

Abstract
TGF-β is a pluripotent cytokine that can have both tumor suppressing and tumor promoting effects on epithelial cells. It is unclear what determines when TGF-β and its signaling pathway act predominantly as a tumor suppressor pathway or as a tumor-promoter pathway and whether TGF-β can have both classes of effects concurrently on a cell. We investigated the effect of TGF-β on anoikis in colorectal cancer cell lines sensitive to TGF-_-mediated growth inhibition to determine if the context of the cells could be one of the factors that would affect whether TGF-β exerts tumor suppressor or oncogene activity on colon cancer cells. We observed variable effects of TGF-_ on anoikis in these cell lines, even though they all are growth-inhibited by TGF-β. Thus, we show that TGF-β has variable effects on anoikis in colon cancer cell lines that likely reflects the effects of concurrent gene mutations in the cancer cells and the activation state of the signaling pathways controlled by these genes.
Keywords: Transforming Growth Factor Beta, colorectal cancer, apoptosis, anoikis
Introduction
The Transforming Growth Factor-β (TGF-β) superfamily comprises a large number of cytokines that regulate tissue homeostasis and development through the regulation of fundamental processes such as cell proliferation, differentiation, motility and programmed cell death. TGF-β has been shown to not only play a central role in development and normal organ function, but also to affect many pathological processes such as cirrhosis, nephrosclerosis, and tumorigenesis (reviewed in (Elliott and Blobe 2005; Massague 1998)).
There is considerable genetic evidence that the TGF-_ signaling pathway is a tumor suppressor pathway in colon epithelial cells. Much of this evidence is derived from studies in human colon cancers demonstrating inactivating mutations in genes encoding proteins involved in TGF-_ signal transduction, including SMAD4 (Hahn et al. 1996; Schutte et al. 1996), SMAD2 (Eppert et al. 1996), and TGFBR2 (Grady et al. 1999; Parsons et al. 1995). Additional evidence for a tumor suppressor role for the TGF-_ signaling pathway is derived from studies of mouse models of cancer. Overexpression of TGF-_1 in mammary epithelial cells and skin keratinocytes suppresses the development of carcinomas (Pierce et al. 1995). Expression of a dominant-negative TGFBR2 (DNIIR) in mammary epithelial cells increases the incidence of mammary tumors (Gorska et al. 2003; Gorska et al. 1998). Furthermore, invasive colon tumors develop in Smad3-/-, Tgfb1-/-, and ApcΔ716 Smad4+/- mice, and mice that lack TGFBR2 in the colon, Fabp4xat-132 Cre Tgfbr2flx/flx mice, are more susceptible to azoxymethane-induced colon neoplasms than are mice with intact TGFBR2 in the colonic epithelium (Biswas et al. 2004; Engle et al. 1999; Engle et al. 2002; Takaku et al. 1998; Zhu et al. 1998).
However, the role of TGF-β signaling during the process of tumor development is complex. Its effects appear to be context-dependent and can even be paradoxical. Results from in vitro and in vivo model systems as well as from epidemiological studies have made evident that the TGF-β signaling pathway can behave both as a tumor suppressor and as a tumor promoter pathway (Derynck et al. 2001; Siegel and Massague 2003). The tumor suppressor mechanisms of TGF-β include, among others, its ability to inhibit cell-cycle progression as well as to induce senescence and apoptosis (Elliott and Blobe 2005; Massague 1998). The tumor promoting capability of this signaling pathway, on the other hand, has been demonstrated by an increase in invasiveness and metastatic potential of mammary neoplasms arising in transgenic mice in which TGF-β1 or the TGF-β type I receptor are overexpressed or constitutively activated in tumor cells (Muraoka-Cook et al. 2004; Siegel and Massague 2003; Swift et al. 2001). Furthermore, the concept that TGF-β signaling can facilitate tumor progression is supported by the observation that some tumor cell lines lose their metastatic potential in xenograft systems upon disruption of the TGF-β signaling pathway (Oft et al. 1998). It also appears that TGF-β signaling may promote tumor progression through effects on the tumor microenvironment by altering the composition of the extracellular matrix or by inducing angiogenesis, as well as through direct effects on the tumor cells by inducing Epithelial to Mesenchymal Transition (EMT) (Oft et al. 1998) or by inhibiting cell death caused by growth factor deprivation (Alazzouzi et al. 2005; Boulay et al. 2002; Jung et al. 2006; Prehn et al. 1994; Samowitz et al. 2002; Shin et al. 2001; Watanabe et al. 2001). Thus, although TGF-β signaling pathway can inhibit tumor formation, it also appears capable of paradoxically promoting invasion and metastasis in established cell lines, especially in the context of breast cancer.
