1 antibody crosslinking of an activating NK receptor (Fig. 4D, left panel). Selectin-deficient NK cells also displayed enhanced degranulation in response to both activating stimuli (Fig. 4D, right panel). In summary, NK cells from selectin-deficient mice appear functional, apart from the in vivo defect in recruitment to tumors.
Figure 4. NK cells from selectin-deficient mice are functional in vitro.
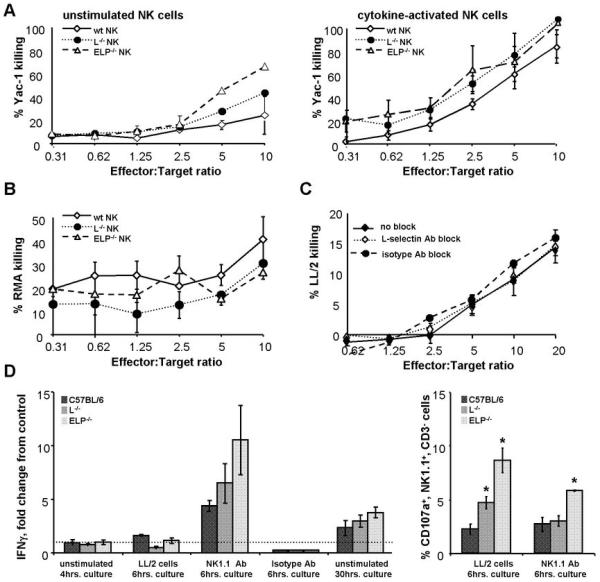
A, B. NK cells were isolated from spleens of selectin-deficient and control C57BL/6 mice. Purified NK cells were co-cultured with varying ratios of tumor target cells in a standard cytotoxicity assay. Cytotoxicity of either freshly isolated NK cells (A, left panel) or NK cells activated overnight in medium containing IL-2 and IL-15 (A, right panel; B, C) was determined. Target killing was measured using the Promega CytoTox 96® Non-Radioactive Cytotoxicity Assay. NK cells from selectin-deficient mice (A, B), as well as NK cells treated with L-selectin-blocking antibody MEL14 or an isotype control antibody prior to and during the assay (C), displayed normal ability to kill tumor targets. D, left panel: IFNγ secretion by purified control and selectin-deficient NK cells was assayed by ELISA. NK cells were either left in culture medium with no additional stimulation for 4hrs. after isolation, stimulated by co-culture with LL/2 tumor cells for 6hrs, stimulated by NK1.1 antibody or an isotype control antibody for 6hrs, or allowed to remain in culture medium for 30hrs. Data are normalized to IFNγ secretion by control NK cells cultured with medium alone for 4hrs. Selectin-deficient NK cells do not demonstrate any statistically significant differences from selectin-replete C57BL/6 NK cells within each group. D, right panel: in addition, NK cells stimulated by LL/2 tumor cells or by the NK1.1 antibody for 6hrs. were assayed for degranulation by CD107a FACS analysis. All analyses were performed for at least 3 animals of each genotype. Results are expressed as bar and line graphs, error bars = SEM
*, p<0.05 by two-tailed Student’s t test.
Subcutaneous tumor growth is enhanced in the absence of NK cells
To test whether loss of NK cells contributes to increased tumor growth, we removed NK cells by two independent methods. Mice were injected with TM-β1 antibody that selectively depletes NK cells for up to 3 weeks (38), or a control RR4-7 antibody of the same isotype that is unreactive to mouse antigens. In a parallel set of experiments, we used GrzA-Ly49A transgenic mice that genetically lack functional NK cells (34, 46). NK depletion by either method was confirmed by monitoring NK cell numbers in the blood (Fig. 5A, B). The antibody depletion was thorough and stable, while a few NK cells were detected in GrzA-Ly49Atg+ mice. However, these cells are reportedly immature and of low cytotoxic potential (34, 46). We then implanted subcutaneous LL/2 tumors into mice lacking NK cells. Depletion of NK cells, either by antibody or genetically, led to significantly enhanced subcutaneous tumor growth in NK-depleted animals as compared with matched controls (Fig. 5C).
