

Abstract
BACKGROUND & AIMS
Helicobacter pylori-induced gastric epithelial cell (GEC) apoptosis is a complex process that includes activation of the tumor suppressor p53. p53-mediated apoptosis involves p53 activation, bax transcription and cytochrome c release from mitochondria. Apurinic/apyrimidinic endonuclease-1 (APE-1) regulates transcriptional activity of p53: H. pylori induce APE-1 expression in human GEC. H. pylori infection increases intracellular calcium ion concentration [Ca2+]i of GEC, which iinduces APE-1 acetylation. We investigated the effects of H. pylori infection and APE-1 acetylation on GEC apoptosis.
METHODS
AGS cells (wild-type or with suppressed APE-1), KATO III cells and cells isolated from gastric biopsies were infected with H. pylori. Effects were examined by immunoblotting, real time RT-PCR, immunoprecipitation, immunofluorescence microscopy, chromatin immunoprecipitation, mobility shift, DNA-binding and luciferase assays.
RESULTS
H. pylori infection increased [Ca2+]i and acetylation of APE-1 in GEC, but the acetylation status of APE-1 did not affect the transcriptional activity of p53. In GEC, expression of a form of APE-1 that could not be acetylated increased total and mitochondrial levels of Bax, and induced release of cytochrome c and fragmentation of DNA; expression of wild-type APE-1 reduced these apoptotic events. We identified a negative calcium response element (nCaRE) in the human bax promoter and found that poly (ADP-ribose) polymerase-1 (PARP-1) recruited the acetylated APE-1/histone deacetylase-1 (HDAC-1) repressor complex to bax nCaRE.
CONCLUSIONS
H. pylori-mediated acetylation of APE-1 suppresses Bax expression; this prevents p53-mediated apoptosis when H. pylori infect GEC.
Half of the world’s population is infected with Helicobacter pylori1 leading to gastritis, duodenal and gastric ulcer, and gastric cancer.2 Severity of disease is associated with the cag (cytotoxin associated gene) pathogenicity island (PAI).3 Interplay between bacterial factors and host signal transduction pathways determine host cell apoptotic or antiapoptotic events.4, 5 H. pylori-mediated apoptosis involves activation of MHC class II,6 p53,7 Fas/FasL system,8 and TNF-related apoptosis inducing ligand (TRAIL).9 H. pylori enhance Bax expression10 and mitochondrial translocation leading to GEC death.11 Antiapoptotic mechanisms induced in H. pylori-infected epithelium may ensure persistence of infection.12
We demonstrated earlier that H. pylori infection and reactive oxygen species augment apurinic/apyrimidinic endonuclease-1 (APE-1) expression in human GEC13 and that it modulates bacterial pathogenesis by controlling chemokine expression in GEC.14 APE-1 is a ubiquitous, multifunctional protein induced in oxidative stress 15, 16 and is involved in base excision repair.17 In addition, APE-1, also named redox factor-1 (Ref-1), reductively activates several transcription factors18, 19 including cell proliferation and apoptosis regulators such as p53.20 p53 is a major transcriptional activator of the proapoptotic genes e.g. bax, PUMA, Noxa and a repressor of antiapoptotic genes such as bcl2.21
Another relatively unexplored role of APE-1 is Ca2+-dependent repression of genes containing negative calcium response elements (nCaREs) in their promoters22, 23. PTH, renin, and APE-1 itself are the only known genes regulated by APE-1-nCaRE interaction. Acetylation on K6/K7 increases affinity of APE-1 for PTH nCaRE and augments binding of HDAC-1 to the nCaRE complex repressing PTH transcription. 24 Complementation experiments show that microinjection of K6R/K7R mutant does not replace the role of wild type APE-1 in preventing of apoptosis.25 Thus, acetylation plays a major role in regulating APE-1 function.
As APE-1 activates p53 but forced overexpression of APE-1 prevents apoptosis,26 we examined this “paradoxical role” of APE-1 in H. pylori-mediated GEC apoptosis. We found induction of p53–mediated apoptotic events in H. pylori-infected GEC that were augmented when APE-1 was suppressed. H. pylori infection enhanced APE-1 acetylation in cultured human GEC and in primary cells isolated from gastric biopsies. We report that the human bax promoter contains an nCaRE and demonstrate that PARP-1 recruits the ac-APE1-HDAC-1 repressor complex to the nCaRE. In spite of the overall induction of bax transactivation in H. pylori infected gastric epithelium ac-APE-1 had a suppressive effect on bax transcription. We conclude that ac-APE-1 functions as a critical molecule in H. pylori infection-induced alterations of GEC homeostasis via regulation of bax.
Materials and Methods
Plasmids
pcDNA-APE-1-Flag, pcDNA-K6R/K7R-APE-1-Flag, pCMV-p300, pCMV-ΔHAT plasmids were used. pcDNA3-Flag-p53, pcDNA3-Flag-ΔNp73 and PG13 reporter plasmid (contains 13 tandem repeats of p53 binding site) were kindly provided by Dr. Alex Zaika, Department of Surgery, Vanderbilt University Medical Center, Nashville, TN, USA. Human bax promoter (−972 to +12) with either WT/mutated nCaRE-B element was cloned in pGL2 basic (Promega).
Cell Culture and Bacterial Strains
AGS cells were grown in Ham’s F/12 (Hyclone) containing 10% FBS (Hyclone). p53-deficient gastric cancer-derived KATO III cells were maintained in RPMI 1640 media (Hyclone) supplemented with 10% heat-inactivated FBS. H. pylori 26695, a cag PAI(+) strain (ATCC), and its isogenic mutant, cag PAI(−) strain 8-1, were maintained on blood agar plates (Becton Dickinson). The bacteria were cultured overnight at 37°C in Brucella broth (GIBCO-BRL) with 10% FBS under microaerophilic conditions before infection.
Human Gastric Epithelial Cell Isolation from Mucosal Biopsy Specimens
Gastric biopsies from the antral gastric mucosa were collected from adult patients undergoing esophagogastroduodenoscopy, according to a University of Virginia Institutional Review Board approved protocol. Epithelial cells were isolated 6, 13, 27 and resuspended in RPMI 1640 containing 10% FBS. 5 × 105 cells were plated in 12 well plates, allowed to adhere for 5 h and then infected with H. pylori 26695 or 8-1 at MOI 300 for 3 h.
Stable APE-1 Knockdown in AGS
APE-1 was stably knocked down using shRNA in AGS cells. We derived stable cell expressing empty pSIRENRetro-Q vector (pSIREN cells) and three other cell lines expressing recombinant pSIRENRetro-Q with APE-1 shRNA (shRNA cells).