In light of studies demonstrating both tumor suppressing and tumor promoting effects of TGF-β, we evaluated the effect of TGF-β on the regulation of anoikis in a panel of colon cancer cell lines that are responsive to TGF-β mediated growth inhibition (Lallemand et al. 2001; Normanno et al. 2004; Ramachandra et al. 2002). A panel of TGF-β responsive cell lines was studied to test the hypothesis that the context of the cells could affect whether the ultimate effect of TGF-β on a cancer cell is tumor suppressing or tumor promoting. The assessment of anoikis, which is the induction of programmed cell death by the loss of cell anchorage on extracellular matrix, was selected because it is one of the fundamental barriers to the metastatic and invasive behavior of tumor cells (Frisch and Francis 1994; Frisch and Screaton 2001; Grossmann 2002). Tumor xenograft models have shown that anoikis-resistant cancer cell lines have increased survival in blood circulation and enhanced capacity to form metastases (Douma S et al. 2004; Duxbury et al. 2004a; Fernandez Y et al. 2002; Zhu et al. 2001). Of interest, we have found that TGF-_ can have concurrent tumor promoting and tumor suppressing effects on the same cancer cells with the ultimate effect depending on the context of the cells and presumably on the concurrent mutations present in the cells (Alazzouzi et al. 2005; Alhopuro et al. 2005; Watanabe et al. 2001).
Materials and methods
Reagents
Luminol (cat# A8511), and p-Coumaric acid (cat# C9008) were purchased from Sigma-Aldrich (St. Louis, MO). The MEK1/2 inhibitor U0126 (cat# 662005), and the PI3K inhibitor LY294002 (cat# 440204) were obtained from Calbiochem (San Diego, CA). TGF-β1 was purchased from R&D Systems (Minneapolis, MN). The antibodies for p-Akt (Ser473) (cat# 9271), Akt (cat# 9272), p-Erk1/2 (Thr202/Tyr204) (cat# 9101), Erk1/2 (cat# 9102), p-Smad2 (Ser465/467) (cat#3101), Smad2/3 (cat#3102), cleaved caspase3 (cat#9661), caspase3 (cat#9662), and PARP (cat#9542) were purchased from Cell Signaling (Beverly, MA). The antibodies for actin (cat# sc-1616), goat anti-mouse IgG-HRP (cat# SC-2005), and goat anti-rabbit IgG-HRP (cat # SC-2004) were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA).
Cell Culture
The colon cancer cell lines HCT116, FET, Moser, GEO and CBS were generously provided by Michael Brattain (Roswell Park Cancer Institute, Buffalo, NY). The HCT116 plus chromosome 3 cell line (designated HCT116+Chr3) was kindly provided by C. Richard Boland (Baylor University Medical Center, Dallas, TX) and John Carethers (University of California San Diego, San Diego, CA). HCT116, HCT116+Chr3, HCT116+Chr3+DN2R, FET and Moser were grown in Dulbecco’s Modified Eagle Medium (Gibco, Grand Island, NY), and CBS and GEO were maintained in McCoy’s 5A Medium (Sigma-Aldrich). All media was supplemented with 10% FBS (HyClone, Logan, UT).
Generation of HCT116+Chr3+DN2R
An isogenic cell line with suppressed TGFBR2 activity was generated by transfecting the HCT116+Chr3 cell line with a plasmid expressing a dominant negative TGFBR2 gene, pIRES-puro2-DN2R, which was constructed by cloning the DN2R transgene, provided by Harold Moses (Vanderbilt University Medical School, Nashville, TN) into the pIRES-puro2 vector (Clontech, Mountain View, CA). The transfection was performed using FuGene (Roche, Indianapolis, IN) following the manufacturer’s protocol. A pool of clones expressing the DN2R construct was selected with 1.5μg/ml puromycin (Sigma-Aldrich) and used in the subsequent experiments.
Thymidine incorporation assay
With the aim of evaluating cell proliferation by means of DNA synthesis, subconfluent cells were treated with TGF-β1 (10ng/ml) in serum free conditions. Forty-six hours after the addition of TGF-β1, 4 μCi/ml (Cook et al.)-Thymidine (Perkin Elmer, Wellesley, MA) was added to the media. The cells were then incubated at 37°C for an 2 additional hours. The cells were fixed with 10% Trichloroacetic Acid (1 ml) for 30 minutes, after which they were solubilized with 0.2N NaOH (300 μl). Aliquots (100 μl) of the cell lysates were resuspended in 4ml of scintillation fluid and then measured with a scintillation counter in triplicates.
Cell Proliferation Assay
3 × 105 cells were seeded into 6-well tissue culture plates in triplicte and then grown for 48 hours with TGF-β (10ng/ml) or vehicle only in serum free media. The cells were then harvested and counted manually using a hemocytometer.
3TP-lux reporter assay
In order to evaluate TGF-β-mediated transcription, tumor-derived cells were transiently transfected with the p3TP-lux reporter (kindly provided by Joan Massagué, Memorial Sloan-Kettering Cancer Center, New York, NY) concomitantly with the pRL-TK reporter construct (Promega, Madison, WI). Subsequently, the cells were treated with TGF-β1 (10ng/ml) , and luciferase activity was evaluated 48h after transfection using the Dual Luciferase Reporter Assay System (Promega) with a Veritas luminometer (Turner Biosystems, Sunnyvale, CA).
Anoikis Assay
For the evaluation of cell death induced by the loss of anchorage, the cells of interest were first plated in regular cell culture vessels with FBS-containing medium at a 50% confluence. Twenty-four hours later, when the cells were properly attached to the plastic, the medium was replaced with serum free media (SFM) and the cells were treated with TGF-β1 (10ng/ml). After 48 hours, the cells were detached from the vessels and resuspended in serum-free media. The cells were then seeded at a concentration of 1×106 cells/ml in Ultra Low Attachment Plates (Corning, Corning, NY), which prevent cell anchorage to the substrate, and treated with TGF-β1 (10ng/ml) or any additional compounds as described in the studies in the results section. After 20 hours in suspension the occurrence of cell death was assessed by Caspase-3 cleavage, PARP cleavage (through western blotting, protocol described below), or DNA fragmentation analysis, ) in which the presence of nucleosomes is photometrically detected by measuring the optical density of the samples at 405nm. (Cell Death Detection ELISA Kit, Roche).