Figure 5. NK cells, but not adaptive immune cells, suppress subcutaneous tumor growth.
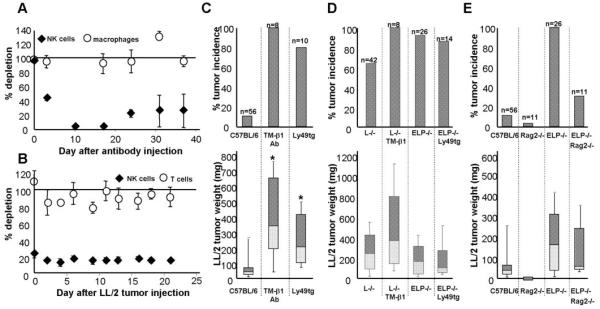
A. As measured by the % NK1.1+/CD3- cells in the blood, mice injected with TM-β1 NK-depleting antibody (1 mg/mouse i.p.) selectively lose NK cells but not macrophages as compared with mice treated with the same dose of an isotype control antibody. NK cell numbers decrease from 3*106 circulating NK cells/mouse on day 0 to 1.2*106 NK cells on day 3 to 6*104 NK cells by day 10 after antibody treatment. B. Similarly, Ly49A-GrzAtg+ mice that are genetically deficient in NK cells, as compared with tg- littermates, remain depleted of NK cells (∼5*105 circulating NK cells/mouse) even after 3 weeks of tumor challenge, while T cells are unaffected. Results are expressed as % NK cells in NK-depleted animals as compared with the appropriate controls ± SEM. Tumor weight was assayed 3 weeks after injection of 1×104 LL/2 cells into: C. C57BL/6 mice depleted of NK cells, either pharmacologically by TM-β1 antibody injection 3 days prior to tumor cell transfer, or genetically in Ly49tg+ mice; D. selectin-deficient mice depleted of NK cells; or E. lacking the adaptive immune system (Rag2-/-). Tumor incidence is reported as % mice with tumors out of total mice of the given genotype that were assayed (n). The weights or sizes of these tumors that grew are displayed as box-whisker plots, the central line denoting the mean, the shaded gray boxes and the lines denoting the 90th, 75th, 25th, and 10th percentiles; plotted below are the corresponding tumor incidences.
*, p<0.05 by two-tailed Student’s t test.
We next wanted to know whether removing NK cells could explain the increased tumor growth in selectin-deficient mice. We therefore depleted NK cells in selectin-deficient mice and monitored tumor growth. Depletion of NK cells from L-/- mice with the TM-β1 antibody did not result in any synergistic enhancement of tumor growth; nor did tumors grow better in ELP-/- GrzA-Ly49Atg+ mice than in ELP-/- animals (Fig. 5D). Therefore, it is likely that the defect in tumor rejection that we observed in selectin-deficient mice is due to a defect in NK cell recruitment. To test whether loss of adaptive immune cells also affects tumor growth in selectin-deficient mice, we repeated the LL/2 tumor growth experiments in Rag2-/- and ELP-/- Rag2-/- mice. In contrast to our results with NK cell depletion, the absence of T, B and NKT cells did not promote tumor growth in control mice and slightly decreased tumor incidence in selectin-deficient mice (Fig. 5E).
NK cell depletion had a similar effect on tumor growth to that seen with loss of selectins. Furthermore, depletion of NK cells from selectin-deficient mice had no additional effect on tumor growth, showing that NK cells are unable to participate in tumor rejection in selectin-null mice, while they do so in selectin-replete controls. This indicates that NK cells are necessary for tumor rejection, and may require selectins in order to function in anti-tumor immunity. The adaptive immune system does not appear to participate in tumor rejection in our system.