Real-Time RT-PCR to Assess APE-1 Suppression
shRNA-mediated stable suppression was analyzed by real-time RT-PCR.
Treatment of Cells
Normal (WT), pSIREN-RetroQ empty vector- or APE-1 shRNA-expressing AGS cells (pSIREN and shRNA cells respectively), freshly isolated GECs or KATO III cells were infected. As described in earlier studies13 multiplicity of infection (MOI) 300 for 3 h was the optimum dose to induce APE-1. We performed an initial dose-response study, which confirmed that MOI 300 was optimum to induce acetylation of APE-1. When required, cells were preincubated with BAPTA-AM (2 or 5 μM) or 100 ng/ml Trichostatin A (both from Sigma) for 1 h followed by coincubation with H. pylori.
Transient Transfections
Cells were transiently transfected with various plasmids including luciferase constructs and PARP-1 siRNA.
Western Blotting and Immunoprecipitation
Protein expression of APE-1, p53, phospho Ser-15 p53, PARP-1, HDACs, poly ADP ribose, cytochrome c, Bcl2 or Bax was assessed by western blot. Co-immunoprecipitation experiments were performed to analyze components that bind to p300 and APE-1.
Indirect Immunofluorescence Microscopy
Bax was detected in cells using immunofluorescence microscopy.
Chromatin Immunoprecipitation (ChlP) Analysis
ChIP assay was performed using a commercial kit from Imgenex.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extracts were prepared from treated cells and EMSA was performed to analyze bax-nCaRE-bound components.
Affinity Purification of nCaRE-Bound Proteins
Streptavidin-coated superparamagnetic beads (Dynal, AS) coated with biotinylated bax nCaRE oligonucleotide were incubated with nuclear extracts prepared from H. pylori 26695-infected or uninfected AGS cells and analyzed.
Cell Death Detection ELISA
DNA fragmentation was checked using a commercially available kit (Cell Death Detection ELISAPLUS, Roche Molecular Biochemicals).
Detailed methods for APE-1 knockdown, real time RT-PCR, transfection, western blotting, immunofluorescence microscopy, EMSA and affinity purification are described in Supplementary Methods Online at www.gastrojournal.org.
RESULTS
p53-Mediated Apoptosis by H. pylori is Enhanced by Downregulation of APE-1 in AGS cells
We established stable APE-1-suppressed AGS cells to better delineate the effect of altering APE-1. We used empty vector (pSIREN), APE-1 shRNA-expressing (shRNA) cells and nontransfected AGS (WT). Figure 1A and Supplementary Figure 1 show shRNA-mediated APE-1 suppression. We previously confirmed that H. pylori cag PAI(+) strain 26695 infection at MOI 300, for 3 h optimally induced APE-1 in GEC.13 This dose also induced p53 phosphorylation in AGS. Western blot of whole cell lysates showed induction of PO4-Ser-15 p53 although total p53 was unchanged by H. pylori 26695 infection (Figure 1B). Similar results were observed with the cag PAI (−) H. pylori strain 8-1 (data not shown). Unless mentioned otherwise, H. pylori strain 26695 was used for all further experiments.
Figure 1.

APE-1 acetylation suppresses Bax expression in H. pylori infected AGS cells.
(A) Basal APE-1 at mRNA (real time RT PCR) and protein level (Western blot) in WT, pSIREN and shRNA cells. Bars depict normalized data (mean ± SEM, n=4), *, p <0.05.
(B) A representative western blot showing p53 phosphorylation in AGS cells infected with MOI 300 of H. pylori for 3h.
(C) The effect of p53 and APE-1 on p53-phosphorylation and Bax and Bcl2 expression was examined by overexpressing empty vector, WT p53, ΔNp73 in pSIREN and shRNA cells (D) Indirect immunofluorescence microscopy determined Bax expression in empty vector, APE-1 or K6R/K7R-expressed shRNA cells infected with H. pylori. Nucleus was stained by DAPI (blue), Bax was stained by Alexa 488-conjugated secondary antibody (green), mitochondria was stained by MitoTracker Red (CMSRos) (red) and merged image of Bax and mitochondria was yellow.
To determine the effect of p53 and APE-1 in H. pylori-infected GEC apoptosis we overexpessed empty vector, WT p53 and ΔNp73 (blocks apoptotic effects of p53) in pSIREN and shRNA cells, followed by H. pylori infection for 6 h at MOI of 300. Western blot of whole cell lysates (Figure 1C) showed that H. pylori augmented Ser-15 phosphorylation of constitutive and overexpressed p53 to the same extent in pSIREN and shRNA cells but this was blocked by ΔNp73. p53 overexpression enhanced Bax expression in shRNA to a greater extent than in pSIREN cells. Bcl2, another p53 target, was induced in pSIREN and shRNA cells by H. pylori with some downregulation by p53 overexpression (Figure 1C and Supplementary Figure 2).
K6R/K7R Nonacetylable Mutant of APE-1 Induces Bax Expression
To determine the effect of APE-1 acetylation on Bax expression in H. pylori-infected GEC, we overexpressed APE-1 or K6R/K7R in shRNA cells and compared the subcellular localization and amount of Bax using immunofluorescence microscopy (Figure 1D). H. pylori at MOI 300 for 6 h induced cytosolic Bax in all groups. Although Bax levels in empty vector and APE-1-transfected cells were similar with mainly nuclear Bax and a sparse amount in the cytosol, K6R/K7R significantly increased cytosolic Bax with some translocated to mitochondria. H. pylori further increased Bax translocation to mitochondria in K6R/K7R transfected cells.
H. pylori Enhance APE-1 Acetylation in GEC
WT, pSIREN and shRNA cells were infected for 3 h at MOI 100, 150, 300 of H. pylori 26695. Western blot of whole cell lysates and densitometric analysis confirmed dose-dependent increases of APE-1 expression with maximal acetylation induced by MOI 300 (Figure 2A). cag PAI(−) 8-1 showed similar response as strain 26695 (data not shown). Supplementary Figure 5 shows similar induction of ac-APE-1, PO4-p53 and Bax in MKN-45, NCI-N87 and Kato III cells.
Figure 2.

H. pylori-induced APE-1 acetylation is a cag PAI-independent event.
(A) WT, pSIREN and shRNA cells were infected with MOI 100, 200, 300 of H. pylori for 3 h and analyzed by western blotting. Graphs summarize the densitometric analyses of APE-1 and ac-APE-1 proteins (n=3).
