

SUMMARY
CD40L on CD4+ T cells has been shown to license dendritic cells (DC) via CD40 to prime CTL responses. Surprisingly, we found that the converse (CD40L on DC) was also important. Anti-CD40L treatment decreases endogenous CTL responses to both OVA and influenza infection even in the absence of CD4+ T cells. DC express CD40L upon stimulation with agonists to TLR 3 and 9. Moreover, influenza infection, which stimulates CTL without help upregulates CD40L on DC, but herpes simplex infection, which elicits CTL through help, does not. CD40L−/− DC are suboptimal both in vivo in bone marrow chimera experiments and in vitro in mixed lymphocyte reactions. In contrast, CD40L−/− CD8+ T cells kill as effectively as wildtype. We conclude that CD40L upregulation on DC promotes optimal priming of CD8+ T cells without CD4+ T cells, providing a mechanism by which pathogens may elicit helper-independent CTL immunity.
INTRODUCTION
CD40L is a member of the TNF superfamily whose expression is tightly regulated (Armitage et al., 1992; Cayabyab et al., 1994; Grewal et al., 1996). Surface expression on CD4+ T cells is detectable within 2 hrs of activation by peptide-pulsed antigen-presenting cells (APC), peaks at 6 hrs and drops by 24 hrs (Lee et al., 2002). Activated CD4+ T cells are the predominant CD40L-bearing population. Modest expression can be found on activated CD8+ T cells, B cells, NK cells, monocytes, Langerhans cells, human thrombocytes and activated DC (DC) (Pinchuk et al., 1996; Salgado et al., 1999; Schonbeck and Libby, 2001). It should be noted however, that low or absent surface staining for CD40L could be misleading for several reasons. Firstly, like FasL, much of the CD40L is stored in secretory lysosomes and is only released to the cell surface upon activation (Koguchi et al., 2007). Secondly, surface CD40L is rapidly endocytosed upon binding to CD40 expressed on cells such as B cells and DC and thirdly, detection of CD40L can be masked by soluble CD40 (van Kooten et al., 1994; Yellin et al., 1994). CD40L on CD4+ T cells is critical in effecting isotype switching by B cells (Gray et al., 1994), ‘licensing’ DC to prime CTL (Bennett et al., 1998; Ridge et al., 1998; Schoenberger et al., 1998; Smith et al., 2004) and generating memory CTL (Borrow et al., 1996; Sun and Bevan, 2003). Despite the vast literature on the function of CD40L on CD4+ T cells (and hence CD40 on DC and B cells), less attention has been devoted to CD40L on the other cells that express it, perhaps because of the modest level of expression.
Although CD4 help is important, helper-independent primary CTL responses are generated against certain infections, including viral infections with ectromelia, vesicular stomatitis virus, human immunodeficiency virus, Epstein-Barr virus, influenza and cytomegalovirus (Andreasen et al., 2000; Buller et al., 1987; Ruedl et al., 1999; Tripp et al., 1995; Zimmerli et al., 2005). Likewise, helper-independent CTL can be generated against Listeria (Hamilton et al., 2001). The ascribed mechanism by which helper-independent CTL were elicited was activation of DC by the microbial infection (Ruedl et al., 1999; Sun and Bevan, 2004) or self help by CD8+ T cells (Hamilton et al., 2001; Wang et al., 2001), although the latter mechanism fails to explain how such cells were stimulated to begin with. Although many microbial pathogens directly activate DC, the basis of why only some pathogens can elicit helper-independent CTL remains unclear.
The use of a blocking anti-CD40L mAb is also of translational interest. The mAb can prolong graft survival dramatically in both mouse and primate transplantation studies (Bucher et al., 2005; Kirk et al., 1999). It can promote non-myeloablative conditioning for the establishment of chimerism (Ito et al., 2006) and it can ameliorate autoimmunity (Bagenstose et al., 2005; Hanninen et al., 2002; Komura et al., 2007). Most such studies have presumed that the anti-CD40L acts on CD4+ T cells alone. We have previously shown that in the absence of CD4 T cells, treatment with anti-CD40L Ab still reduced CTL response to alloantigens (Zhan et al., 2000). One recent report has also shown that in the absence of CD4+ T cells CD40L is still important for the priming of transgenic CD8+ T cells (Hernandez et al., 2007) and suggested a role for CD40L on CD8+ T cells.
No previous studies have explored the converse phenomenon where CD40L on DC may trigger CD40 on T cells. We found that endogenous in vivo CTL responses to cell-associated ovalbumin (OVA) were sensitive to CD40L blockade even in the absence of CD4 help. We then set out to understand the mechanism of anti-CD40L treatment and to determine the role of CD40L in the absence of CD4+ T cells, including its expression on CD8+ T cells and DC. Results from both in vitro mixed lymphocyte reactions and in vivo mixed bone marrow chimera experiments directed us to focus on the role of CD40L being expressed on DC and the correlation between CD40L upregulation by selected TLR agonists and viruses and their ability to facilitate helper-independent CTL responses. It should also be noted that our studies primarily relate to primary CTL induction; the parameters for the generation of memory may be different.
RESULTS
Anti-CD40L treatment decreases the endogenous in vivo cytotoxic T cell (CTL) response
We initially used the endogenous CTL response to OVA as our model system. Anti-CD40L treatment was administered intraperitoneally on the day of immunization with cell associated OVA (viz. OVA-coated spleen cells). Seven days later, in vivo CTL activity was assessed by comparing the numbers of peptide loaded targets versus unloaded targets. As expected, we found that anti-CD40L or anti-CD4 treatment decreased the endogenous CTL response (Fig 1A). Moreover, in the absence of CD4+ T cells, anti-CD40 antibody could substitute for help and restore CTL killing (Fig 1A) as had been shown previously (Bennett et al., 1998; Schoenberger et al., 1998). The agonistic anti-CD40 (FGK45) is thought to simulate CD4 help to license DC for CTL priming. Initially we thought that anti-CD40L acted by interfering with CD4+ T cell licensing of DC and thus had assumed that anti-CD40L would have no effect in any model that induced CTL without CD4+ T cells.
Figure 1. Treatment with Anti-CD40L decreases the CTL response even in the absence of CD4+ T cells.

Mice, wildtype (A), GK transgenic mice (B) and MHC class II knockout mice (C) respectively, were immunized with 2×107 irradiated OVA-coated spleen cells with 1μg LPS I.V. On the same day as immunization, 400μg of anti-CD40L (MR1) or control hamster IgG I.P. was administered. After 7–8 days mice were given I.V. 2×107 CFSEhigh labeled peptide pulsed cells and CFSElow labeled control unpulsed cells in equal numbers. After 24 hours spleens were analyzed by flow cytometry. Percentage lysis is calculated by the reduction in the CFSEhigh labeled peptide pulsed target cells compared with unprimed mice. CD4 deficient mice (A) and (B) were treated with 100μg of anti-CD40 (FGK45) I.P. on the day of immunization. **p=0.0076 and 0.0072 respectively; Mann-Whitney U Test.
To investigate this, we used two different mouse models that lack CD4+ T cells. In the first model, we used GK mice, which secrete anti-CD4 (GK1.5) antibody transgenically, effectively depleting the mouse of all peripheral CD4+ T cells (Zhan et al., 2004). In the second model, we analyzed MHC class II KO mice, which are unable to positively select for CD4+ T cells (Cosgrove et al., 1991). Again, we used anti-CD40 to facilitate priming of CTL responses to OVA-coated spleen cells. Surprisingly in both models, our results showed that even in the absence of CD4+ T cells, the anti-CD40L Ab treatment was still able to suppress the CTL response (Fig. 1B,C). This indicated that the antibody treatment played another role, apart from blocking CD4 help and led us to investigate what other cell type is being targeted by this treatment.