NK-mediated tumor rejection in vivo requires selectins
To test whether selectins are required for NK cell-mediated tumor rejection, we used a tumor rejection model that is strongly dependent on NK cells. The B16 C57BL/6 melanoma cell line does not express NK cell ligands and is fairly resistant to NK cell killing. However, B16 cells transfected with Rae1, a ligand to an activating NK cell receptor, NKG2D, are rejected specifically by NK cells (12). We generated matched B16 cell lines to test NK cell function in vivo: a control cell line (B16-puro) and a cell line stably expressing Rae1ε (B16-Rae1ε). Rae1ε expression in the matched cell lines was confirmed by FACS analysis (Fig. 6A).
Figure 6. Selectin-deficient NK cells are defective in tumor target cell rejection in vivo.
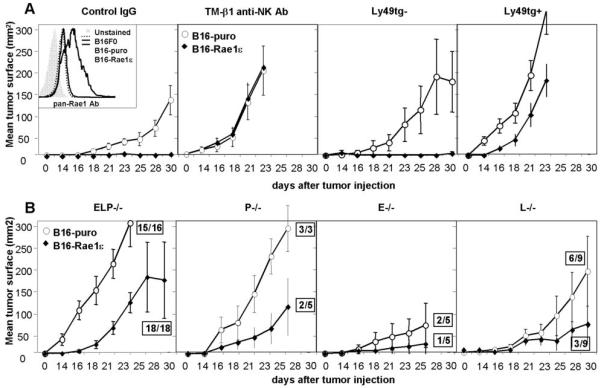
A. Inset: B16F0 tumor cells were stably infected with either a construct carrying the Rae1ε NK cell-activating antigen (B16-Rae1ε cells) or with a vector control (B16-puro cells). Rae1ε antigen expression was confirmed by FACS. 1×104 B16-Rae1ε or B16-puro cells were injected s.c. into mice treated with control or NK-depleting TM-β1 antibody, or into Ly49A-GrzAtg+ and control tg- animals. Tumor growth was assayed by measuring tumor length (L) and width (W) with calipers every other day and multiplying the values together to obtain tumor surface area (L*W=surface area, mm2). B. 1×104 B16-Rae1ε or B16-puro cells were injected s.c. into selectin-deficient mice. Boxed numbers indicate number of mice that developed a tumor out of the total number of mice assayed. Results are expressed as line graphs of mean tumor surface area of 5-18 individual mice, error bars = SEM
When injected into control mice, B16-puro cells grew sizeable tumors in all animals, while B16-Rae1ε cells were completely rejected, as expected. Also, as previously reported (12), rejection was NK cell-specific, since both cell lines gave rise to tumors in GrzA-Ly49Atg+ mice or in mice treated with the TM-β1 antibody (Fig. 6A). When injected into ELP-/- mice, however, B16-Rae1ε tumors were not rejected (18/18 mice). These results demonstrate a profound defect in NK cell-mediated tumor rejection in selectin-deficient mice (Fig. 6B). Of the single selectin knockouts, L-/- mice and P-/- mice displayed partial defects in B16-Rae1ε tumor rejection, while E-/- mice did not show a significant defect.
These data confirm that selectin-deficient mice have a clear defect in NK-mediated tumor rejection in vivo. They also demonstrate that both L-selectin and P-selectin contribute to NK cell anti-tumor function in vivo, while E-selectin does not contribute significantly.
Discussion
In this study we used subcutaneous tumor models to explore the role of selectins in tumor surveillance by innate immune cells. Existing evidence indicated that, in the absence of selectins, subcutaneous growth of human tumor cell lines in Rag2-/- mice is enhanced, and that the innate immune system is likely responsible for selectin-dependent tumor suppression (33). Here we report increased growth of several syngeneic tumor cell lines of different origins in selectin-deficient mice, on several genetic backgrounds, confirming that selectin-dependent tumor suppression is a reproducible and important phenomenon. This phenotype was most apparent in the absence of all three selectins. Of the single selectin knockouts, L-selectin contributed most to tumor suppression, P-selectin contributed somewhat, and E-selectin did not contribute significantly. The predominant role of L-selectin in tumor suppression suggested that leukocytes are the key mediators of the observed phenotype.