(B) Comparison of APE-1 acetylation by western blot analysis of whole cell lysates of gastric epithelial cells isolated from gastric biopsies from uninfected human subjects and infected in vitro for 3 h with cag PAI(+) (26695) and cag PAI(−) (8-1) H. pylori strains.
(C) Densitometric analysis of immunoblots identical to the experiment represented in panel A and depicts normalized APE-1 and ac-APE-1 (mean ± SEM, n=4) in cag PAI(+) and cag PAI(−) H. pylori-infected cells. Bar graphs normalized as percent increase over uninfected controls (p < 0.05 for all bars).
(D) Ratios of ac-APE-1 to total APE-1 protein in biopsy-derived GEC from uninfected (n=4) and three naturally H. pylori infected subjects, *, p <0.05.
H. pylori Induce APE-1 Acetylation in GEC Isolated from Human Gastric Biopsy Specimens
To determine the effect of H. pylori infection in native gastric epithelium, we isolated epithelial cells from H. pylori-infected and uninfected human gastric biopsy samples. Representative western blots show constitutive APE-1 expression in uninfected GEC and short term in vitro infection with H. pylori increased the levels of both total and acetylated-APE-1 (Figure 2B) but APE-1 expression and acetylation were cag PAI-independent events (Figure 2C). Expression of ac-APE-1 in cells from three naturally H. pylori–infected subjects was 8-fold higher (P<0.05) than those from uninfected individuals (n=4) (Figure 2D)
Intracellular Calcium Ion Concentration [Ca2+]i Mediates p300-Driven Acetylation of APE-1 in H. pylori-Infected GEC
shRNA cells were transfected with empty vector or APE-1 plasmid and infected with H. pylori for 3 h at a MOI 300. Infection enhanced APE-1 protein in both empty vector and APE-1-transfected cells (Figure 3A and Supplementary Figure 3). Acetylation was detected in APE-1-overexpressed cells, which was further enhanced by H. pylori. Live H. pylori have been shown to cause transient increase in [Ca2+]i in human gastric epithelial mucous cells.28 Pretreatment of cells with Ca2+i-chelator BAPTA-AM for 1 h, followed by exposure to media or H. pylori in APE-1 expressed shRNA cells decreased ac-APE-1 suggesting a role of Ca2+i in APE-1 acetylation. Ca2+i-chelation had no effect on total APE-1 level.
Figure 3.

H. pylori-induced increase in [Ca2+]i promotes APE-1′s interaction with p300 in AGS cells.
(A) Comparison of empty vector and APE-1 overexpressed shRNA cells infected with H. pylori ± BAPTA-AM pretreatment to assess the effect of Ca2+i on APE-1 expression and acetylation. Summary of densitomety analyses (n=3) is shown in Supplementary Figure 3.
(B) Immunoprecipitation of whole cell lysates from untreated or BAPTA-AM pretreated, cag PAI(+) or cag PAI(−) H. pylori-infected AGS cells with p300 antibody and western analysis using lys 6 ac-APE-1 antibody.
(C) Same as shown in Figure 3(B) except that ac-lysine antibody was used for detection.
(D) Western analysis of APE-1-immunoprecipitate from infected or uninfected AGS.
(E) Effect of Ca2+i- chelation with BAPTA-AM and HDAC inhibition with TSA on APE-1 acetylation as assessed by western analysis.
(F) Western analysis of lysates of cells cotransfected with APE-1 WT or K6R/K7R mutant and WT p300 or ΔHAT constructs in shRNA cells ± H. pylori infection.
The transcriptional coactivators p300/CBP regulate transcription factors via acetylation. We examined whether p300 is involved in H. pylori-induced acetylation of APE-1 in AGS. Immunoprecipitation of whole cell lysates with p300 antibody followed by western blot with Lys 6 ac-APE-1 (Figure 3B) or acetyl lysine antibody (Figure 3C) showed an increased level of ac-APE-1 in H. pylori-infected cells, which was inhibited by BAPTA-AM. Reprobing with APE-1 antibody confirmed that Ca2+i mediated the interaction of APE-1 with p300 as BAPTA-AM reduced the amount of APE-1 immunoprecipitated by p300 antibody (Figure 3B, Figure 3C). These experiments also demonstrated that APE-1 acetylation is a cag PAI-independent event. APE-1-p300 interaction after H. pylori infection was further demonstrated by immunoprecipitation of whole cell lysates with APE-1 antibody followed by western blot with p300 antibody (Figure 3D). Reprobing with ac-lys antibody established that H. pylori infection resulted in an interaction of p300 with APE-1, possibly leading to enhanced acetylation of APE-1. Trichostatin A (TSA), an inhibitor of HDACs, induced more acetylation than H. pylori (Figure 3E) indicating that the steady level of ac-APE-1 is regulated by both p300 and HDACs after infection. TSA had no effect on total APE-1 levels. To assess HAT activity of p300 in APE-1 acetylation, we cotransfected shRNA cells with empty vector or APE-1 plasmids along with either p300 or p300 HAT domain-deleted mutant (ΔHAT). APE-1 acetylation occurred only when WT p300 and APE-1 were both overexpressed in shRNA cells and H. pylori further enhanced APE-1 acetylation (Figure 3F). H. pylori-mediated APE-1 acetylation was specific for Lys 6/7 residues since p300 failed to acetylate K6R/K7R even after H. pylori challenge (Figure 3 F).
H. pylori Induce Formation of a Multiprotein Repressor Complexat bax nCaRE
Our promoter analysis identified an nCaRE-B like sequence in the human bax promoter (−626 to −612 from the translation initiation site). Underlined regions indicate the nCaRE region (Figure 4A). In vivo binding of APE-1 to the bax nCaRE was analyzed by ChIP assay using APE-1 antibody. The PCR product of DNA in the APE-1 immunocomplex corresponding to the human bax promoter flanking the nCaRE sequence demonstrated specific binding of APE-1 which was not present in the PCR product corresponding to the 5′ far upstream region (Figure 4 B). Supplementary Figure 4 represents real-time PCR of the immunoprecipitated bax nCaRE region. Ca2+i-dependent binding was confirmed by a BAPTA-AM mediated decrease in the amount of immunoprecipitate. Furthermore, increased complex formation after TSA treatment indicates enhanced promoter binding of APE-1 after acetylation. EMSA showed a substantial increase in the complex formation after H. pylori infection (Figure 4C). As shown, two major nCaRE oligo-protein complexes were formed. Competition with unlabeled nCaRE oligo diminished both upper and lower bands. The specificity of binding was further confirmed by using nCaRE-mutant oligo (Lane 4), unlabeled WT competitor oligo (Lane 5) and nCaRE-mutated oligo (Lane 6). Preincubation of the nuclear extract with APE-1 antibody (Lane 7) significantly reduced the amount of the complexes suggesting APE-1 as a component of the complexes. A greater level of complex was observed with pSIREN cell nuclear lysate compared to shRNA cells (Lanes 8-11) emphasizing APE-1′s interaction with the bax nCaRE. Individual overexpression of APE-1 and of p300 in shRNA cells increased the H. pylori-induced binding of nuclear proteins to the nCaRE oligo and their combined overexpression further increased complex formation (data not shown).