Western Blotting
For evaluation of protein expression during anoikis western blotting was used. Cells were maintained in suspension as described above, harvested, lysed with RIPA buffer supplemented with a complete protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktails 1 and 2 (Sigma-Aldrich), and then used for SDS-PAGE. PVDF membranes (Pierce, Rockford, IL) were incubated with the antibodies of interest under the conditions suggested by the maker of each antibody, and the reaction was detected by chemiluminescence using autoradiographic methods.
Statistical analysis
All experiments were performed in triplicate and repeated at least twice. The results in the figures are represented as means ± S.E. The Student’s t-test was used to compare the means of the treatment groups and a p-value ≤ 0.05 was considered statistically significant.
Results
TGF-β responsiveness is restored in HCT116 upon chromosome 3 transfer
The DNA mismatch repair (MMR) deficient human colon carcinoma cell line HCT116 is resistant to TGF-β due to biallelic mutational inactivation of TGFBR2. An MMR proficient clone of HCT116 has been made through the transfer of chromosome 3 into this cell line, which reconstitutes not only the MMR gene MLH1 but also TGFBR2, restoring TGF-β responsiveness in the HCT116+Chr3 cell line (Koi et al. 1994). Our analysis revealed that in contrast to the parental HCT116 cell line, HCT116+Chr3 displays TGF-_ mediated activation of the 3TP-lux reporter and is growth inhibited by TGF-_ (Figure 1). These results demonstrate that TGF-β sensitivity and signal transduction is effectively restored in the HCT116+Chr3 cell line. Moreover, given that recent data suggest that the level of expression of TGFBR2 can affect a cell’s response to TGF-_, we propose that this cell line is a well defined reagent for studying the effects of restoring TGF-β signaling in TGF-_ resistant cells, because only one functional copy of TGFBR2 has been introduced and the reconstituted TGFBR2 is in its native chromatin environment, which should minimize spurious effects caused by overexpression of TGFBR2 (Shimanuki et al. 2007).
Figure 1. TGF-β1 induces transcriptional activity of the 3TP-lux reporter construct and growth arrest in HCT116+Chr3.

A. Parental cell line HCT116, as well as HCT116+Chr3 and HCT116+Chr3+DN2R cells were transfected with the 3TP-lux and pRL-TK reporter constructs, and treated with TGF-β1 (10ng/ml). TGF-_ mediated induction of the 3TP-Lux reporter is only apparent in the HCT116+Chr3 cell line. B. The effect of TGF-_ on DNA synthesis was evaluated in HCT116, HCT116+Chr3 and HCT116+Chr3+DN2R treated with TGF-β1 (10ng/ml) for 48h. All experiments were performed in triplicate. The bars correspond to S.E. TGF-_ suppression of thymidine incorporation is present only in the HCT116+Chr3 cell line. C. The effect of TGF-β on cell proliferation was assessed in HCT116, HCT116+Chr3 and HCT116+Chr3+DN2R treated as in 2B by measuring cell counts using a hematocytometer. All experiments were performed in triplicate. The bars correspond to S.E. TGF-β considerably reduces the proliferation of HCT116+Chr3 cells. Autocrine TGF-β signaling considerably reduces the proliferation of HCT116+Chr3 cells. The asterisks indicate statistically significant differences, p-value ≤ 0.05 as determined by the Student’s t-test.
Although TGF-_1 inhibits proliferation in HCT116+Chr3, TGF-β1 paradoxically protects the HCT116+Chr3 cell line from anoikis
TGF-β signaling is involved in the regulation of multiple cellular responses disrupted during the process of tumor formation that can be potentially tumor promoting or tumor suppressing. After demonstrating that TGF-_ has inhibitory effects on proliferation in HCT116+Chr3, we assessed the effect of TGF-_ on anoikis to determine whether these effects were also consistent with tumor suppressor activities. We chose to study anoikis because TGF-_ signaling has been shown to both promote and suppress anoikis in cancer cell lines (Lallemand et al. 2001; Normanno et al. 2004). Deregulation of anoikis is of particular interest because it contributes to the malignant behavior of many types of neoplastic cells by allowing anchorage-independent survival and facilitating colonization of secondary sites. Thus, with the aim of evaluating the effect of TGF-β1 on HCT116+Chr3 on this type of cell death, HCT116, HCT116+Chr3, and HCT116+Chr3 transduced with a dominant negative mutant of TGFBR2 (HCT116+Chr3+DN2R) were maintained under conditions of no attachment with or without exogenous TGF-β1. After twenty hours in suspension, DNA fragmentation and PARP cleavage were evaluated using the Cell Death Detection ELISA assay and western blotting, respectively. Unexpectedly, we observed autocrine TGF-β1 protects HCT116+Chr3 from anoikis and this protective influence is further enhanced by the addition of exogenous TGF-β1 (Figure 2).
Figure 2. TGF-β1 protects HCT116+Chr3 cells from anoikis.