Selectins play a role in immune cell recruitment to tissues and to lymphoid organs. Exploration of immune cell recruitment to Matrigel-embedded tumors in selectin-deficient mice led to several important findings. First, we established that all mice, regardless of the presence or absence of selectins, exhibited a tumor-specific immune response. Second, we found fewer macrophages, DCs and NK cells recruited to tumors in the absence of selectins. The defect in NK cell recruitment in the absence of all three selectins, or of L-selectin alone, was particularly striking. To explore the possibility that other aspects of NK cell biology are also affected in the absence of selectins, we confirmed that NK cells are present in selectin-deficient mice in normal numbers, appear to develop normally, and to be functional in in vitro anti-tumor cytotoxicity and in cytokine secretion assays. Since NK cells are known to be tumoricidal, seemed normal in the absence of selectins, but failed to infiltrate tumors in selectin-deficient mice, they became our top candidates for mediating selectin-dependent tumor suppression.
NK cells proved essential for tumor suppression in our system, as growth of several subcutaneously injected tumor cell lines was enhanced in two different NK cell-deficient mouse models. In contrast, the adaptive immune system did not actively promote tumor immunity. Tumor growth in Rag2-/- mice lacking B, T and NKT cells was, if anything, slightly inhibited. The slight inhibition of tumor growth in Rag2-/- animals could be due to several factors, including increased NK cell numbers in Rag2-/- mice, and lack of regulatory T cells, which could normally act to suppress tumor immunity and have recently been shown to suppress NK cell function (47). To test directly the function of NK cells in selectin-deficient mice, we used a subcutaneous tumor model where tumor rejection is strongly dependent on NK cells. Selectin-deficient mice demonstrated a severe defect in NK cell-dependent elimination of tumor targets in vivo, establishing that selectins are necessary for this process.
Taken together, our data provide evidence that selectins are necessary for tumor elimination; that NK cells are necessary for tumor elimination; and that NK cells require selectins to clear tumor targets in vivo. We also found that, in the absence of selectins, NK cells are defective in entering the tumor. The simplest model suggested by these findings is that in our system NK cells require selectins, particularly L-selectin, and selectin ligands in order to interact with tumor vasculature, enter the tumor, and promote its rejection. Our findings support and extend previous reports, in which it has been demonstrated that in L-selectin-deficient mice, NK cell numbers around primary or metastatic tumors are reduced (48).
The work described here leaves some open questions. One important caveat to the proposed model is that it is unclear whether the defect in NK cell function observed in selectin-deficient mice is cell-intrinsic, or partly attributable to defects in other selectin-dependent cell(s) that regulate NK cell recruitment or function. NK cells are not the only innate immune cell type affected by the absence of selectins, as we also observed some defects in tumor-specific recruitment of macrophages and DCs in selectin-deficient mice. Thus, NK cells and their recruitment are necessary but may not be sufficient to explain selectin-dependent tumor suppression.
In addition, it is worth noting that while tumor growth seemed to be strongly dependent on NK cells in our system, other immune cells may be more important in different tumor models. Several factors could influence the immune response to tumors in our model. First, we inject fully transformed tumor cells, in a procedure that is concomitant with some tissue damage. A naturally arising tumor that proceeds through all stages of transformation in vivo may well interact differently with its microenvironment and with the immune system than our tumor model does. Second, tumor growth in our system is assayed on the timescale of 3 weeks, which is a relatively short period. NK cells are an early-response immune cell; it is possible that in models where tumor growth is slower the adaptive immune response would have time to develop and would contribute to tumor rejection. Lastly, the location and anatomical origin of the tumor may influence the immune response to that tumor. Nonetheless, our results clearly demonstrate an important contribution of selectins and NK cells to tumor rejection. It would therefore be interesting to address the role of NK cells and selectins in spontaneous tumor models.