CD40L is expressed on DC
The expression of CD40L on CD4+ T cells has been well documented (Grewal et al., 1996). However, the expression of CD40L on other cells has been less well characterized. CD40L expression on DC has been reported for human blood DC (Pinchuk et al., 1996), mouse Langerhans cells (Salgado et al., 1999), mouse lung DC (Masten et al., 1997) and plasmacytoid DC (Kuwajima et al., 2006). In preliminary studies, we found that surface staining of CD40L on splenic CD8+ T cells and DC (both resting and activated) was unconvincing. To further explore CD40L expression, we next used intracellular staining, as we surmised that accumulation of CD40L after the addition of monensin during the procedure might avail easier detection. Lymph node cells obtained from naïve wildtype C57BL/6 (B6) or CD40L−/− mice were cultured for 4 hours in the presence of PMA and Ionomycin, permeabilized and stained for intracellular stores of CD40L. DC were simply cultured overnight in media, which is known to induce activation (Wilson et al., 2006). CD40L, as expected, stained strongly on CD4+ T cells (Fig 2A), compared with either CD40L−/− controls (Fig 2A) or with control antibody staining (data not shown). CD8+ T cells showed virtually no staining of CD40L (Fig 2B). We found that CD40L was also expressed quite strongly on cultured splenic DC (Fig 2C). Given that CD40 is also found on activated wildtype CD8+ T cells ((Bourgeois et al., 2002; Sun and Bevan, 2004); Fig 2D) and DC, it was feasible that CD40L expressed by DC may interact with CD40 on T cells, in addition to the previously described interaction of CD40L on T cells interact with CD40 on DC.
Figure 2. CD40L is expressed on DC and CD8+ T cells modestly express CD40.

Lymph node cells from naïve C57BL/6 mice (grey filled) or CD40L−/− mice (black) were stimulated in vitro for 4 hours with PMA and Ionomycin. Cells were stained for CD4 and CD8 and intracellular CD40L. (A) is gated on CD4+ cells and shows strong expression of CD40L. (B) gated CD8+ cells shows modest expression of CD40L. (C) DC were purified from spleens of naïve C57BL/6 mice (grey filled) or CD40L−/− mice (black) and cultured overnight prior to intracellular staining.
(D) Lymph node cells from C57BL/6 mice were analyzed immediately or were activated with PMA and Ionomycin. They were then stained with CD8 and CD40 and analyzed using flow cytometry.
CD40L−/− DC have reduced function in priming CD8+ T cells
To explore further the role of CD40L being expressed by DC, we tested whether this CD40L was required for priming CD8+ T cells in an endogenous CTL assay. To discriminate between the role of CD40L on DC and that of CD8+ T cells, we generated mixed bone marrow chimeras that would allow us to examine independently the different cell types expressing CD40L. Irradiated mice were reconstituted with equal parts of bm1 and CD40L−/− bone marrow. Bm1 mice differ from B6 mice in that H-2Kbm1 has three mutations in the H-2Kb molecule that disallows presentation of the OVA peptide SIINFEKL (Nikolic-Zugic and Carbone, 1990). The resultant mice will have two sets of DC. One set can present OVA, but is CD40L deficient. The other set cannot present SIINFEKL, but is capable of expressing CD40L. In this situation, any CTL priming must be due to the CD40L deficient DC. To examine CTL priming in the absence of CD4 help, reconstituted mice were CD4-depleted and treated with agonistic anti-CD40 Ab. To exclude the possibility that the bm1 T cells are unable to kill, mixed chimeras of bm1 and Rag1−/− (RAG) bone marrow (1:1 ratio) were generated. Such mice will have bm1 T cells and a set of wildtype B6 DC (that can present OVA and express CD40L). Our results (Fig 3A) show that there was a significant decrease in killing in bm1/CD40L−/− chimeric mice compared with the chimeric bm1/RAG controls. Thus the CD40L−/− DC are less capable of eliciting CTL priming.
Figure 3. CD40L−/− APC have reduced function in priming CD8+ T cells in vivo CD4 deficient models.

Mice, either (A) bm1 reconstituted or (B) and (C) RAG reconstituted mice were treated with 0.5mg GK1.5 prior to immunizing with 2×107 irradiated ova coated spleen cells with 1g LPS I.V. and 0.1mg anti-CD40 (FGK45). Staining with a non-competing RM4–4 Ab had shown that the CD4 depletion was complete. After 7–8 days 2×107 CFSEhigh labeled peptide pulsed cells and CFSElow labeled control unpulsed cells in equal numbers. After 24 hours spleens analyzed by flow cytometry. Percentage lysis is calculated by the reduction in the CFSEhigh labeled peptide pulsed target cells compared with unprimed mice. *** p= < 0.0002, Mann-Whitney U Test.
Elimination of DC by diphtheria toxin in CD40L−/− and CD11cDTR mixed bone marrow chimeras reduces CTL killing
To show that DC were critical in this CD40L-dependent antigen presentation, we generated chimeric mice with CD11cDTR (Probst et al., 2005) and CD40L−/− BM. The CD11cDTR mice express a fusion protein of the grivet monkey diptheria toxin receptor and green fluorescent protein (GFP) under the control of the CD11c promoter. Therefore, all mice are generated similarly and the reconstitution of DC with CD40L−/− vs CD11cDTR CD40L+ genotype can easily be tracked by GFP and CD11c expresssion (CD11c+GFP− vs CD11c+GFP+). Diptheria toxin (DT) was administered to one group (100ng per 20g mouse on day −1, 0 and 1 relative to immunization; n=5) and compared to untreated mice. All mice were depleted of CD4+ T cells with 0.5mg GK1.5 prior to immunizing with 2×107 irradiated OVA-coated spleen cells I.V. and 0.1mg anti-CD40 (FGK45) I.P. After 7 days, 2×107 CFSEhigh labelled peptide pulsed cells and CFSElow labelled control cells in equal numbers were injected I.V. Spleens were analyzed 24 hours later in this in vivo CTL assay. As shown in Fig 4b, the DC in these mixed BM chimeras comprise 32–40% CD11cDTR and 60–68% CD40L−/−. Thus the majority of DC are of the CD40L−/− genotype and therefore if anything there is a bias to show presentation function by the CD40L−/− DC. The depletion of GFP+ CD11cDTR DC (CD40L+/+ genotype) upon administration of DT (Fig 4b) resulted in a substantial reduction (>50%) in CTL killing (Fig 4a; p=0.021). This confirms that DC expressing CD40L is a critical component of CTL priming in the absence of CD4+ T cells.
Figure 4. Elimination of DC by diphtheria toxin in CD40L−/− and CD11cDTR mixed bone marrow chimeras reduces CTL killing.

Chimeric mice were generated by using CD11cDTR BM and CD40L−/− BM. Diptheria toxin (DT) was administered to one group (n=5) prior to immunization and compared to untreated mice (n=5). Mice were treated with 0.5mg GK1.5 prior to immunizing with 2×107 irradiated OVA-coated spleen cells I.V. and 0.1mg anti-CD40 (FGK45). After 7 days, 2×107 CFSEhigh labelled peptide pulsed cells and CFSElow labelled control cells in equal numbers were injected I.V. Spleens were analyzed 24 hours later. Percentage lysis was calculated by the reduction in the peptide pulsed target cells compared to unprimed mice. In (b) DC were enriched using a nycodenz gradient and stained for CD11c. As DTR was made as a fusion of DTR and GFP, DC from CD11cDTR mice express GFP. Thus the DC comprise 35% CD11cDTR and 65% CD40L−/−. Moreover, the GFP+ CD11cDTR DC are depleted upon administration of DT.