The cells were treated with TGF-β1 (10ng/ml) in serum free conditions in regular tissue culture flasks. After 48h, 1×106 cells/ml were placed in suspension in “Ultra Low Attachment Clusters ”(Costar) with the same concentration of TGF-β1. A. DNA fragmentation was assessed with the “Cell Death Assay ”after the cells had been in suspension for twenty hours. The assays were run in triplicate. The bars represent S.E. The asterisks indicates a statistically significant difference, p-value ≤ 0.05 as determined by the Student’s t-test. B. The expression of p-Smad-2, Smad2/3, and PARP was evaluated in HCT116, HCT116+Chr3, and HCR116+Chr3+DN2R after 20h in suspension.
TGF-β1 has varying effects on colorectal cancer cell lines that display sensitivity to TGF-_1 mediated growth inhibition
In light of our findings on the effect of TGF-_ on anoikis in the HCT116+Chr3, we next assessed the effect of TGF-_ on anoikis in a set of TGF-_ sensitive colorectal cancer cell lines in order to determine how commonly this phenomenon might occur in colon cancer. We evaluated the occurrence of cell death upon the loss of anchorage in the human colorectal cancer lines GEO, Moser, FET and CBS through analysis of DNA fragmentation and caspase-3 activation. Our analysis revealed that, even though TGF-β induces the activation of the Smad signaling pathway and inhibits proliferation in all of these cell lines, TGF-β’s effects on anoikis in the cell lines are diverse. We observed that although exogenous TGF-β protects CBS from undergoing anoikis, it increases the amount of cell death in FET lacking anchorage. Furthermore, we found that treatment with TGF-β does not have a significant effect on anoikis in GEO and Moser (Figure 3).
Figure 3. Effect of TGF-β1 treatment on colorectal cancer cell lines deprived from anchorage.
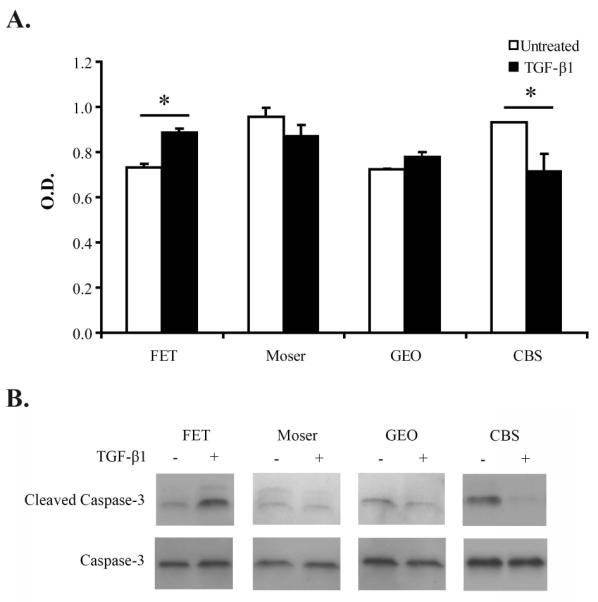
The TGF-β-responsive colon cancer cell lines FET, Moser, GEO and CBS were treated as described in figure 2. A. Results from the Cell Death Detection ELISA assay (Roche) demonstrate that TGF-_ induces apoptosis in FET, reduces apoptosis in CBS and has no significant effect on GEO or Moser . The asterisks indicate statistically significant differences, two-tailed Student’s t-test P ≤ 0.005. B. Immunoblot results for cleaved caspase 3. These results demonstrate similar findings compared to those from the Cell Death Detection ELISA. and validate the findings of the ELISA assay.
Smad-independent pathways are activated by exogenous TGF-β1 in HCT116+Chr3 cells when they are in an anchorage free state
The paradoxical and varying effects of TGF-β in terms of apoptosis regulation emphasize the relevance of the cellular context in the response of cancer cells to this cytokine. Since this contextual effect may be due to differences between the cell lines with regard to post-TGF-_ receptor signal pathway activation, we studied whether differences in the effects of TGF-_ on anoikis between the cell lines could be attributed to differences in activation of signaling pathways by TGF-β1. We first evaluated the PI3K/Akt pathway in the HCT116+Chr3 cell line because this pathway has been implicated in the protective effect of TGF-β1 against apoptosis (Shin et al. 2001). The addition of exogenous TGF-β1 induces AKT phosphorylation in HCT116+Chr3 grown in suspension, and inhibition of the TGF-β signaling pathway by the DN2R dominant negative TGFBR2 transgene inhibits AKT phosphorylation (Figure 4A). Additionally, we found that inhibition of autocrine TGF-β signaling in HCT116+Chr3, by the DN2R dominant negative TGFBR2 transgene, dramatically reduces the basal levels of phosphorylated ERK1/2, another protein associated with cell survival (Figure 4B).
Figure 4. TGF-β-induced activation of the MAPK/ERK and PI3K/AKT pathways in HCT116+Chr3 is necessary for protection against anoikis.

A. Western blot analysis of phosphorylated ERK1/2 (Thr202/Tyr204) and total ERK1/2 in cells after treatment with TGF-β1. B. Western blot analysis of phosphorylated AKT (S473) and total AKT in cells treated with TGF-β1. C. Cell death analysis for HCT116+Chr3 and HCT116+Chr3+DN2R deprived from substrate for 24h and treated with LY294002 (10μM), U0126 (5μM) and TGF-β1 (10ng/ml). These results are representative of several assays. Each experiment was done in triplicate. The bars indicate S.E. The asterisks indicate statistically significant difference, p-value ≤ 0.05 as determined by the Student’s t-test (two-tailed).
