

Abstract
Protein tyrosine phosphatases (PTPs) are central players in many different cellular processes and their aberrant activity is associated with multiple human pathologies. In this review, we present current knowledge on the PTPRR subfamily of classical PTPs that is expressed in neuronal cells and comprises receptor-type (PTPBR7, PTP-SL) as well as cytosolic (PTPPBSγ-37, PTPPBSγ-42) isoforms. The two receptor-type isoforms PTPBR7 and PTP-SL both localize in late endosomes and the Golgi area. PTPBR7, however, is additionally localized at the cell surface and on early endosomes. During cerebellar maturation, PTPBR7 expression in developing Purkinje cells ceases and is replaced by PTP-SL expression in the mature Purkinje cells. All PTPRR isoforms contain a kinase interacting motif that makes them mitogen-activated protein kinase phosphatases. The distinct subcellular localization of the different PTPRR isoforms may reflect differential roles in growth-factor-induced MAPK-mediated retrograde signaling cascades. Studies in PTPRR-deficient mice established that PTPRR isoforms are physiological regulators of MAPK phosphorylation levels. Surprisingly, PTPRR-deficient mice display defects in motor coordination and balancing skills, while cerebellar morphological abnormalities, which are often encountered in ataxic mouse models, are absent. This is reminiscent of the phenotype observed in a handful of mouse mutants that have alterations in cerebellar calcium ion homeostasis. Elucidation of the molecular mechanisms by which PTPRR deficiency imposes impairment of cerebellar neurons and motor coordination may provide candidate molecules for hereditary cerebellar ataxias that still await identification of the corresponding disease genes.
Electronic supplementary material
The online version of this article (doi:10.1007/s12311-008-0088-y) contains supplementary material, which is available to authorized users.
Keywords: Calcium signaling, Endosomes, Ultrastructural localization, Live cell imaging, Motor coordination, Signal transduction
Introduction
Reversible tyrosine phosphorylation is a powerful mechanism to regulate protein-mediated processes that steer the proliferation, differentiation and functioning of cells and the development, homeostasis and functioning of multicellular organisms. Protein tyrosine phosphatases (PTPs) and protein tyrosine kinases are the enzyme classes that are instrumental in controlling the spatial and temporal ratio of tyrosine-phosphorylated and non-phosphorylated targets and thus coordinately regulate cellular responses to intra- and extracellular cues. Tyrosine phosphatases can be roughly divided into two distinct groups: the truly phosphotyrosine-specific PTPs on the one hand and the dual-specificity phosphatases (DSPs) on the other [1, 2]. DSPs can dephosphorylate phosphoserine and phosphothreonine in addition to phosphotyrosine residues, and some are also active against phospholipids [3]. The classical, phosphotyrosine-specific PTPs consist of a subgroup that possesses a transmembrane domain, a receptor-like extracellular region and one or two catalytic PTP domains and a second subgroup that is non-transmembrane and mostly cytoplasmic in location [4]. The non-transmembrane PTPs and DSPs all contain a single catalytic PTP domain. This ~250-residue-long PTP domain is highly conserved and contains an essential cysteine residue within its active site that is used in a cyteinyl-phosphate enzyme intermediate during dephosphorylation [5].
How PTP activity is regulated is still subject of intense investigation, but mechanisms include alternative mRNA splicing, modulation of steady state levels, posttranslational modification (including phosphorylation), dimerization and/or subcellular confinement [6]. With regard to the physiological functions of individual PTPs, also much more work needs to be done. Many studies at the cellular level are hampered by the very low endogenous expression levels of PTPs and their firm catalytic activity that is some log orders of magnitude over that of kinases. In combination, this may cause overexpression artifacts in transfected cell-based readouts, and thus, it should come as no surprise that most information on PTP functioning resulted from studies of mammalian pathologies or via exploitation of genetically modified animal models [4]. In this paper, we will review the current knowledge on the neuronal, phosphotyrosine-specific PTP isoforms that are encoded by the gene Ptprr in mouse and PTPRR in man. This gene and its products may be of particular relevance to cerebellar function, as rodent postnatal cerebellar Purkinje cells express a unique PTPRR isoform (PTP-SL) and Ptprr knockout mice display an ataxia phenotype. We will focus on the ways that confine and regulate PTPRR enzymatic activity, most notably the distinct subcellular locations displayed by the various isoforms, and will elaborate on the motor coordination defect displayed by PTPRR-deficient mice.