Furthermore, selectins are likely not the only adhesion molecules that are required for NK cell migration to the tumor. Chemokines and chemokine receptors, as well as integrins, may also be necessary. Of the chemokines, CX3C chemokine (fractalkine) and its receptor CX3CR1 have been implicated in NK cell recruitment to tumors (49). Of the integrins, one publication associated αLβ2 and αMβ2 integrins with NK cell tumor migration (48). Previous work from our laboratory also implicates αVβ3 and αVβ5 integrins in tumor suppression (33). It will be interesting to investigate the contribution of other adhesion molecules to tumor surveillance by NK cells.
Lastly, L-selectin is also important for immune cell traffic to lymphoid organs. Our findings do not address this aspect of NK cell migration, but it has been reported that NK cells can use L-selectin to enter lymph nodes in cancer (50). Although the main proposed role for NK cells at the lymph node is in regulating the adaptive immune response, and the adaptive immune system does not appear to control tumor rejection in our system, we cannot rule out the possibility that defects in NK cell traffic to lymphoid organs may play a role in tumor overgrowth in selectin-deficient mice.
The results reported here contribute towards a more complete understanding of tumor immunology, particularly in the poorly understood areas of immune cell migration in cancer and of innate immune response to cancer. We have examined the role of one family of adhesion molecules, the selectins, in mouse models of tumorigenesis, and determined that selectins promote tumor immune surveillance in our experimental system. We have also uncovered some of the mechanisms by which selectins control tumor growth by demonstrating that NK cell recruitment and in vivo tumor-suppressive functions are defective in the absence of selectins. These results strongly implicate selectin-mediated recruitment of NK cells in tumor surveillance. Understanding the migration and recruitment mechanisms of immune cells in cancer, while challenging, is both scientifically interesting and potentially clinically valuable, raising the possibility of targeting the immune response specifically to the tumor.
Acknowledgements
We thank Dr. Toshiyuki Tanaka for providing the TM-β1 hybridoma cells; Dr. Herman Eisen for RR4-7 hybridoma cells; Dr. Ajit Varki for the MC38GFP cells; Dr. Wayne Yokoyama for the GrzA-Ly49A transgenic mice; Dr. David Raulet for the Rae1ε construct; and the MIT Division of Comparative Medicine and the Koch Institute FACS and histology facilities for their help.
Financial support:
NIH, National Cancer Institute grants: U54CA126515 and U54CA112967
Howard Hughes Medical Institute
OS - Ludwig Center at MIT; Biology Department MIT training grant
ALH - UK Research Council Fellow
ROH - Investigator, Howard Hughes Medical Institute
Footnotes
Conflicts of interest: Authors declare no conflicts of interest.
Bibliography
- 1.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 2.Soloski MJ. Recognition of tumor cells by the innate immune system. Curr Opin Immunol. 2001;13:154–62. doi: 10.1016/s0952-7915(00)00198-9. [DOI] [PubMed] [Google Scholar]
- 3.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 4.Stagg J, Johnstone RW, Smyth MJ. From cancer immunosurveillance to cancer immunotherapy. Immunol Rev. 2007;220:82–101. doi: 10.1111/j.1600-065X.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- 5.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117:1137–46. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vakkila J, Lotze MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol. 2004;4:641–8. doi: 10.1038/nri1415. [DOI] [PubMed] [Google Scholar]
- 7.Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–61. doi: 10.1111/j.1600-065X.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- 8.Conti P, Castellani ML, Kempuraj D, et al. Role of mast cells in tumor growth. Ann Clin Lab Sci. 2007;37:315–22. [PubMed] [Google Scholar]