Figure 4.
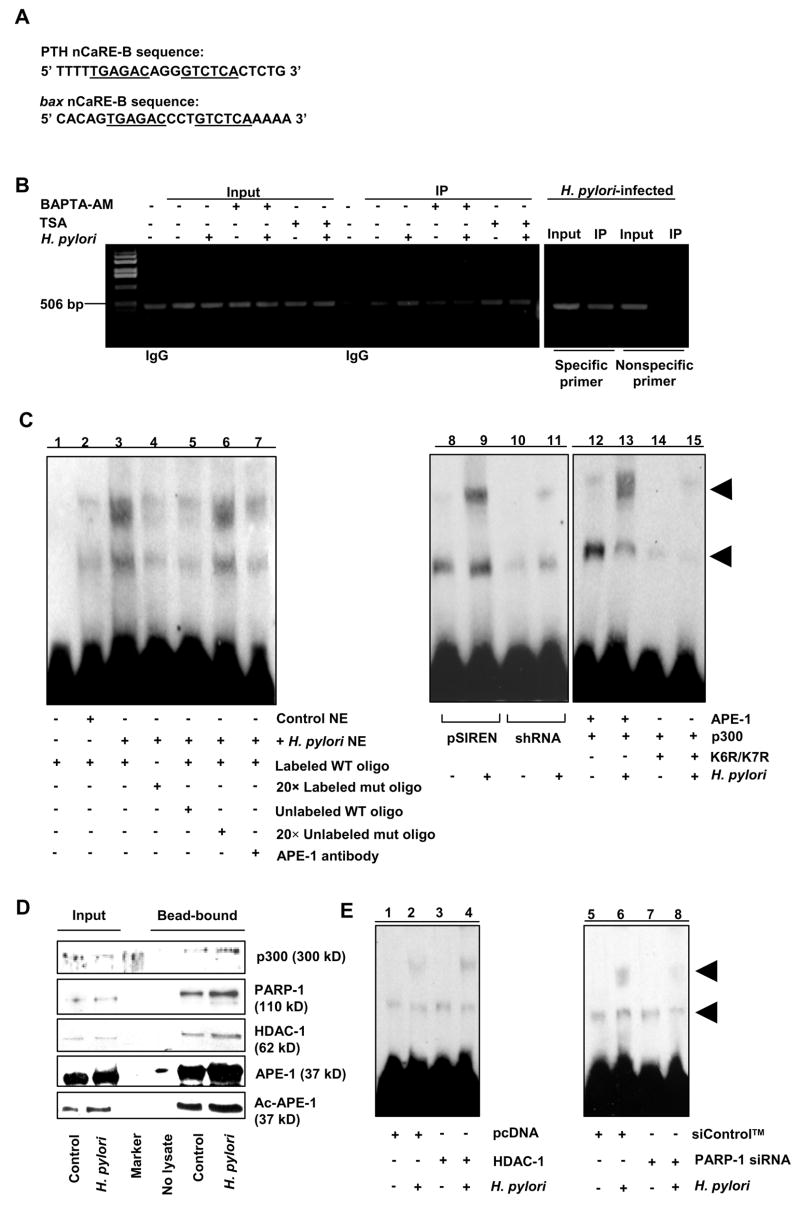
Components of the multiprotein complex that bind to bax promoter nCaRE.
(A) The nCaRE-B sequences of the human PTH and bax promoter.
(B) ChIP analysis of APE-1 immunocomplex for nCaRE-containing bax promoter. Right panel, specificity of APE-1 binding to bax nCaRE by comparing 5′ far upstream region with nCaRE-flanking region of the bax promoter. Quantitative real-time PCR analysis is shown in Supplementary Figure 4.
(C) EMSA to evaluate the binding of APE-1 and p300 to bax nCaRE oligo. NE= Nuclear extract.
(D) Western analysis of proteins bound to streptavidin beads coated with nCaRE oligo.
(E) EMSA to assess the binding of HDAC-1 and PARP-1 to nCaRE oligo.
In contrast, K6R/K7R did not induce complex formation even in the presence of p300 indicating the requirement of APE-1 acetylation for its binding to the nCaRE (Figure 4C, lanes 12–15). Streptavidin-coated magnetic beads were used to further analyze bax nCaRE oligo-bound complex. Analysis of bead-bound proteins by western blot (Figure 4D) confirmed the presence of p300, APE-1, HDAC-1 and PARP-1. The presence of PARP-1 was reconfirmed by mass spectrometry. nCaRE-bound HDAC-1 and PARP-1 levels were increased by H. pylori. We found that HDAC-1 overexpression followed by H. pylori infection stimulated formation of the upper EMSA band to a much larger extent suggesting that the larger complex contained HDAC-1 (Figure 4E, lanes 1–4). PARP-1 suppression by siRNA reduced level of complex implicating PARP-1 as an nCaRE-bound component (Figure 4E, lanes 5–8).
Immunoprecipitation of whole cell lysates with PARP-1 antibody followed by western blotting analysis demonstrated that H. pylori enhanced interaction of PARP-1 with HDAC-1, p300 and APE-1 or ac-APE-1 in AGS (Figure 5A). Figure 5B shows that H. pylori-induced binding was abrogated by PARP-1-suppression even after infection indicating that PARP-1 is crucial for recruitment of the protein complex to the nCaRE. None of these proteins were bound to the mutated nCaRE oligo. As neither APE-1 nor p300 can directly bind to nCaRE,24 we sought to determine if PARP-1 directly binds to nCaRE oligo. Incubation of recombinant human PARP-1 with WT and mutated biotinylated nCaRE oligo-coated streptavidin beads showed direct binding of PARP-1 with only WT oligo indicating that PARP-1 had specific binding affinity for WT bax nCaRE even in the absence of other components of the multiprotein complex (Figure 5C). These observations led us to postulate that PARP-1 recruits the protein complex to the nCaRE. H. pylori-induced acetylation of APE-1 by p300 recruits HDAC-1 to the complex thereby repressing bax transcription.
Figure 5.