Next, we assessed whether TGF-β-induced activation of the MAPK/ERK and PI3K/AKT pathways mediates the protective effect of TGF-_1 against anoikis. We treated HCT116+Chr3 with the pharmacological inhibitors U0126 and LY294002 in order to block the activity of MEK1/2 and PI3K, respectively. Anoikis increased in HCT116+Chr3 after treatment with both inhibitors. The increase in apoptosis was more pronounced after MAPK/ERK pathway inhibition compared to PI3K inhibition, and blockade of the MAPK/ERK and PI3K/AKT pathways concurrently had an additive effect on impairing TGF-β-mediated prevention of anoikis (Figure 4C). Interestingly, TGF-β1 could still partially prevent cell death in HCT116+Chr3 even in the presence of both U0126 and LY294002, suggesting the existence of MEK1/2- and PI3K-independent mechanisms in TGF-β-mediated resistance against cell death. Treatment of HCT116+Chr3+DN2R with LY294002 alone, or in combination with U0126, increased the amount of cell death to a greater extent than in HCT116+Chr3 (Figure 4C). Thus, it appears that autocrine TGF-β signaling can partially prevent cell death after loss of anchorage even in the presence of MEK1/2 and PI3K inhibitors. In summary, our results suggest that even though the activation of the PI3K/AKT and MAPK/ERK pathways is necessary for TGF-β to maximally protect HCT116+Chr3 against anoikis, modulation of these two signaling cascades is likely not the only mechanism through which TGF-β exerts its protective effect.
SMAD and non-SMAD pathway activation in FET, Moser, GEO and CBS and TGF-β’s effect on anoikis
Our analysis of the expression of activated ERK1/2 and AKT showed that they are involved in TGF-β-mediated inhibition of anoikis in HCT116+Chr3. Consequently, we examined the status of these pathways in other colorectal cell lines in order to determine the specificity of TGF-β’s induction of these pathways in the setting of anoikis. This analysis revealed that TGF-β1 does not induce the activation of ERK1/2 or AKT in Moser or GEO (Figure 5), which correlates with the lack of a protective effect of TGF-β1 on anoikis in these cell lines. Conversely, we found that although TGF-β treatment promotes anoikis in FET and prevents it in CBS, exogenous TGF-β1 treatment induces the activation of ERK1/2 but not AKT in these two cell lines (Figure 5). The paradoxical observation in FET and CBS cells suggests then that additional signaling pathways beyond MAPK-ERK and PI3K/AKT are involved with mediating the effect of TGF-_ on anoikis in these two colorectal cancer lines resulting in opposing responses to TGF-β stimulation in the context of lack of anchorage.
Figure 5. Effect of TGF-β1 treatment on caspase-3, ERK1/2, AKT and SMAD2 activation in colorectal cancer cell lines deprived from anchorage.
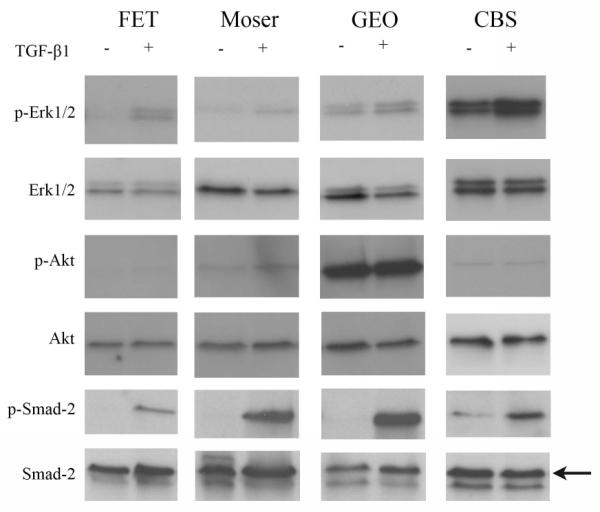
Immunoblot assay results for caspase-3, ERK1/2, AKT and SMAD2 and their active forms in FET, Moser, GEO, and CBS maintained in suspension for 20 hours with exogenous TGF-β1 (10ng/ml) are shown. Activation of SMAD2 is present in all of the cell lines, but only FET and CBS display increased ERK1/2 phosphorylation after TGF-_1 treatment.
Discussion
Survival signals are conveyed to epithelial cells upon the activation of cell surface receptors by components of the extracellular matrix (ECM) or by cell surface proteins of neighboring cells (Frisch and Francis 1994). Cell death triggered by the lack of appropriate cell-cell or cell-extracellular matrix contacts, called anoikis, is a mechanism for maintaining tissue architecture; and in the context of cancer progression, it represents a barrier for tumor invasion and metastasis (Frisch and Francis 1994; Frisch and Screaton 2001; Grossmann 2002). Tumor cells that have an increased ability to survive in conditions of detachment appear to have an enhanced capacity to metastasize in in vivo systems. Tumor cells less susceptible to anoikis due to alterations in proteins involved in integrin signaling (Duxbury et al. 2004b), cell-cell adhesion (Duxbury et al. 2004a) or the mitochondrial cell death pathway (Yawata et al. 1998) have greater metastatic potential.