- 9.DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007;9:212. doi: 10.1186/bcr1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowe D, Jorizzo J, Hutt MS. Tumour-associated eosinophilia: a review. J Clin Pathol. 1981;34:1343–8. doi: 10.1136/jcp.34.12.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim R, Emi M, Tanabe K, Arihiro K. Potential functional role of plasmacytoid dendritic cells in cancer immunity. Immunology. 2007;121:149–57. doi: 10.1111/j.1365-2567.2007.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165–71. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci U S A. 2001;98:11521–6. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 16.Smyth MJ, Hayakawa Y, Takeda K, Yagita H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer. 2002;2:850–61. doi: 10.1038/nrc928. [DOI] [PubMed] [Google Scholar]
- 17.Smyth MJ, Swann J, Hayakawa Y. Innate tumor immune surveillance. Adv Exp Med Biol. 2007;590:103–11. doi: 10.1007/978-0-387-34814-8_7. [DOI] [PubMed] [Google Scholar]
- 18.Takeda K, Akira S. Roles of Toll-like receptors in innate immune responses. Genes Cells. 2001;6:733–42. doi: 10.1046/j.1365-2443.2001.00458.x. [DOI] [PubMed] [Google Scholar]
- 19.Cooper MA, Fehniger TA, Fuchs A, Colonna M, Caligiuri MA. NK cell and DC interactions. Trends Immunol. 2004;25:47–52. doi: 10.1016/j.it.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 20.Kalinski P, Giermasz A, Nakamura Y, et al. Helper role of NK cells during the induction of anticancer responses by dendritic cells. Mol Immunol. 2005;42:535–9. doi: 10.1016/j.molimm.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 21.Martin-Fontecha A, Thomsen LL, Brett S, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5:1260–5. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 22.Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol. 2007;7:329–39. doi: 10.1038/nri2073. [DOI] [PubMed] [Google Scholar]
- 23.Woan K, Reddy V. Potential therapeutic role of natural killer cells in cancer. Expert Opin Biol Ther. 2007;7:17–29. doi: 10.1517/14712598.7.1.17. [DOI] [PubMed] [Google Scholar]
- 24.Gregoire C, Chasson L, Luci C, et al. The trafficking of natural killer cells. Immunol Rev. 2007;220:169–82. doi: 10.1111/j.1600-065X.2007.00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris MA, Ley K. Trafficking of natural killer cells. Curr Mol Med. 2004;4:431–8. doi: 10.2174/1566524043360609. [DOI] [PubMed] [Google Scholar]
- 26.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–56. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 27.Sperandio M. Selectins and glycosyltransferases in leukocyte rolling in vivo. Febs J. 2006;273:4377–89. doi: 10.1111/j.1742-4658.2006.05437.x. [DOI] [PubMed] [Google Scholar]
- 28.Vestweber D, Blanks JE. Mechanisms that regulate the function of the selectins and their ligands. Physiol Rev. 1999;79:181–213. doi: 10.1152/physrev.1999.79.1.181. [DOI] [PubMed] [Google Scholar]
- 29.Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte rolling and extravasation are severely compromised in P selectin-deficient mice. Cell. 1993;74:541–54. doi: 10.1016/0092-8674(93)80055-j. [DOI] [PubMed] [Google Scholar]
- 30.Arbones ML, Ord DC, Ley K, et al. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. 1994;1:247–60. doi: 10.1016/1074-7613(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 31.Frenette PS, Mayadas TN, Rayburn H, Hynes RO, Wagner DD. Double knockout highlights value of endothelial selectins. Immunol Today. 1996;17:205. doi: 10.1016/0167-5699(96)80555-x. [DOI] [PubMed] [Google Scholar]