CD40−/− but not CD40L−/− CD8+ T cells have reduced in vivo killing
RAG mice were sublethally irradiated (300 cGy) and reconstituted with either CD40L−/− (Fig 3B) or CD40−/− (Fig 3C) bone marrow. Mice were depleted of CD4+ T cells prior to immunizing with OVA-coated spleen cells and anti-CD40 Ab. Using CFSE labeled peptide coated cells to detect differential in vivo lysis, we showed that CD40L−/− CD8+ T cells but not CD40−/− CD8+ T cells can be primed to become CTL (Fig 3B,C).
CD40L−/− DC are inefficient at T-cell priming, but CD40L−/− T cells are able to be stimulated in vitro
To further test the role of CD40L expression by DC on T-cell priming, we investigated an in vitro system, viz. mixed lymphocyte reactions (MLR) between BALB/c and B6 cells, using naïve CD8+ T cells as responders. Purified (97–99% purity) CD11c+ cells were used as APC and the proliferation of mismatched purified CD8+ T cells was measured by thymidine incorporation after 3 days. The addition of anti-CD40L to the MLR caused a significant decrease in proliferation of CD8+ T cells (Fig 5A), supporting our earlier findings of its effect on CTL generation in vivo. Whereas CD40L−/− CD8+ T cells behaved like wildtype CD8+ T cells (Fig 4B), CD40L−/− DC are poor stimulators compared to wildtype DC (Fig 5C). Reciprocally, CD40−/− CD8+ T cells showed decreased proliferation, consistent with a role of CD40L on DC (Fig 5D). Hence for CD8+ T cell responders in MLR, it is important for DC to have CD40L and for CD8+ T cell responders to have CD40.
Figure 5. CD40L−/− DC are inefficient at eliciting MLR.

2×105 purified CD8+ T cells were cultured with allogeneic purified CD11c+ DC for 3 days prior to the addition of [H3] thymidine. Proliferation was measured by the incorporation of [H3] thymidine above that of syngeneic cultures. (A) B6 T cells with BALB/c DC or BALB/c T cells with B6 DC are inhibited by the addition of anti-CD40L to the cultures (B) CD40L−/− T cells respond equally as well as wildtype. (C) CD40L−/− DC are poorer presenters to BALB/c T cells (D) CD40−/− T cells proliferate to a lesser extent (**p=0.003, Mann Whitney U Test). Mean + SEM are shown.
CD40L on DC is upregulated upon activation with TLR 3 and 9 agonists
Our results above indicated that CD40L on DC was important in priming anti-CD40 potentiated CTL responses. This led us to investigate whether anti-CD40 might upregulate CD40L on DC. We cultured DC for 8 hr with anti-CD40 Ab. This shortened timepoint was necessary, because longer incubation of DC (e.g. overnight) resulted in their activation (Wilson et al., 2006) and CD40L upregulation (Fig 2C). Indeed, we found that CD40L on DC was upregulated by anti-CD40 Ab (Fig 6A). This raised the question whether other DC-activating agents like TLR agonists (mimicking infection) could also upregulate CD40L on DC. To test this, a panel of TLR agonists were used, including pam2cys (TLR2), poly I:C (TLR3), LPS (TLR4), Loxoribine (TLR7) and CpG (TLR9). We observed expression of CD40L when adding TLR3 agonists (poly I:C) or TLR9 agonists for an 8 hour timepoint (Fig 6B). In contrast, agonists against TLR2 (Pam2cys), 4 (LPS) and 7 (Loxoribine) despite activating DC as shown by CD86 expression (supplementary Fig 1), did not upregulate CD40L (Fig 6B).
Figure 6. CD40L is upregulated in DC by TLR 3 and TLR 9 agonists and these agonists can potentiate CD40L dependent CTL responses.

DC were purified (CD11c+) and cultured for 8 hours with (A) anti-CD40 monoclonal antibody (FGK45) or (B) a panel of agonists for TLR 2, 3, 4, 7 and 9. Histograms show expression of intracellular CD40L on gated CD11c+ cells from C57BL/6 mice (black) relative to CD40L−/− mice (grey). (C) GK (CD4 deficient) mice were immunized with 2×107 irradiated OVA coated spleen cells with 1μg LPS I.V. and either 0.1mg anti-CD40, 40nmols CpG, 20nmols Pam2Cys, 1mg Loxoribine or 100μg Poly I:C. Anti-CD40L or hamster IgG, 400μg, was also given on the day of immunization. After 7 days 2×107 CFSEhigh labeled peptide pulsed cells and CFSElow labeled control unpulsed cells in equal numbers. After 24 hours spleens analyzed by flow cytometry. Percentage lysis is calculated by the reduction in the CFSEhigh labeled peptide pulsed target cells compared with unprimed mice.
The potentiation of helper-independent CTL responses by TLR3 and 9 agonists is dependent on CD40L upregulation
As shown above, anti-CD40 can upregulate CD40L on DC and facilitate CTL priming in the absence of CD4+ T cells. As TLR3 and TLR9 agonists could also upregulate CD40L on DC just like anti-CD40 (Fig 6B), we wanted to determine whether these agonists could also potentiate helper-independent CTL responses. We showed that in CD4 T cell-deficient mice (GK mice), the only TLR ligands to allow helper-independent CTL induction were those that upregulated CD40L, viz. poly I:C and CpG (black columns; Fig 6C). Moreover, this induction was dependent on CD40L, as anti-CD40L dramatically reduced the CTL response (white columns; Fig 6C).
Helper-independent pathogens may license APC by upregulating CD40L
As agonists to TLR 3 and 9 can activate APC to potentiate helper-independent CTL responses, we investigated whether a pathogen itself is able to have the same function. It has been known that some pathogens are able to elicit immune responses in the absence of CD4+ T cells (Behrens et al., 2004; Shedlock and Shen, 2003; Sun and Bevan, 2003) and that this was due to such viruses activating DC (Wu and Liu, 1994). However, no differential activation between viruses that elicit CTL without help versus those that do not has been forthcoming. Thus, we questioned whether the upregulation of CD40L is required for these helper-independent responses. To investigate this, we chose two viruses: influenza whose primary effector CTL responses are mainly helper-independent (Allan et al., 1990; Tripp et al., 1995) and herpes simplex virus (HSV) whose CTL responses are mainly helper-dependent (for the intravenous and intradermal route (Smith et al., 2004); for flank infection, supplementary Fig 2). Mice depleted of CD4+ T cells were infected intranasally with influenza virus and treated with either anti-CD40L or control IgG. The CTL response against the influenza NP peptide pulsed cells was reduced in the treated mice (Fig 7Ai &ii). The number of NP specific CD8+ T cells was also dramatically reduced as shown with tetramer staining (Fig 7Aiii). Thus, we hypothesized that part of helper-independence for influenza-induced CTL immunity is the virus’s ability to upregulate CD40L on APC. Indeed, we showed that influenza infection of DC upregulated CD40L expression in vitro whereas HSV did not (Fig 7B), even though both viruses upregulated CD86 (Fig 7B). Therefore we conclude that upregulation of CD40L on APC is one mechanism by which some pathogens are able to elicit helper-independent effector CTL.
Figure 7. Influenza can upregulate CD40L on the dendritic cell and generate CD40L partially dependent CTLs.