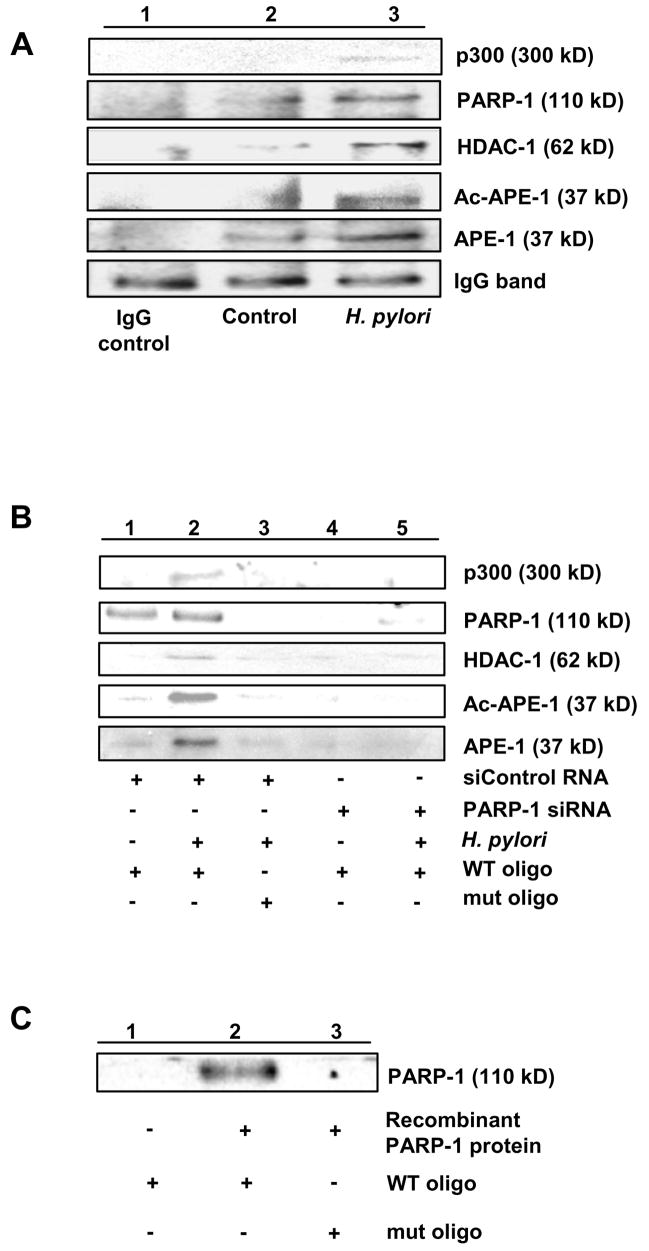
PARP-1 directly binds to bax nCaRE and is a component of nCaRE-bound protein complex.
(A) Enhanced coimmunoprecipitation of APE-1, p300 and HDAC-1 with PARP-1 in H. pylori-infected AGS.
(B) H. pylori infection-induced APE-1-p300-HDAC-1-PARP-1 complex specifically binds to WT nCaRE oligo, but not to mutated oligo, assessed by elution of bound proteins and immunoblotting with respective antibodies. siRNA- mediated PARP-1 suppression decreased the binding of other members to the nCaRE.
(C) Direct and specific binding of purified PARP-1 protein to nCaRE oligo.
Ac-APE-1 Suppresses bax Transcription
Dual luciferase assays were performed with H. pylori-infected (6 h) or uninfected AGS. Enhanced bax transactivation was observed in nCaRE-mutated AGS cells compared to WT confirming the negative regulatory activity of nCaRE on bax transcription. (Figure 6A). Transient transfection of WT bax luciferase construct along with either empty vector or APE-1 or K6R/K7R mutant in shRNA cells demonstrated that luciferase activity after infection in APE-1 transfected cells was lower than empty vector or K6R/K7R mutant-transfected group suggesting a repressor function of ac-APE-1 in bax transactivation (Figure 6B). Human/murine bax promoters contain p53 binding sites and are regulated by the tumor suppressor p53 and its coactivators.29, 30 APE-1 transactivates p53 and regulates p53 activation in a redox-dependent and independent manner.20, 31 APE-1 and K6R/K7R equally enhanced p53 transcriptional activation (Figure 6C).
Figure 6.

APE-1 acetylation and PARP-1 suppression negatively regulates H. pylori-mediated bax transcription.
(A) Bar graph of dual luciferase activities (mean ± SEM, n=4) driven by WT and nCaRE mutated bax promoter in AGS cells with or without H. pylori infection. *, p <0.05.
(B) Bar graph of dual luciferase activities (mean ± SEM, n=3) to compare the effect of WT and nonacetylable mutant (K6R/K7R) expression and H. pylori infection on bax promoter in shRNA cells.
(C) Bar graph of dual luciferase assays (mean ± SEM, n=3) to compare the effect of APE-1 acetylation and H. pylori on DNA binding activity of p53. *, p <0.05.
(D) Dual luciferase assays (mean ± SEM, n=3) to evaluate effect of PARP-1 suppression on H. pylori-induced bax transcription in AGS cells, *, p <0.05.
(E) Luciferase assay on samples (mean ± SEM, n=3) prepared from AGS cells showing the negative regulation of bax transcription by Ca2+i.
To address the role of PARP-1 on bax luciferase activity, we compared PARP-1-suppressed AGS cells with siControl™ RNA-expressed cells with and without H. pylori (Figure 6 D). Our results indicate that PARP-1 has a regulatory role in bax transcription. Chelation of Ca2+i enhanced luciferase activity thus establishing the negative regulation of bax transcription by Ca2+i (Figure 6E).
APE-1 Suppresses Bax Expression and Reduces Apoptosis in H. pylori Infected GEC
To further evaluate the repressor activity of APE-1 on Bax expression, we transfected p53-deficient gastric epithelial KATO III cells with empty vector/APE-1 plasmid and infected with H. pylori 26695 for 3, 6 or 12 h. Western blot (Figure 7A) shows that after H. pylori infection, APE-1-overexpressing KATO III cells expressed less Bax than empty vector-transfected cells and a longer time of infection was required for Bax expression in these cells.
Figure 7.