Our studies indicate that in a subset of colorectal cancer cell lines, the MAPK/ERK and the PI3K/AKT signaling pathways can be regulated by TGF-β and that these pathways can induce increased resistance to anoikis. Moreover, in light of our demonstration in HCT116+Chr3 that TGF-β inhibits cell proliferation when the cells are grown attached, we have provided evidence that the cellular context determines the effects TGF-β exerts on colorectal cancer lines, and that paradoxically, TGF-β can both inhibit cell growth and induce resistance to anoikis in the same cell line. Our results suggest then that by increasing resistance to anoikis, TGF-β signaling may enhance the capacity of some colorectal cancer cells to invade and colonize secondary sites. This finding supports previous data that indicate that late-stage events in colorectal cancer progression may be promoted by cell-autonomous TGF-β signaling (Oft et al. 1998).
Our results also highlight that TGF-β’s effects on tumor progression are context-dependent. We demonstrated that even though TGF-β causes growth inhibition of HCT116+Chr3, CBS, Moser, GEO, and FET, this cytokine induces different responses to anoikis in these colorectal cancer lines. It is likely that the diversity in the responses is due to the effect of concurrent oncogene or tumor suppressor gene mutations in these cell lines even though they are all derived from human colorectal cancers. Accordingly, we established that TGF-β inhibits cell death upon detachment in HCT116+Chr3 and that this protective effect is associated with the activation of the MAPK/ERK and the PI3K/AKT pathways. In contrast, the protective effect in CBS is associated only with TGF-β-induced activation of MAPK/ERK. Also, we found that exogenous TGF-β has no effect on the occurrence of anoikis in Moser or GEO cell lines, which we propose is related to the inability of TGF-β to modulate the activity of the MAPK/ERK or PI3K/AKT pathways in these cell lines.
TGF-β signaling has been shown to exert a protective effect against apoptosis triggered by different stimuli in various cell systems through a variety of molecular mechanisms. TGF-β can protect microglia cells from FasL-induced death by a mechanism dependent on the activity of MEK (Schlapbach et al. 2000). In contrast, neuronal cells are protected from apoptosis caused by Ca2+ overloading or growth factor deprivation by TGF-β-induced upregulation of Bcl-2 expression (Prehn et al. 1994). Furthermore, TGF-β can prevent cell death upon growth factor withdrawal in mammary epithelial cells in an AKT-dependent fashion (Shin et al. 2001).
We now provide evidence that the MAPK/ERK and PI3K/AKT pathways are involved in TGF-β’s effects on anoikis in colorectal cancer cell lines. The relevance of the MAPK/ERK and PI3K/AKT cascades in protection against anoikis has been described in a wide variety of model systems. Oncogenic forms of RAS (Khwaja et al. 1997), RAF and AKT (Boisvert-Adamo and Aplin 2006; Schulze et al. 2001) can promote resistance against anoikis in epithelial cells. It is also clear that the specific role of these pathways in the regulation of the protective effects is cell-type dependent. Furthermore, it appears that TGF-β signaling interacts with these two survival pathways, both in Smad-dependent and Smad-independent manners, and in concert, they can modulate the responses of tumor cells to diverse apoptotic stimuli (reviewed in (Sanchez-Capelo 2005)).
In conclusion, our studies have revealed some of the molecular mechanisms through which cell-autonomous TGF-β signaling may promote colon cancer progression. Furthermore, these studies highlight the relevance of the cellular context in the determination of the cell responses to TGF-β stimulation. Hence, if the TGF-β signaling pathway is to be considered for potential therapeutic strategies, it appears likely that the cellular context of the cancer cells will need to be taken into consideration when such treatments are developed and used clinically.
Acknowledgements
This work was supported by RO1 CA115513, VA Dept. of R&D Presidential Early Career Award for Scientists and Engineers, and Mallinckrodt Scholar Award from the Edward Mallinckrodt Jr Foundation (to WMG). We thank Mary Aakre, Vanderbilt University, Nashville, TN, for her valuable technical assistance for the development of the thymidine incorporation assays.