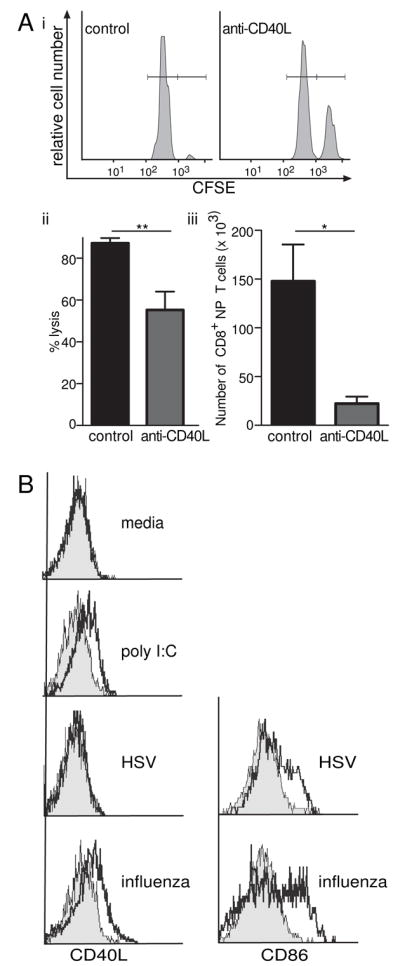
(A) Mice treated with GK1.5 (anti-CD4) were infected with 30 pfu intranasal influenza (PR8 strain); on the same day 400μg Anti-CD40L or IgG treatment was administered. 6 days later mice were injected with 2×107 CFSEhigh labeled peptide (influenza NP366–374) pulsed cells and CFSElow labeled control unpulsed cells. After 24 hours spleens analyzed by flow cytometry. (i) Histograms show higher proportion of CFSEhigh cells in anti-CD40L treated mice. (ii) Percentage lysis is calculated by the reduction in the CFSEhigh labeled peptide pulsed target cells compared with uninfected mice, column graph shows mean and SEM of percentage lysis of target cells. **p=0.0027, Mann-Whitney U Test. (iii) Mediastinal lymph nodes (MSLN) were taken from CD4 depleted influenza infected mice at day 5 and stained with CD8/NP tetramer. The bar graph shows total number of CD8+ NP specific T cells in the MSLN. *p=0.0286, Mann-Whitney U test. (B) CD11c+ DC were purified and infected for 1 hour with 5 pfu/cell of either PR8 influenza or HSV. DC were then cultured for 6 hours prior to surface staining of CD11c and CD86 and intracellular staining of CD40L. Histograms are gated on CD11c+ cells. CD40L histograms show B6 (black line) overlayed onto CD40L−/− DC (grey filled). CD86 histograms show HSV- or influenza-infected (black line) cells overlayed onto uninfected DC (grey filled).
DISCUSSION
CD40/CD40L interactions have long been associated with DC maturation (O’Sullivan and Thomas, 2003), although the majority of investigations have approached this from the angle of DC displaying CD40. As both CD40L and CD40 are found in CD4+ T cells, DC (highlighted in this study) and CD8+ T cells, there are many potential permutations for amplifying the response within or between each cell type. Moreover such potential interactions may differ among various situations e.g. between initial CTL induction vs. memory generation. In this study we have provided evidence that CD40L is required on DC for optimal priming of naïve CD8+ T cells to become CTL. Our results show that anti-CD40L treatment decreases the endogenous in vivo cytotoxic T cell response to OVA. Anti-CD40L treatment is most effective when used on the day of immunization, which inferred interference at the priming/expansion phase rather than the effector phase. Although the importance of CD40L expression by CD4+ T cells and CD40 by DC in licensing DC for CTL priming is not in dispute, little attention has been drawn to the possible role for CD40L expression by DC in helper-independent responses. In using two different models of CD4 deficient mice we have shown that anti-CD40L treatment decreases the CTL response in the absence of CD4+ T cells. Thus, in the absence of CD4+ T cells, anti-CD40L treatment must be targeting another cell type that expresses CD40L. Upon examination of CD40L expression on various cell types we discovered that CD40L is upregulated on DC. This CD40L seems to be functionally required as in vivo CTL assays in bone marrow chimeras showed that when the DC are CD40L−/−, killing is reduced. Likewise in vitro MLRs show that CD40L−/− DC do not prime T cells as effectively as wildtype DC, whereas CD40L−/− CD8+ T cells proliferate just as efficiently as wildtype CD8+ T cells. CD40−/−CD8+ T cells however, in both in vivo and in vitro assays are less efficacious than wildtype or CD40L−/− CD8+ T cells. CD40 expression by T cells, especially activated ones, has been reported previously (Bourgeois et al., 2002; Munroe and Bishop, 2007), although the role for CD40 on CD8+ T cells remained undefined. It has been shown to have a co-stimulatory function, augmenting in vitro responses to agonists against CD3 and CD28 (Munroe and Bishop, 2007). Our work may give insight into an undefined pathway where the CD40 on these CD8+ T cells interact with the CD40L on the DC. Our hypothesis that CD40L on DC is important in helper-independent responses does not discredit the notion that CD40 may also play a role in some situations. Hernandez et al. (Hernandez et al., 2007) had ascribed a role of CD40L on CD8+ T cells, much of that work used the P-14 TCR transgenic system (and lymphocytic choriomeningitis virus gp33 peptide on H-2Db). In contrast, we have found no evidence that CD40L is important on CD8+ T cells in our endogenous models as CD40L−/− CD8+ T cells are able to have normal cytotoxic activity in the presence of CD40L+ DC. We have also looked at TCR-transgenic models viz OT-I and found OT-I T cells are not reliant on CD40L for proliferation or killing (data not shown). Given the recent findings that clonal size is important in determining outcomes it may not be surprising that there may be differences between TCR-transgenic systems versus endogenous systems (Ford et al., 2007; Mintern et al., 2002). One of the key components of the Hernandez paper was the CD40L−/− P-14 T cells proliferated poorly compared with wildtype P-14 T cells in vivo. Interestingly, anti-CD40 stimulation availed these CD40L−/− T cells to proliferate and this suggests that CD8+ T cells do not require CD40L to proliferate. Our proposal that anti-CD40 Ab can upregulate CD40L on DC to prime CD8+ T cells would explain this result.
We therefore propose that CD40L on the DC interacts with the CD40 on CD8+ T cells and is crucial for helper-independent CTL responses. As CD40 is expressed on other immune cells including CD4+ T cells, B cells, endothelial cells, thymic epithelial cells and DC themselves (Gray et al., 2006; Quezada et al., 2004; van Kooten and Banchereau, 2000), whether CD40L on DC may interact with CD40 of all these cell types invites further elucidation. The CD40L on DC can be induced in vitro upon activation with TLR3 and TLR9 agonists. TLR3 and 9 agonists can also be used to generate helper-independent CTL responses; this can be dramatically blocked by anti-CD40L. However agonists to TLR2, 4 and 7 failed to upregulate CD40L and were unable to deliver signals to prime T cells in vivo. Thus to generate helper-independent CTL responses, TLR3 and 9 agonists can upregulate CD40L expression on DC and promote CTL priming. TLR3 and 9 agonists (but not TLR2 or 4 agonists) have previously been shown to facilitate cross-presentation in vitro (Datta et al., 2003). Moreover, the addition of TLR3 or TLR9 agonists to cationic liposomes can elicit in vivo cross-priming of CD8+ T cells independent of CD4+ T cell help (Zaks et al., 2006). Our findings now offer a mechanistic explanation for their findings, i.e. upregulation of CD40L on DC by agonists to TLR3 and TLR9 promotes CD8+ T cell priming.