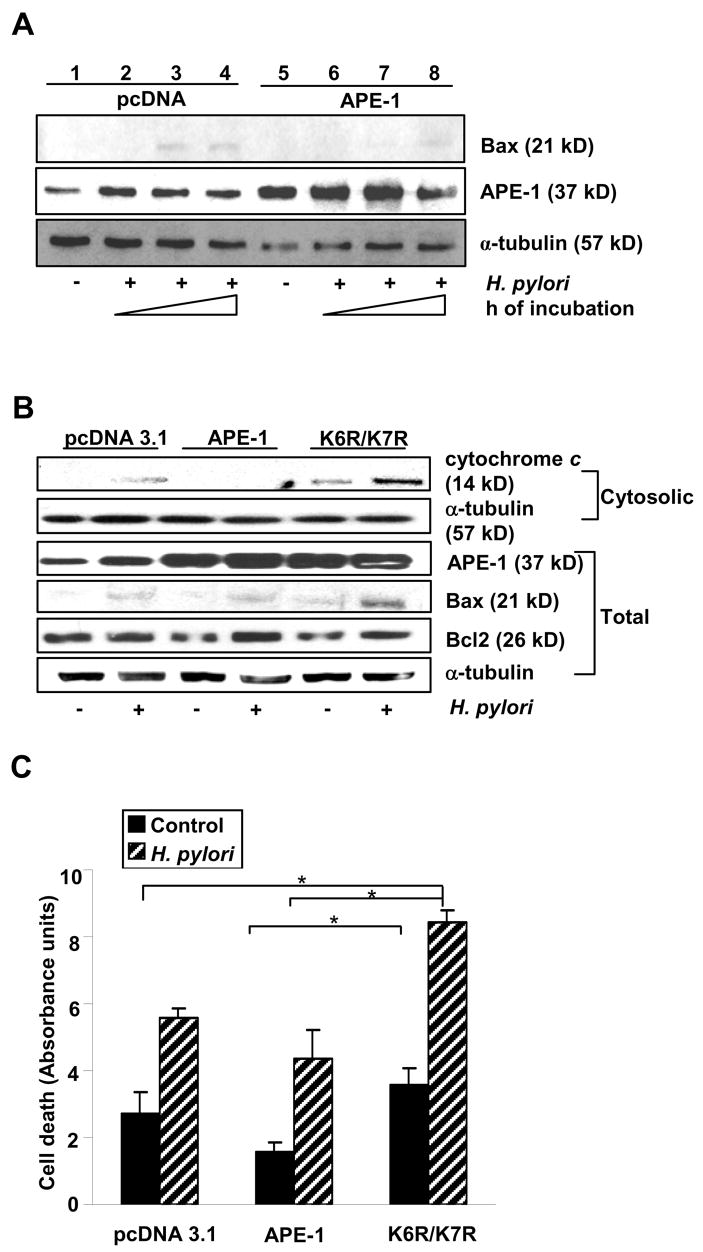
APE-1 acetylation protects GEC from H. pylori-induced Bax expression and downstream apoptotic events.
(A) Kato III cells transfected with empty vector or APE-1 were infected with H. pylori for 3, 6 and 12 h. Bax expression was less in APE-1-overexpressed cells.
(B) shRNA cells transfected with empty vector, APE-1 or K6R/K7R plasmids were infected with H. pylori for 8 h. Western blot of whole cell lysates showed maximum Bax:Bcl2 ratio and cytochrome c release in K6R/K7R transfected cells. Highest Bcl2 expression was noted in APE-1 transfected, H. pylori-infected cells.
(C) shRNA cells were transfected with empty vector, APE-1 or K6R/K7R plasmids. H. pylori infection for 12 h resulted in optimal DNA-fragmentation in K6R/K7R transfected shRNA cells but WT APE-1 suppressed H. pylori-mediated DNA fragmentation as depicted in summarized (mean ± SEM, n=3) bar graph. *, p <0.05.
The observation of nCaRE-mediated suppression of bax transcription led us to assess the effect of APE-1 acetylation on Bax. shRNA cells were transfected with empty vector, APE-1 or K6R/K7R plasmids and infected with H. pylori. Western blot analysis of whole cell lysates demonstrated that H. pylori-induced Bax expression was greater in K6R/K7R-transfected shRNA cells than in WT APE-1-overexpressed shRNA cells (Figure 7B) again confirming negative regulation of Bax by APE-1.
Finally, we assessed DNA fragmentation using an ELISA that detects exposed histones in empty vector, APE-1 or K6R/K7R-expressed shRNA cells ± H. pylori (12 h). Our data (Figure 7 C) confirmed that although H. pylori induced DNA-fragmentation in all transfected groups, APE-1 overexpression counteracted while K6R/K7R enhanced that effect. A representative western blot from a parallel experiment (Figure 7B) showed that K6R/K7R induced cytochrome c release which was futher enhanced by H. pylori. The finding of a maximal Bax:Bcl2 ratio in whole cell lysates from K6R/K7R-overexpressed cells further established that blocking acetylation of APE-1 increases H. pylori-infected GEC susceptibility to apoptotic death. Figure 8 summarizes our findings.
Figure 8.

APE-1 acetylation suppresses H. pylori-mediated apoptosis in gastric epithelial cells (GEC) by negative regulation of bax transcription. H. pylori enhance mitochondrial ROS generation and induce [Ca2+ promote nuclear translocation of APE-1. Ca2+ ]i. ROS AND Ca 2+i induces p300 binding to nuclear APE-1 leading to APE-1 acetylation. In parallel, p53, a major transcriptional activator of bax is activated by H. pylori and APE-1. We propose that in H. pylori-infected GEC, ac-APE-1 binds to the nCaRE present in the bax promoter and attracts HDAC-1 thus repressing bax transcription mediated by p53; PARP-1 recruits the repressor complex to the bax nCaRE. P=phosphorylated, PLC=phospholipase C, PLA2=phospholipase A2.
Discussion
H. pylori infection is a key risk factor in the development of chronic gastritis, peptic ulceration, and gastric carcinoma. The host gastric epithelium plays an important role in H. pylori-mediated pathogenesis and disease manifestations as imbalances between cell death and proliferation may lead to peptic ulcer and gastric carcinogenesis.4 Our findings of Bax induction in the H. pylori-infected GEC are in keeping with earlier studies which reported that H. pylori induce Bax in gastric epithelium.32, 33 However, our novel observation that a bacterial infection can induce APE-1 acetylation, which suppresses the extent of Bax expression and apoptosis in H. pylori infected epithelium, adds a new level of complexity to understanding the interaction of H. pylori with host cells.