References
- Alazzouzi H, Alhopuro P, Salovaara R, Sammalkorpi H, Jarvinen H, Mecklin J-P, Hemminki A, Schwartz S, Jr., Aaltonen LA, Arango D. SMAD4 as a Prognostic Marker in Colorectal Cancer. Clin Cancer Res. 2005;11(7):2606–2611. doi: 10.1158/1078-0432.CCR-04-1458. [DOI] [PubMed] [Google Scholar]
- Alhopuro P, Alazzouzi H, Sammalkorpi H, Davalos V, Salovaara R, Hemminki A, Jarvinen H, Mecklin JP, Schwartz S, Jr., Aaltonen LA. SMAD4 levels and response to 5-fluorouracil in colorectal cancer. Clin Cancer Res. 2005;11(17):6311–6. doi: 10.1158/1078-0432.CCR-05-0244. others. [DOI] [PubMed] [Google Scholar]
- Biswas S, Chytil A, Washington K, Romero-Gallo J, Gorska AE, Wirth PS, Gautam S, Moses HL, Grady WM. Transforming Growth Factor β Receptor Type II Inactivation Promotes the Establishment and Progression of Colon Cancer. Cancer Res. 2004;64(14):4687–4692. doi: 10.1158/0008-5472.CAN-03-3255. [DOI] [PubMed] [Google Scholar]
- Boisvert-Adamo K, Aplin AE. B-RAF and PI-3 kinase signaling protect melanoma cells from anoikis. Oncogene. 2006;25(35):4848–56. doi: 10.1038/sj.onc.1209493. [DOI] [PubMed] [Google Scholar]
- Boulay JL, Mild G, Lowy A, Reuter J, Lagrange M, Terracciano L, Laffer U, Herrmann R, Rochlitz C. SMAD4 is a predictive marker for 5-fluorouracil-based chemotherapy in patients with colorectal cancer. Br J Cancer. 2002;87(6):630–4. doi: 10.1038/sj.bjc.6600511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook T, Gebelein B, Mesa K, Mladek A, Urrutia R. Molecular cloning and characterization of TIEG2 reveals a new subfamily of transforming growth factor-beta-inducible Sp1-like zinc finger-encoding genes involved in the regulation of cell growth. J Biol Chem. 1998;273(40):25929–36. doi: 10.1074/jbc.273.40.25929. [DOI] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A. TGF-_ signaling in tumor suppression and cancer progression. Nat Genet. 2001;29(2):117. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper D. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430(7003):1034–9. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. CEACAM6 gene silencing impairs anoikis resistance and in vivo metastatic ability of pancreatic adenocarcinoma cells. Oncogene. 2004a;23(2):465–73. doi: 10.1038/sj.onc.1207036. [DOI] [PubMed] [Google Scholar]
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Focal adhesion kinase gene silencing promotes anoikis and suppresses metastasis of human pancreatic adenocarcinoma cells. Surgery. 2004b;135(5):555–62. doi: 10.1016/j.surg.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Elliott RL, Blobe GC. Role of transforming growth factor Beta in human cancer. J Clin Oncol. 2005;23(9):2078–93. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- Engle S, Hoying J, Boivin G, Ormsby I, Gartside P, Doetschman T. Transforming growth factor ß1 suppresses nonmetastatic colon cancer at an early stage in tumorigenesis. Cancer Research. 1999;59:3379–3386. [PubMed] [Google Scholar]
- Engle SJ, Ormsby I, Pawlowski S, Boivin GP, Croft J, Balish E, Doetschman T. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res. 2002;62(22):6362–6. [PubMed] [Google Scholar]
- Eppert K, Scherer S, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui L-C, Bapat B, Gallinger S, Andrulis I. MADR2 maps to 18q21 and encodes a TGFß-regulated MAD-related protein that is functionally mutated in colorectal cancer. Cell. 1996;86:543–552. doi: 10.1016/s0092-8674(00)80128-2. others. [DOI] [PubMed] [Google Scholar]
- Fernandez Y, Gu B, Martinez A, Torregrosa A, Sierra A. Inhibition of apoptosis in human breast cancer cells: role in tumor progression to the metastatic state. Int J Cancer. 2002;101(4):317–326. doi: 10.1002/ijc.10628. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994;124(4):619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13(5):555–62. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- Gorska AE, Jensen RA, Shyr Y, Aakre ME, Bhowmick NA, Moses HL. Transgenic mice expressing a dominant-negative mutant type II transforming growth factor-beta receptor exhibit impaired mammary development and enhanced mammary tumor formation. Am J Pathol. 2003;163(4):1539–49. doi: 10.1016/s0002-9440(10)63510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorska AE, Joseph H, Derynck R, Moses HL, Serra R. Dominant-negative interference of the transforming growth factor beta type II receptor in mammary gland epithelium results in alveolar hyperplasia and differentiation in virgin mice. Cell Growth Differ. 1998;9(3):229–38. [PubMed] [Google Scholar]
- Grady WM, Myeroff LL, Swinler SE, Rajput A, Thiagalingam S, Lutterbaugh JD, Neumann A, Brattain MG, Chang J, Kim S-J. Mutational inactivation of transforming growth factor-beta receptor type II in microsatellite stable colon cancers. Cancer Res. 1999;59(2):320–324. others. [PubMed] [Google Scholar]
- Grossmann J. Molecular mechanisms of“detachment-induced apoptosis--Anoikis”. Apoptosis. 2002;7(3):247–60. doi: 10.1023/a:1015312119693. [DOI] [PubMed] [Google Scholar]
- Hahn S, Schutte M, Hoque A Shamsul, Moskaluk C, da Costa L, Rozenblum E, Weinstein C, Fischer A, Yeo C, Hruban R. DPC4, a candidate tumor supressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. others. [DOI] [PubMed] [Google Scholar]