As TLR agonists mimic microbial infection, we surmised that pathogens that promote CTL priming in a helper-independent fashion may do so by upregulating CD40L on DC. Viruses can infect both human and murine DC leading to activation and upregulation of co-stimulatory molecules such as CD40 and CD80/86 (Harui et al., 2006; Jain et al., 2007; Montoya et al., 2005; Saurwein-Teissl et al., 1998; Wu and Liu, 1994). CD40L as a costimulatory marker on DC has not been investigated in this capacity, presumably because most workers assume that CD40L is primarily a T cell molecule. We chose two infectious models: intranasal influenza, which is helper-independent (Allan et al., 1990; Tripp et al., 1995) and HSV flank infection, which is mostly dependent on CD4 help (supplementary Fig 2), just like for the intravenous or footpad route (Smith et al., 2004). CD40L on DC was upregulated by influenza but not by HSV, whereas both infections upregulated CD86 on DC. Moreover, the helper-independent CTL responses by influenza were substantially reduced by anti-CD40L treatment (Fig 7A).
The mechanism of how viruses like ectromelia, vesicular stomatitis virus, human immunodeficiency virus, Epstein-Barr virus, influenza and cytomegalovirus can potentiate helper independent CTL responses in the absence of CD4 help remained unclear despite much exploration (Hamilton et al., 2001; Ruedl et al., 1999; Sun and Bevan, 2004; Wang et al., 2001). Our work provides at least one mechanism by which some viruses can elicit CTL responses in a CD4 deficient environment. Many viruses, such as HIV (Lore et al., 2002; Trimble et al., 2000), MCMV (Mintern et al., 2006), HPV (Ortiz-Sanchez et al., 2007), HCMV (Moutaftsi et al., 2002) and HSV (Mikloska et al., 2001) have developed evasion strategies that downregulate co-stimulatory markers on both T cells and DC. This evasion mechanism leads to decreased T cell mediated immunity to the virus, allowing chronic infections to manifest. Interestingly, HIV has been shown to downregulate CD40L on T cells (Subauste et al., 2007), thus it would be interesting to speculate that CD40L on DC may also be downregulated. Correcting such a deficiency (e.g. by TLR agonists) could potentially lead to increased immunity in the absence of CD4+ T cells in HIV+ patients. There is ongoing work investigating the creation of viruses or cell lines expressing CD40L (Bereta et al., 2004; Dotti et al., 2001; Mehling et al., 2001; Ostrowski et al., 2000; Tomihara et al., 2008) for either vaccine strategies or eradication of tumours using cell based therapies. Our findings that CD40L upregulation on DC is involved in generating CTL responses to influenza virus may lead to increased understanding of how these therapies may be tailored.
In conclusion, we show that CD40L upregulation on DC allows the generation of helper-independent CTLs. This CD40L upregulation also explains why only certain TLR agonists can promote helper-independent CTLs and provides a mechanism by which certain viruses can stimulate helper-independent CTL.
EXPERIMENTAL PROCEDURES
Mice
Mice aged 6 to 8 weeks were used in all experiments. CD4 deficient mice GK5 (transgenic for anti-CD4 monoclonal antibody (Zhan et al., 2004)), CD40L deficient mice (CD154-H) (Xu et al., 1994), MHC class II knockout mice (Cosgrove et al., 1991), CD11cDTR mice (Probst et al., 2005), Rag1−/− (Mombaerts et al., 1992) CD40−/− and MHC class I:C.H-2bm1 (called bm1) mice were maintained in specific pathogen free condition in the animal facilities of Walter and Eliza Hall Institute of Medical Research. All mice were used according to the regulations of the Institute Animal Ethics Committee.
DC preparations
DC were harvested from spleens as previously described (Vremec et al., 1992). Spleens were finely diced and added to a collagenase-DNase solution. Digestion occurred for 25mins prior to adding EDTA. After sieving, diluting and centrifuging, the pellet was then resuspended in 5mL of Nycodenz/4 spleens and overlayed onto a further 5mL Nycodenz in a polypropylene tube. 2mL of FCS-EDTA was overlayed onto the splenic cell layer. The gradient was then centrifuged at 4000rpm without brake for 15 minutes. The low-density cells were collected and washed with EDTA-BSS-FCS before cell counts were performed. Depletion antibody cocktail containing antibodies against CD3, Thy1, GR1, B220 and Ery was added to negatively select for DC. Dynabeads sheep anti-rat IgG magnetic beads (Dynal Biotech, Oslo, Norway) were washed in EDTA-BSS-FCS and added at 5 beads per cell for 20 minutes at 4°C on a slowly rotating mixer. Cells were then depleted via negative selection on a magnet (Miltenyi Biotec, Bergisch Gladbach, Germany), collecting the supernatant and performing the depletion twice for extra purity. Cells were washed in EDTA-BSS-FCS, counted and checked via flow cytometry for yield.
Intracellular staining
Cells were stained intracellularly for CD40L. T cells from B6 mice or CD40L−/− mice were cultured 4×106 cells/ml in a 24 well plate in RPMI with 10% FCS in 5% incubator. PMA (25ng/ml) and Ionomycin (1 μg/ml) for 3 hours. Monensin (Golgistop, Becton Dickinson, Franklin Lakes, NJ) at 0.6μl/ml was added to the cultures to prevent transport out of the cells. Cells were washed and stained with surface markers prior to fixing. Cells were fixed with 100μl Cytofix (Becton Dickinson) for 20 minutes at 4°C. Cells were permeabilised in saponin (PermWash, Becton Dickinson) before staining with biotinylated CD40L and allophycocyanin-streptavidin (BDBiosciences, San Jose, CA).
In vivo CTL assays
Mice were immunized with 2×107 irradiated OVA coated spleen cells from either a B6 or bm1 donor with 1μg LPS I.V. 7 days later mice were given I.V. 2×107 CFSEhigh labeled SIINFEKL (Mimotopes, Clayton, Australia) peptide pulsed cells and CFSElow labeled control unpulsed cells in equal ratios. 18–24 hours later spleens were analyzed by flow cytometry.
Generation of chimeric mice
Lethal irradiation consisted of two doses of 550 cGy (γ-irradiation; 60Co source) 2 hr apart. Femoral bone marrow, 1×106 cells were injected I.V. into the recipient mice within hours of irradiation. Neomycin sulfate was added prophylactically to the drinking water for four weeks post irradiation.
Mice were treated with 100μl of anti-Thy1 antibody (T24) I.P. day one post reconstitution to eliminate residual host T cells. Chimeric mice were used experimentally 6–8 weeks later.
Mixed lymphocyte reactions
Spleens and lymph nodes were isolated from C57BL/6, BALB/c or CD40L−/− mice. DC and CD8+ T cells were purified by MACS (Miltenyi Biotech) positive selection. 2×105 T cells, as responders, were cultured with allogeneic DC, as stimulators at concentrations of 1:100. Cells were cultured in a 96 well round bottom plate in MT-RPMI-10% FCS with 55μM 2-mercapto-ethanol and incubated with 5% CO2. Syngeneic stimulators and responders were used as background controls. [H3] thymidine was added 2 or 3 days later. Cells were harvested 18 hours after the addition of [H3] thymidine and proliferation was measured as CPM over background.
Antibodies and TLR agonists
Anti-CD40L (MR1), anti-CD40 (FGK45) and anti-CD4 (GK1.5) were purified by in-house facilities. Flow cytometry antibodies were purchased from BDBiosciences. Loxoribine and Polyinosinic-Polycytidilyc Acid (Poly I:C) were purchased from Invivogen, San Diego, CA; Lipopolysaccharide (LPS) from E. coli 0111:B4 was purchased from Sigma, St Louis, MO. Dr. David Jackson, University of Melbourne, generously provided Pam2Cys.