In this study we have identified an nCaRE-B in the human bax promoter and show that H. pylori-induced increases in [Ca2+]i augment APE-1 acetylation in GEC by enhancing the APE-1-p300 interaction. Enhanced binding of ac-APE-1 to bax nCaRE recruits more HDAC-1 to the nCaRE and thus leads to reduced bax expression. Acetylation at Lys6/Lys7 mediated by p300 was previously shown to enhance APE-1′s ability to bind to the nCaRE of the PTH promoter.24 In our study, we found that H. pylori-induced rise in [Ca2+]i regulates APE-1 acetylation. As HDAC inhibition induces APE-1 acetylation and augments APE-1 binding to the nCaRE, we postulate that acetylation is the prime regulator of APE-1 binding to the bax nCaRE. Although 45% endogenous APE-1 protein is present in shRNA cells, ectopic expression of a nonacetylable APE-1 mutant enhances bax transactivation compared to the empty vector or WT APE-1 overexpressing cells indicating the significance of ac-APE-1 in regulation of bax. We reproduced our results in GEC freshly isolated from biopsies and infected in vitro with H. pylori. Importantly, we show that natural (chronic) H. pylori infection in the human host also results in enhanced APE-1 acetylation. Thus, our study provides evidence of the mechanisms underlying the antiapoptotic properties of APE-1.
In spite of the negative regulatory role of ac-APE-1 on the bax gene, H. pylori induce bax transactivation. This phenomenon may be explained by the activation of a nuclear phosphoprotein, p53. p53-mediated apoptotic effects involving Bax include bax transactivation, Bax translocation from the cytosol to mitochondrial membranes, cytochrome c release from mitochondrial intermembrane space, caspase-9 activation, followed by the activation of downstream effector caspases.34 Bcl-2 protects from Bax-mediated apoptotic death.34 p53 is phosphorylated at several sites35 with phosphorylation indicative of p53 activation. Ser-15 phosphorylated p53 upregulates Bax and downregulates Bcl2.35 Consistent with those results, we show that H. pylori-mediated p53 activation induces Bax and downregulates Bcl-2 in GEC. Interestingly, p53-mediated Bax induction is much greater in APE-1-suppressed AGS. Our results further demonstrate that wild type but not nonacetylable APE-1 expression lowers Bax levels in p53 deficient Kato III cells after H. pylori infection. Although we do not see an influence of APE-1 acetylation on p53 transcriptional activity measured by PG13-reporter assay, it should be noted that blocking APE-1 acetylation enhances expression and mitochondrial translocation of Bax as well as cytochrome c release. Hence, we conclude that H. pylori-induced GEC apoptosis is regulated by a balanced interplay between p53 activation and APE-1 acetylation. Although these observations fit well with a previous study in which removal of APE-1 initiated cell death which was not prevented by introduction of K6R/K7R APE-1,25 our results with K6R/K7R require further investigation to establish how acetylation-deficient APE-1 induces mitochondrial translocation of Bax and apoptosis.
Earlier studies showed that APE-1 does not bind directly to the nCaRE and requires additional factors to bind.36, 37 We show here that PARP-1 directly binds to the bax nCaRE oligo and confirm that PARP-1 recruits the ac-APE-1-HDAC-1 repressor complex to bax nCaRE, thus repressing bax transcription. Through direct binding to histones as well as to promoter/enhancer complexes, PARP-1 can regulate transcription.38 Polyadenylation catalyzed by PARP-1 has been reported to inhibit transcription factors.39 We found that H. pylori infection does not play a role in acetylation or self poly ADP ribosylation of PARP-1 (data not shown). Further studies will determine whether PARP-1 binding to nCaRE is specific for GEC or occurs in all cell types.
In summary, our work elaborates the role of ac-APE-1 in gene regulation during bacterial infection. So far, more than 300 posttranslational mechanisms have been identified to participate in different physiological processes and an imbalance in acetylation/deacetylation has been implicated in various disease states.40 We have identified a critical protective role of APE-1 acetylation in Bax-mediated GEC apoptosis by H. pylori. As there are some reports that APE-1 levels are altered in gastric neoplasia,41, 42 our finding that H. pylori infection enhances APE-1 acetylation in gastric epithelial cells suggests that the acetylation status of APE-1 may influence development of gastric cancer. Future studies are required to assess how mutation of APE-1 at its N-terminal Lys residues causes mitochondrial translocation of Bax and induction of host cell apoptosis to prevent potential loss of their niche thereby increasing the opportunity to permit chronic or persistent infection.
Acknowledgments
Supported by NIH RO1 DK61769 and an AGA Funderburg Research Scholar Award
Abbreviations used in this paper
- Ac-APE-1
acetylated apurinic/apyrimidinic endonuclease-1
- [Ca2+]
intracellular calcium ion concentration
- cag
cytotoxin associated gene
- GEC
gastric epithelial cells
- HAT
histone acetyl transferase
- HDAC
histone deacetylase
- nCaRE
negative calcium response element
- PAI
pathogenicity island
- PARP-1
poly (ADP-ribose) polymerase-1
- WT
wild type
Footnotes
All authors declare that they have no conflict of interest to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117:60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 3.Bourzac KM, Guillemin K. Helicobacter pylori-host cell interactions mediated by type IV secretion. Cell Microbiol. 2005;7:911–919. doi: 10.1111/j.1462-5822.2005.00541.x. [DOI] [PubMed] [Google Scholar]
- 4.Mimuro H, Suzuki T, Nagai S, et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe. 2007;2:250–263. doi: 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Wroblewski LE, Peek RM., Jr Orchestration of dysregulated epithelial turnover by a manipulative pathogen. Cell Host Microbe. 2007;2:209–211. doi: 10.1016/j.chom.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 6.Fan XJ, Crowe SE, Behar S, et al. The effect of class II MHC expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: A mechanism for Th1 cell-mediated damage. J Exp Med. 1998;187:1659–1669. doi: 10.1084/jem.187.10.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei J, O’Brien D, Vilgelm A, et al. Interaction of Helicobacter pylori with gastric epithelial cells is mediated by the p53 protein family. Gastroenterology. 2008;134:1412–1423. doi: 10.1053/j.gastro.2008.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houghton JM, Bloch LM, Goldstein M, et al. In vivo disruption of the fas pathway abrogates gastric growth alterations secondary to Helicobacter infection. J Infect Dis. 2000;182:856–864. doi: 10.1086/315788. [DOI] [PubMed] [Google Scholar]