- 32.Robinson SD, Frenette PS, Rayburn H, et al. Multiple, targeted deficiencies in selectins reveal a predominant role for P-selectin in leukocyte recruitment. Proc Natl Acad Sci U S A. 1999;96:11452–7. doi: 10.1073/pnas.96.20.11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taverna D, Moher H, Crowley D, Borsig L, Varki A, Hynes RO. Increased primary tumor growth in mice null for beta3- or beta3/beta5-integrins or selectins. Proc Natl Acad Sci U S A. 2004;101:763–8. doi: 10.1073/pnas.0307289101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim S, Iizuka K, Aguila HL, Weissman IL, Yokoyama WM. In vivo natural killer cell activities revealed by natural killer cell-deficient mice. Proc Natl Acad Sci U S A. 2000;97:2731–6. doi: 10.1073/pnas.050588297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shinkai Y, Rathbun G, Lam KP, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–67. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 36.Borsig L, Wong R, Hynes RO, Varki NM, Varki A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc Natl Acad Sci U S A. 2002;99:2193–8. doi: 10.1073/pnas.261704098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokoyama WM, Kim S. Analysis of individual natural killer cell responses. Methods Mol Biol. 2008;415:179–96. doi: 10.1007/978-1-59745-570-1_11. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka T, Kitamura F, Nagasaka Y, Kuida K, Suwa H, Miyasaka M. Selective long-term elimination of natural killer cells in vivo by an anti-interleukin 2 receptor beta chain monoclonal antibody in mice. J Exp Med. 1993;178:1103–7. doi: 10.1084/jem.178.3.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleinman HK, McGarvey ML, Hassell JR, et al. Basement membrane complexes with biological activity. Biochemistry. 1986;25:312–8. doi: 10.1021/bi00350a005. [DOI] [PubMed] [Google Scholar]
- 40.Kleinman HK, McGarvey ML, Liotta LA, Robey PG, Tryggvason K, Martin GR. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry. 1982;21:6188–93. doi: 10.1021/bi00267a025. [DOI] [PubMed] [Google Scholar]
- 41.Andre P, Spertini O, Guia S, et al. Modification of P-selectin glycoprotein ligand-1 with a natural killer cell-restricted sulfated lactosamine creates an alternate ligand for L-selectin. Proc Natl Acad Sci U S A. 2000;97:3400–5. doi: 10.1073/pnas.040569797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frey M, Packianathan NB, Fehniger TA, et al. Differential expression and function of L-selectin on CD56bright and CD56dim natural killer cell subsets. J Immunol. 1998;161:400–8. [PubMed] [Google Scholar]
- 43.Uksila J, Salmi M, Butcher EC, Tarkkanen J, Jalkanen S. Function of lymphocyte homing-associated adhesion molecules on human natural killer and lymphokine-activated killer cells. J Immunol. 1997;158:1610–7. [PubMed] [Google Scholar]
- 44.Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annu Rev Immunol. 2004;22:405–29. doi: 10.1146/annurev.immunol.22.012703.104711. [DOI] [PubMed] [Google Scholar]
- 45.Loza MJ, Zamai L, Azzoni L, Rosati E, Perussia B. Expression of type 1 (interferon gamma) and type 2 (interleukin-13, interleukin-5) cytokines at distinct stages of natural killer cell differentiation from progenitor cells. Blood. 2002;99:1273–81. doi: 10.1182/blood.v99.4.1273. [DOI] [PubMed] [Google Scholar]
- 46.Kim S, Song YJ, Higuchi DA, et al. Arrested natural killer cell development associated with transgene insertion into the Atf2 locus. Blood. 2006;107:1024–30. doi: 10.1182/blood-2005-04-1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ralainirina N, Poli A, Michel T, et al. Control of NK cell functions by CD4+CD25+ regulatory T cells. J Leukoc Biol. 2007;81:144–53. doi: 10.1189/jlb.0606409. [DOI] [PubMed] [Google Scholar]
- 48.Yamada M, Yanaba K, Hasegawa M, et al. Regulation of local and metastatic host-mediated anti-tumour mechanisms by L-selectin and intercellular adhesion molecule-1. Clin Exp Immunol. 2006;143:216–27. doi: 10.1111/j.1365-2249.2005.02989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lavergne E, Combadiere B, Bonduelle O, et al. Fractalkine mediates natural killer-dependent antitumor responses in vivo. Cancer Res. 2003;63:7468–74. [PubMed] [Google Scholar]
- 50.Chen S, Kawashima H, Lowe JB, Lanier LL, Fukuda M. Suppression of tumor formation in lymph nodes by L-selectin-mediated natural killer cell recruitment. J Exp Med. 2005;202:1679–89. doi: 10.1084/jem.20051473. [DOI] [PMC free article] [PubMed] [Google Scholar]