Viral Infections
All viruses were stored at −70°C in PBS as single use aliquots. The influenza strain used was Influenza A/Puerto Rico/8/34 (Mt Sinai, H1N1) (PR8). Titre of the original influenza virus AF17B = 3.1 × 107 plaque forming units (pfu)/ml. B6 mice were infected with 30 pfu per mouse of the above strain of influenza intranasally in a volume of 20μl. Purified DC were infected by incubating at 37°C for 45 min with virus at a concentration of 5 pfu/cell. The suspension was swirled periodically. DC were washed three times in KDS-RPMI-FCS to ensure any excess virus was removed prior to incubation for five hours to upregulate surface markers. HSV flank infections were performed as previously described (Allan et al., 2006; van Lint et al., 2004).
Tetramer Staining
Mediastinal lymph nodes were removed from mice infected with influenza 5 days prior. Cells were prepared into a single cell solution and incubated with NP tetramer-PE, 1 in 200, for 40 minutes at room temperature. Cells were then stained for CD8, 30 minutes on ice and analysed via flow cytometry.
Statistical Methods
Data are shown as mean ± SEM. Non-normally distributed data were compared with unpaired Mann Whitney U Tests.
Supplementary Material
Acknowledgments
A Melbourne Research Scholarship, provided by the University of Melbourne, supports S.J. This work was supported by grants from the National Health & Medical Research Council of Australia and Juvenile Diabetes Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Allan W, Tabi Z, Cleary A, Doherty PC. Cellular events in the lymph node and lung of mice with influenza. Consequences of depleting CD4+ T cells. J Immunol. 1990;144:3980–3986. [PubMed] [Google Scholar]
- Andreasen SO, Christensen JE, Marker O, Thomsen AR. Role of CD40 ligand and CD28 in induction and maintenance of antiviral CD8+ effector T cell responses. J Immunol. 2000;164:3689–3697. doi: 10.4049/jimmunol.164.7.3689. [DOI] [PubMed] [Google Scholar]
- Armitage RJ, Fanslow WC, Strockbine L, Sato TA, Clifford KN, Macduff BM, Anderson DM, Gimpel SD, Davis-Smith T, Maliszewski CR, et al. Molecular and biological characterization of a murine ligand for CD40. Nature. 1992;357:80–82. doi: 10.1038/357080a0. [DOI] [PubMed] [Google Scholar]
- Bagenstose LM, Agarwal RK, Silver PB, Harlan DM, Hoffmann SC, Kampen RL, Chan CC, Caspi RR. Disruption of CD40/CD40-ligand interactions in a retinal autoimmunity model results in protection without tolerance. J Immunol. 2005;175:124–130. doi: 10.4049/jimmunol.175.1.124. [DOI] [PubMed] [Google Scholar]
- Behrens G, Li M, Smith CM, Belz GT, Mintern J, Carbone FR, Heath WR. Helper T cells, dendritic cells and CTL Immunity. Immunol Cell Biol. 2004;82:84–90. doi: 10.1111/j.1440-1711.2004.01211.x. [DOI] [PubMed] [Google Scholar]
- Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Bereta M, Bereta J, Park J, Medina F, Kwak H, Kaufman HL. Immune properties of recombinant vaccinia virus encoding CD154 (CD40L) are determined by expression of virally encoded CD40L and the presence of CD40L protein in viral particles. Cancer gene therapy. 2004;11:808–818. doi: 10.1038/sj.cgt.7700762. [DOI] [PubMed] [Google Scholar]
- Borrow P, Tishon A, Lee S, Xu J, Grewal IS, Oldstone MB, Flavell RA. CD40L-deficient mice show deficits in antiviral immunity and have an impaired memory CD8+ CTL response. The Journal of experimental medicine. 1996;183:2129–2142. doi: 10.1084/jem.183.5.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 2002;297:2060–2063. doi: 10.1126/science.1072615. [DOI] [PubMed] [Google Scholar]
- Bucher P, Gang M, Morel P, Mathe Z, Bosco D, Pernin N, Wekerle T, Berney T, Buhler LH. Transplantation of discordant xenogeneic islets using repeated therapy with anti-CD154. Transplantation. 2005;79:1545–1552. doi: 10.1097/01.tp.0000163505.63159.69. [DOI] [PubMed] [Google Scholar]
- Buller RM, Holmes KL, Hugin A, Frederickson TN, Morse HC., 3rd Induction of cytotoxic T-cell responses in vivo in the absence of CD4 helper cells. Nature. 1987;328:77–79. doi: 10.1038/328077a0. [DOI] [PubMed] [Google Scholar]
- Cayabyab M, Phillips JH, Lanier LL. CD40 preferentially costimulates activation of CD4+ T lymphocytes. J Immunol. 1994;152:1523–1531. [PubMed] [Google Scholar]
- Cosgrove D, Gray D, Dierich A, Kaufman J, Lemeur M, Benoist C, Mathis D. Mice lacking MHC class II molecules. Cell. 1991;66:1051–1066. doi: 10.1016/0092-8674(91)90448-8. [DOI] [PubMed] [Google Scholar]
- Datta SK, Redecke V, Prilliman KR, Takabayashi K, Corr M, Tallant T, DiDonato J, Dziarski R, Akira S, Schoenberger SP, Raz E. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J Immunol. 2003;170:4102–4110. doi: 10.4049/jimmunol.170.8.4102. [DOI] [PubMed] [Google Scholar]
- Dotti G, Savoldo B, Takahashi S, Goltsova T, Brown M, Rill D, Rooney C, Brenner M. Adenovector-induced expression of human-CD40-ligand (hCD40L) by multiple myeloma cells. A model for immunotherapy. Experimental hematology. 2001;29:952–961. doi: 10.1016/s0301-472x(01)00668-3. [DOI] [PubMed] [Google Scholar]
- Ford ML, Koehn BH, Wagener ME, Jiang W, Gangappa S, Pearson TC, Larsen CP. Antigen-specific precursor frequency impacts T cell proliferation, differentiation, and requirement for costimulation. The Journal of experimental medicine. 2007;204:299–309. doi: 10.1084/jem.20062319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray D, Siepmann K, Wohlleben G. CD40 ligation in B cell activation, isotype switching and memory development. Semin Immunol. 1994;6:303–310. doi: 10.1006/smim.1994.1039. [DOI] [PubMed] [Google Scholar]
- Gray DH, Seach N, Ueno T, Milton MK, Liston A, Lew AM, Goodnow CC, Boyd RL. Developmental kinetics, turnover, and stimulatory capacity of thymic epithelial cells. Blood. 2006;108:3777–3785. doi: 10.1182/blood-2006-02-004531. [DOI] [PubMed] [Google Scholar]
- Grewal IS, Foellmer HG, Grewal KD, Xu J, Hardardottir F, Baron JL, Janeway CA, Jr, Flavell RA. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science. 1996;273:1864–1867. doi: 10.1126/science.273.5283.1864. [DOI] [PubMed] [Google Scholar]
- Hamilton SE, Tvinnereim AR, Harty JT. Listeria monocytogenes infection overcomes the requirement for CD40 ligand in exogenous antigen presentation to CD8(+) T cells. J Immunol. 2001;167:5603–5609. doi: 10.4049/jimmunol.167.10.5603. [DOI] [PubMed] [Google Scholar]