- 9.Neu B, Rad R, Reindl W, et al. Expression of tumor necrosis factor- alpha -related apoptosis-inducing ligand and its proapoptotic receptors is down-regulated during gastric infection with virulent cagA+/vacAs1+ Helicobacter pylori strains. J Infect Dis. 2005;191:571–578. doi: 10.1086/427510. [DOI] [PubMed] [Google Scholar]
- 10.Konturek PC, Pierzchalski P, Konturek SJ, et al. Helicobacter pylori induces apoptosis in gastric mucosa through an upregulation of Bax expression in humans. Scand J Gastroenterol. 1999;34:375–383. doi: 10.1080/003655299750026380. [DOI] [PubMed] [Google Scholar]
- 11.Ashktorab H, Frank S, Khaled AR, et al. Bax translocation and mitochondrial fragmentation induced by Helicobacter pylori. Gut. 2004;53:805–813. doi: 10.1136/gut.2003.024372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maeda S, Yoshida H, Mitsuno Y, et al. Analysis of apoptotic and antiapoptotic signalling pathways induced by Helicobacter pylori. Mol Pathol. 2002;55:286–293. doi: 10.1136/mp.55.5.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding SZ, O’Hara AM, Denning TL, et al. Helicobacter pylori and H2O2 increases AP endonuclease-1/redox factor-1 expression in human gastric epithelial cells. Gastroenterology. 2004;127:845–858. doi: 10.1053/j.gastro.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 14.O’Hara AM, Bhattacharya A, Bai J, et al. Interleukin-8 induction by Helicobacter pylori in human gastric epithelial cells is dependent on apurinic/apyrimidinic endonuclease-1/redox factor-1. J Immunol. 2006;177:7990–7999. doi: 10.4049/jimmunol.177.11.7990. [DOI] [PubMed] [Google Scholar]
- 15.Grosch S, Fritz G, Kaina B. Apurinic endonuclease (Ref-1) is induced in mammalian cells by oxidative stress and involved in clastogenic adaptation. Cancer Res. 1998;58:4410–4416. [PubMed] [Google Scholar]
- 16.Ramana CV, Boldogh I, Izumi T, et al. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci. 1998;95:5061–5065. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demple B, Sung JS. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair (Amst) 2005 doi: 10.1016/j.dnarep.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 19.Tell G, Damante G, Caldwell D, et al. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 20.Jayaraman L, Murthy KGK, Zhu C, et al. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes & Development. 1997;11:558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- 21.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 22.Okazaki T, Chung U, Nishishita T, et al. A redox factor protein, ref1, is involved in negative gene regulation by extracellular calcium. J Biol Chem. 1994;269:27855–27862. [PubMed] [Google Scholar]
- 23.Fuchs S, Philippe J, Corvol P, et al. Implication of Ref-1 in the repression of renin gene transcription by intracellular calcium. J Hypertens. 2003;21:327–335. doi: 10.1097/00004872-200302000-00024. [DOI] [PubMed] [Google Scholar]
- 24.Bhakat KK, Izumi T, Yang SH, et al. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003;22:6299–6309. doi: 10.1093/emboj/cdg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Izumi T, Brown DB, Naidu CV, et al. Two essential but distinct functions of the mammalian abasic endonuclease. Proc Natl Acad Sci U S A. 2005;102:5739–5743. doi: 10.1073/pnas.0500986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Angkeow P, Deshpande SS, Qi B, et al. Redox factor-1: an extra-nuclear role in the regulation of endothelial oxidative stress and apoptosis. Cell Death Differ. 2002;9:717–725. doi: 10.1038/sj.cdd.4401025. [DOI] [PubMed] [Google Scholar]
- 27.Ye G, Barrera C, Fan XJ, et al. Expression of B7–1 and B7–2 costimulatory molecules by human gastric epithelial cells. Potential role in CD4+ T cell activation during Helicobacter pylori infection. J Clin Invest. 1997;99:1628–1636. doi: 10.1172/JCI119325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marlink KL, Bacon KD, Sheppard BC, et al. Effects of Helicobacter pylori on intracellular Ca2+ signaling in normal human gastric mucous epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2003;285:G163–G176. doi: 10.1152/ajpgi.00257.2002. [DOI] [PubMed] [Google Scholar]
- 29.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 30.Thornborrow EC, Manfredi JJ. One mechanism for cell type-specific regulation of the bax promoter by the tumor suppressor p53 is dictated by the p53 response element. J Biol Chem. 1999;274:33747–33756. doi: 10.1074/jbc.274.47.33747. [DOI] [PubMed] [Google Scholar]
- 31.Gaiddon C, Moorthy NC, Prives C. Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J. 1999;18:5609–5621. doi: 10.1093/emboj/18.20.5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moss SF. Cellular markers in the gastric precancerous process. Aliment Pharmacol Ther. 1998;12 Suppl 1:91–109. doi: 10.1111/j.1365-2036.1998.00002.x. [DOI] [PubMed] [Google Scholar]
- 33.Yamasaki E, Wada A, Kumatori A, et al. Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J Biol Chem. 2006;281:11250–11259. doi: 10.1074/jbc.M509404200. [DOI] [PubMed] [Google Scholar]
- 34.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 35.Li DW, Liu JP, Schmid PC, et al. Protein serine/threonine phosphatase-1 dephosphorylates p53 at Ser-15 and Ser-37 to modulate its transcriptional and apoptotic activities. Oncogene. 2006;25:3006–3022. doi: 10.1038/sj.onc.1209334. [DOI] [PubMed] [Google Scholar]
- 36.Chung U, Igarashi T, Nishishita T, et al. The interaction between Ku antigen and REF1 protein mediates negative gene regulation by extracellular calcium. J Biol Chem. 1996;271:8593–8598. doi: 10.1074/jbc.271.15.8593. [DOI] [PubMed] [Google Scholar]
- 37.Kuninger DT, Izumi T, Papaconstantinou J, et al. Human AP-endonuclease 1 and hnRNP-L interact with a nCaRE-like repressor element in the AP-endonuclease 1 promoter. Nucleic Acids Res. 2002;30:823–829. doi: 10.1093/nar/30.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraus WL, Lis JT. PARP goes transcription. Cell. 2003;113:677–683. doi: 10.1016/s0092-8674(03)00433-1. [DOI] [PubMed] [Google Scholar]
- 39.D’Amours D, Desnoyers S, D’Silva I, et al. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342 ( Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 40.Hodawadekar SC, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene. 2007;26:5528–5540. doi: 10.1038/sj.onc.1210619. [DOI] [PubMed] [Google Scholar]
- 41.Wang GS, Wang MW, Wu BY, et al. A gene encoding an apurinic/apyrimidinic endonuclease-like protein is up-regulated in human gastric cancer. World J Gastroenterol. 2003;9:1196–1201. doi: 10.3748/wjg.v9.i6.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Futagami S, Hiratsuka T, Shindo T, et al. Expression of apurinic/apyrimidinic endonuclease-1 (APE-1) in H. pylori-associated gastritis, gastric adenoma, and gastric cancer. Helicobacter. 2008;13:209–218. doi: 10.1111/j.1523-5378.2008.00605.x. [DOI] [PubMed] [Google Scholar]