- Jung B, Smith E, Doctolero R, Gervaz P, Alonso J, Miyai K, Keku T, Sandler R, Carethers J. Influence of target gene mutations on survival, stage and histology in sporadic microsatellite unstable colon cancers. International Journal of Cancer. 2006;118(10):2509–2513. doi: 10.1002/ijc.21710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. Embo J. 1997;16(10):2783–93. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N’-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54(16):4308–4312. [published erratum appears in Cancer Res 1995 Jan 1;55(1):201] [PubMed] [Google Scholar]
- Lallemand F, Mazars A, Prunier C, Bertrand F, Kornprost M, Gallea S, Roman-Roman S, Cherqui G, Atfi A. Smad7 inhibits the survival nuclear factor kappaB and potentiates apoptosis in epithelial cells. Oncogene. 2001;20(7):879–84. doi: 10.1038/sj.onc.1204167. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF-_ Signal Transduction. Annual Review of Biochemistry. 1998;67(1):753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Muraoka-Cook RS, Kurokawa H, Koh Y, Forbes JT, Roebuck LR, Barcellos-Hoff MH, Moody SE, Chodosh LA, Arteaga CL. Conditional overexpression of active transforming growth factor beta1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res. 2004;64(24):9002–11. doi: 10.1158/0008-5472.CAN-04-2111. [DOI] [PubMed] [Google Scholar]
- Normanno N, De Luca A, Maiello MR, Bianco C, Mancino M, Strizzi L, Arra C, Ciardiello F, Agrawal S, Salomon DS. CRIPTO-1: a novel target for therapeutic intervention in human carcinoma. Int J Oncol. 2004;25(4):1013–20. [PubMed] [Google Scholar]
- Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8(23):1243–52. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res. 1995;55(23):5548–50. [PubMed] [Google Scholar]
- Pierce D, Gorska A, Chytil A, Meise K, Page D, Coffey R, Moses H. Mammary tumor suppression by transforming growth factor ß1 transgene expression. Proceedings of the National Academy of Science USA. 1995;92:4254–4258. doi: 10.1073/pnas.92.10.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehn JHM, Bindokas VP, Marcuccilli CJ, Krajewski S, Reed JC, Miller RJ. Regulation of Neuronal Bc12 protein expression and calcium homeostasis by transforming growth factor type {beta} confers wide-ranging protection on rat hippocampal Neurons. PNAS. 1994;91(26):12599–12603. doi: 10.1073/pnas.91.26.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandra M, Atencio I, Rahman A, Vaillancourt M, Zou A, Avanzini J, Wills K, Bookstein R, Shabram P. Restoration of transforming growth factor Beta signaling by functional expression of smad4 induces anoikis. Cancer Res. 2002;62(21):6045–51. [PubMed] [Google Scholar]
- Samowitz WS, Curtin K, Neuhausen S, Schaffer D, Slattery ML. Prognostic implications of BAX and TGFBRII mutations in colon cancers with microsatellite instability. Genes Chromosomes Cancer. 2002;35(4):368–71. doi: 10.1002/gcc.10125. [DOI] [PubMed] [Google Scholar]
- Sanchez-Capelo A. Dual role for TGF-[beta]1 in apoptosis. Cytokine & Growth Factor Reviews. 2005;16(1):15. doi: 10.1016/j.cytogfr.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Schlapbach R, Spanaus KS, Malipiero U, Lens S, Tasinato A, Tschopp J, Fontana A. TGF-beta induces the expression of the FLICE-inhibitory protein and inhibits Fas-mediated apoptosis of microglia. Eur J Immunol. 2000;30(12):3680–8. doi: 10.1002/1521-4141(200012)30:12<3680::AID-IMMU3680>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Schulze A, Lehmann K, Jefferies HB, McMahon M, Downward J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev. 2001;15(8):981–94. doi: 10.1101/gad.191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte M, Hruban R, Hedrick L, Cho K, Nadasdy G, Weinstein C, Bova G, Isaacs W, Cairns P, Nawroz H. DPC4 gene in various tumor types. Cancer Research. 1996;56:2527–2530. others. [PubMed] [Google Scholar]
- Shimanuki T, Hara T, Furuya T, Imamura T, Miyazono K. Modulation of the functional binding sites for TGF-beta on the type II receptor leads to suppression of TGF-beta signaling. Oncogene. 2007;26(23):3311–20. doi: 10.1038/sj.onc.1210123. [DOI] [PubMed] [Google Scholar]
- Shin I, Bakin AV, Rodeck U, Brunet A, Arteaga CL. Transforming growth factor beta enhances epithelial cell survival via Akt-dependent regulation of FKHRL1. Mol. Biol. Cell. 2001;12(11):3328–3339. doi: 10.1091/mbc.12.11.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel P, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3(11):807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- Swift LL, Farkas MH, Major AS, Valyi-Nagy K, Linton MF, Fazio S. A recycling pathway for resecretion of internalized apolipoprotein E in liver cells. J Biol Chem. 2001;276(25):22965–70. doi: 10.1074/jbc.M100172200. [DOI] [PubMed] [Google Scholar]
- Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin M, Taketo M. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92:645–656. doi: 10.1016/s0092-8674(00)81132-0. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Wu T-T, Catalano PJ, Ueki T, Satriano R, Haller DG, Benson AB, Hamilton SR. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344(16):1196–1206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawata A, Adachi M, Okuda H, Naishiro Y, Takamura T, Hareyama M, Takayama S, Reed JC, Imai K. Prolonged cell survival enhances peritoneal dissemination of gastric cancer cells. Oncogene. 1998;16(20):2681–6. doi: 10.1038/sj.onc.1201792. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Richardson J, Parada L, Graff J. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–714. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Sanchez-Sweatman O, Huang X, Wiltrout R, Khokha R, Zhao Q, Gorelik E. Anoikis and metastatic potential of cloudman S91 melanoma cells. Cancer Res. 2001;61(4):1707–1716. [PubMed] [Google Scholar]