- Hanninen A, Martinez NR, Davey GM, Heath WR, Harrison LC. Transient blockade of CD40 ligand dissociates pathogenic from protective mucosal immunity. J Clin Invest. 2002;109:261–267. doi: 10.1172/JCI13720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harui A, Roth MD, Vira D, Sanghvi M, Mizuguchi H, Basak SK. Adenoviral-encoded antigens are presented efficiently by a subset of dendritic cells expressing high levels of alpha(v)beta3 integrins. Journal of leukocyte biology. 2006;79:1271–1278. doi: 10.1189/jlb.1105694. [DOI] [PubMed] [Google Scholar]
- Hernandez MG, Shen L, Rock KL. CD40-CD40 ligand interaction between dendritic cells and CD8+ T cells is needed to stimulate maximal T cell responses in the absence of CD4+ T cell help. J Immunol. 2007;178:2844–2852. doi: 10.4049/jimmunol.178.5.2844. [DOI] [PubMed] [Google Scholar]
- Ito H, Takeuchi Y, Shaffer J, Sykes M. Anti-CD40L monoclonal antibodies can replace anti-CD4 monoclonal antibodies for the nonmyeloablative induction of mixed xenogeneic chimerism. Transplantation. 2006;82:251–257. doi: 10.1097/01.tp.0000226147.69877.6f. [DOI] [PubMed] [Google Scholar]
- Jain P, Ahuja J, Khan ZK, Shimizu S, Meucci O, Jennings SR, Wigdahl B. Modulation of dendritic cell maturation and function by the Tax protein of human T cell leukemia virus type 1. Journal of leukocyte biology. 2007;82:44–56. doi: 10.1189/jlb.1006641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk AD, Burkly LC, Batty DS, Baumgartner RE, Berning JD, Buchanan K, Fechner JH, Jr, Germond RL, Kampen RL, Patterson NB, et al. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in nonhuman primates. Nat Med. 1999;5:686–693. doi: 10.1038/9536. [DOI] [PubMed] [Google Scholar]
- Koguchi Y, Thauland TJ, Slifka MK, Parker DC. Preformed CD40 ligand exists in secretory lysosomes in effector and memory CD4+ T cells and is quickly expressed on the cell surface in an antigen-specific manner. Blood. 2007;110:2520–2527. doi: 10.1182/blood-2007-03-081299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komura K, Fujimoto M, Yanaba K, Matsushita T, Matsushita Y, Horikawa M, Ogawa F, Shimizu K, Hasegawa M, Takehara K, Sato S. Blockade of CD40-CD40 ligand interactions attenuates skin fibrosis and autoimmunity in the tight-skin mouse. Ann Rheum Dis. 2007 doi: 10.1136/ard.2007.073387. [DOI] [PubMed] [Google Scholar]
- Kuwajima S, Sato T, Ishida K, Tada H, Tezuka H, Ohteki T. Interleukin 15-dependent crosstalk between conventional and plasmacytoid dendritic cells is essential for CpG-induced immune activation. Nat Immunol. 2006;7:740–746. doi: 10.1038/ni1348. [DOI] [PubMed] [Google Scholar]
- Lee BO, Haynes L, Eaton SM, Swain SL, Randall TD. The biological outcome of CD40 signaling is dependent on the duration of CD40 ligand expression: reciprocal regulation by interleukin (IL)-4 and IL-12. The Journal of experimental medicine. 2002;196:693–704. doi: 10.1084/jem.20020845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lore K, Sonnerborg A, Brostrom C, Goh LE, Perrin L, McDade H, Stellbrink HJ, Gazzard B, Weber R, Napolitano LA, et al. Accumulation of DC-SIGN+CD40+ dendritic cells with reduced CD80 and CD86 expression in lymphoid tissue during acute HIV-1 infection. AIDS (London, England) 2002;16:683–692. doi: 10.1097/00002030-200203290-00003. [DOI] [PubMed] [Google Scholar]
- Masten BJ, Yates JL, Pollard Koga AM, Lipscomb MF. Characterization of accessory molecules in murine lung dendritic cell function: roles for CD80, CD86, CD54, and CD40L. Am J Respir Cell Mol Biol. 1997;16:335–342. doi: 10.1165/ajrcmb.16.3.9070619. [DOI] [PubMed] [Google Scholar]
- Mehling A, Loser K, Varga G, Metze D, Luger TA, Schwarz T, Grabbe S, Beissert S. Overexpression of CD40 ligand in murine epidermis results in chronic skin inflammation and systemic autoimmunity. The Journal of experimental medicine. 2001;194:615–628. doi: 10.1084/jem.194.5.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikloska Z, Bosnjak L, Cunningham AL. Immature monocyte-derived dendritic cells are productively infected with herpes simplex virus type 1. Journal of virology. 2001;75:5958–5964. doi: 10.1128/JVI.75.13.5958-5964.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintern JD, Davey GM, Belz GT, Carbone FR, Heath WR. Cutting edge: precursor frequency affects the helper dependence of cytotoxic T cells. J Immunol. 2002;168:977–980. doi: 10.4049/jimmunol.168.3.977. [DOI] [PubMed] [Google Scholar]
- Mintern JD, Klemm EJ, Wagner M, Paquet ME, Napier MD, Kim YM, Koszinowski UH, Ploegh HL. Viral interference with B7-1 costimulation: a new role for murine cytomegalovirus fc receptor-1. J Immunol. 2006;177:8422–8431. doi: 10.4049/jimmunol.177.12.8422. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Montoya M, Edwards MJ, Reid DM, Borrow P. Rapid activation of spleen dendritic cell subsets following lymphocytic choriomeningitis virus infection of mice: analysis of the involvement of type 1 IFN. J Immunol. 2005;174:1851–1861. doi: 10.4049/jimmunol.174.4.1851. [DOI] [PubMed] [Google Scholar]
- Moutaftsi M, Mehl AM, Borysiewicz LK, Tabi Z. Human cytomegalovirus inhibits maturation and impairs function of monocyte-derived dendritic cells. Blood. 2002;99:2913–2921. doi: 10.1182/blood.v99.8.2913. [DOI] [PubMed] [Google Scholar]
- Munroe ME, Bishop GA. A costimulatory function for T cell CD40. J Immunol. 2007;178:671–682. doi: 10.4049/jimmunol.178.2.671. [DOI] [PubMed] [Google Scholar]
- Nikolic-Zugic J, Carbone FR. The effect of mutations in the MHC class I peptide binding groove on the cytotoxic T lymphocyte recognition of the Kb-restricted ovalbumin determinant. European journal of immunology. 1990;20:2431–2437. doi: 10.1002/eji.1830201111. [DOI] [PubMed] [Google Scholar]
- O’Sullivan B, Thomas R. CD40 and dendritic cell function. Crit Rev Immunol. 2003;23:83–107. doi: 10.1615/critrevimmunol.v23.i12.50. [DOI] [PubMed] [Google Scholar]
- Ortiz-Sanchez E, Chavez-Olmos P, Pina-Sanchez P, Salcedo M, Garrido E. Expression of the costimulatory molecule CD86, but not CD80, in keratinocytes of normal cervical epithelium and human papillomavirus-16 positive low squamous intraepithelial lesions. Int J Gynecol Cancer. 2007;17:571–580. doi: 10.1111/j.1525-1438.2007.00904.x. [DOI] [PubMed] [Google Scholar]
- Ostrowski MA, Justement SJ, Ehler L, Mizell SB, Lui S, Mican J, Walker BD, Thomas EK, Seder R, Fauci AS. The role of CD4+ T cell help and CD40 ligand in the in vitro expansion of HIV-1-specific memory cytotoxic CD8+ T cell responses. J Immunol. 2000;165:6133–6141. doi: 10.4049/jimmunol.165.11.6133. [DOI] [PubMed] [Google Scholar]
- Pinchuk LM, Klaus SJ, Magaletti DM, Pinchuk GV, Norsen JP, Clark EA. Functional CD40 ligand expressed by human blood dendritic cells is up-regulated by CD40 ligation. J Immunol. 1996;157:4363–4370. [PubMed] [Google Scholar]
- Probst HC, Tschannen K, Odermatt B, Schwendener R, Zinkernagel RM, Van Den Broek M. Histological analysis of CD11c-DTR/GFP mice after in vivo depletion of dendritic cells. Clinical and experimental immunology. 2005;141:398–404. doi: 10.1111/j.1365-2249.2005.02868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol. 2004;22:307–328. doi: 10.1146/annurev.immunol.22.012703.104533. [DOI] [PubMed] [Google Scholar]
- Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Ruedl C, Kopf M, Bachmann MF. CD8(+) T cells mediate CD40-independent maturation of dendritic cells in vivo. The Journal of experimental medicine. 1999;189:1875–1884. doi: 10.1084/jem.189.12.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado CG, Nakamura K, Sugaya M, Tada Y, Asahina A, Koyama Y, Irie S, Tamaki K. Functional CD40 ligand is expressed on epidermal Langerhans cells. Journal of leukocyte biology. 1999;66:281–285. doi: 10.1002/jlb.66.2.281. [DOI] [PubMed] [Google Scholar]
- Saurwein-Teissl M, Zisterer K, Schmitt TL, Gluck R, Cryz S, Grubeck-Loebenstein B. Whole virus influenza vaccine activates dendritic cells (DC) and stimulates cytokine production by peripheral blood mononuclear cells (PBMC) while subunit vaccines support T cell proliferation. Clinical and experimental immunology. 1998;114:271–276. doi: 10.1046/j.1365-2249.1998.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Schonbeck U, Libby P. The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci. 2001;58:4–43. doi: 10.1007/PL00000776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, Belz GT. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat Immunol. 2004;5:1143–1148. doi: 10.1038/ni1129. [DOI] [PubMed] [Google Scholar]
- Subauste CS, Subauste A, Wessendarp M. Role of CD40-dependent down-regulation of CD154 in impaired induction of CD154 in CD4(+) T cells from HIV-1-infected patients. J Immunol. 2007;178:1645–1653. doi: 10.4049/jimmunol.178.3.1645. [DOI] [PubMed] [Google Scholar]
- Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Bevan MJ. Cutting edge: long-lived CD8 memory and protective immunity in the absence of CD40 expression on CD8 T cells. J Immunol. 2004;172:3385–3389. doi: 10.4049/jimmunol.172.6.3385. [DOI] [PubMed] [Google Scholar]
- Tomihara K, Kato K, Masuta Y, Nakamura K, Uchida H, Sasaki K, Tanaka T, Huang J, Hiratsuka H, Hamada H. Gene transfer of CD40-ligand to dendritic cells stimulates interferon-gamma production to induce growth arrest and apoptosis of tumor cells. Gene therapy. 2008;15:203–213. doi: 10.1038/sj.gt.3303056. [DOI] [PubMed] [Google Scholar]
- Trimble LA, Shankar P, Patterson M, Daily JP, Lieberman J. Human immunodeficiency virus-specific circulating CD8 T lymphocytes have down-modulated CD3zeta and CD28, key signaling molecules for T-cell activation. Journal of virology. 2000;74:7320–7330. doi: 10.1128/jvi.74.16.7320-7330.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripp RA, Sarawar SR, Doherty PC. Characteristics of the influenza virus-specific CD8+ T cell response in mice homozygous for disruption of the H-2lAb gene. J Immunol. 1995;155:2955–2959. [PubMed] [Google Scholar]
- van Kooten C, Banchereau J. CD40-CD40 ligand. Journal of leukocyte biology. 2000;67:2–17. doi: 10.1002/jlb.67.1.2. [DOI] [PubMed] [Google Scholar]
- van Kooten C, Gaillard C, Galizzi JP, Hermann P, Fossiez F, Banchereau J, Blanchard D. B cells regulate expression of CD40 ligand on activated T cells by lowering the mRNA level and through the release of soluble CD40. European journal of immunology. 1994;24:787–792. doi: 10.1002/eji.1830240402. [DOI] [PubMed] [Google Scholar]
- van Lint A, Ayers M, Brooks AG, Coles RM, Heath WR, Carbone FR. Herpes simplex virus-specific CD8+ T cells can clear established lytic infections from skin and nerves and can partially limit the early spread of virus after cutaneous inoculation. J Immunol. 2004;172:392–397. doi: 10.4049/jimmunol.172.1.392. [DOI] [PubMed] [Google Scholar]
- Vremec D, Zorbas M, Scollay R, Saunders DJ, Ardavin CF, Wu L, Shortman K. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. The Journal of experimental medicine. 1992;176:47–58. doi: 10.1084/jem.176.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Norbury CC, Greenwood R, Bennink JR, Yewdell JW, Frelinger JA. Multiple paths for activation of naive CD8+ T cells: CD4-independent help. J Immunol. 2001;167:1283–1289. doi: 10.4049/jimmunol.167.3.1283. [DOI] [PubMed] [Google Scholar]
- Wilson NS, Behrens GM, Lundie RJ, Smith CM, Waithman J, Young L, Forehan SP, Mount A, Steptoe RJ, Shortman KD, et al. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol. 2006;7:165–172. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- Wu Y, Liu Y. Viral induction of co-stimulatory activity on antigen-presenting cells bypasses the need for CD4+ T-cell help in CD8+ T-cell responses. Curr Biol. 1994;4:499–505. doi: 10.1016/s0960-9822(00)00110-x. [DOI] [PubMed] [Google Scholar]
- Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, Flavell RA. Mice deficient for the CD40 ligand. Immunity. 1994;1:423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Yellin MJ, Sippel K, Inghirami G, Covey LR, Lee JJ, Sinning J, Clark EA, Chess L, Lederman S. CD40 molecules induce down-modulation and endocytosis of T cell surface T cell-B cell activating molecule/CD40-L. Potential role in regulating helper effector function. J Immunol. 1994;152:598–608. [PubMed] [Google Scholar]
- Zaks K, Jordan M, Guth A, Sellins K, Kedl R, Izzo A, Bosio C, Dow S. Efficient immunization and cross-priming by vaccine adjuvants containing TLR3 or TLR9 agonists complexed to cationic liposomes. J Immunol. 2006;176:7335–7345. doi: 10.4049/jimmunol.176.12.7335. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Brown LE, Deliyannis G, Seah S, Wijburg OL, Price J, Strugnell RA, O’Connell PJ, Lew AM. Responses against complex antigens in various models of CD4 T-cell deficiency: surprises from an anti-CD4 antibody transgenic mouse. Immunologic research. 2004;30:1–14. doi: 10.1385/IR:30:1:001. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Corbett AJ, Brady JL, Sutherland RM, Lew AM. CD4 help-independent induction of cytotoxic CD8 cells to allogeneic P815 tumor cells is absolutely dependent on costimulation. J Immunol. 2000;165:3612–3619. doi: 10.4049/jimmunol.165.7.3612. [DOI] [PubMed] [Google Scholar]
- Zimmerli SC, Harari A, Cellerai C, Vallelian F, Bart PA, Pantaleo G. HIV-1-specific IFN-gamma/IL-2-secreting CD8 T cells support CD4-independent proliferation of HIV-1-specific CD8 T cells. Proc Natl Acad Sci U S A. 2005;102:7239–7244. doi: 10.1073/pnas.0502393102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.