Protein Tyrosine Phosphatase Receptor-Type R Isoforms
PTPRR gene orthologues are present in many vertebrates and homologous genes can still be found in distant species like Drosophila (Table 1) [7–10]. Interestingly, like in human and rat, the mouse Ptprr gene gives rise to multiple neuronal PTPRR protein isoforms [11, 12]. Four different transcripts (represented by PTPBR7, PTP-SL, PTPPBSγ+, and PTPPBSγ− cDNAs) have been identified [7, 13, 14] that result from the differential use of three distinct transcription start sites [12, 15] and the alternative inclusion of a 117-nucleotide sequence stretch in the unique 5′ UTR of the shortest mRNA (PTPPBSγ [7]. In situ hybridization studies, Northern blot experiments, and reverse transcription polymerase chain reaction (RT-PCR) analyses have gathered RNA expression data for these individual transcripts. PTPBR7 messengers are expressed during early embryogenesis in spinal ganglia and Purkinje cells (PCs). PTP-SL transcripts are not present during prenatal development, but postnatally, PTP-SL transcripts replace the decreasing PTPBR7 levels in PCs [15]. PTPBR7 expression persists in all other brain regions after birth. Trace amounts of PTP-SL messengers are present in midbrain, brainstem, and cortex, but firm expression is exclusively in adult cerebellar PCs (Fig. 1a). Intriguingly, this cell-specific expression pattern is fully conserved in rat [8, 9]. Mouse PTPPBSγ variants are expressed at extremely low levels throughout the brain. In fact, they represent the only Ptprr transcripts that have been detected in non-neuronal tissues; trace amounts have been detected in the gastrointestinal tract and in developing cartilage [7, 12]. Unfortunately, most cell lines in culture do not express PTPRR-encoding transcripts. Thus far, only in the rat neuroendocrine PC12 cells has a Ptprr-derived transcript, the one that encodes the receptor-type protein isoform PCPTP1, been detected [9, 16].
Table 1.
PTPRR nomenclature in various species
| Species | Homo sapiens | Mus musculus | Rattus norvegicus | Drosophila melanogaster |
|---|---|---|---|---|
| Gene | PTPRR | Ptprr | Ptprr | PTP-ER |
| (position) | 12q15 | 10A2 | 7q22 | 2(57F) |
| Protein isoforms | PTPPBSα | PTPBR7 | PCPTP1 | – |
| PTPPBSβ | PTP-SL | PCPTP1-Ce | – | |
| PTPPBSγ | PTPPBSγ42 | – | PTP-ER | |
| – | PTPPBSγ37 | – | – | |
| PTPPBSδ | – | – | – |
Fig. 1.

PTPRR isoforms are expressed in the brain and interact with MAPKs. a PTPRR mRNA levels in adult mouse brain. A sagittal section demonstrating high transcript levels in the hippocampal area and most notably cerebellar Purkinje cells, extracted from the Allen Brain Atlas [72]. b Schematic representation of the different mouse PTPRR protein isoforms. Protein names and lengths, in amino acid residue numbers (aa), are on the left. SP signal peptide, HR hydrophobic region, TM transmembrane region, KIM kinase interacting motif, PTP protein tyrosine phosphatase domain. c MAPK localization in the cytoplasm and into the nucleus depends on the balance between growth factor activation and KIM-containing PTP inhibition. Activated PKA will prevent the PTPRR–MAPK association by phosphorylation of the KIM domain [34]. RTK receptor tyrosine kinase, MAPKKK MAPKK kinase, MAPKK MAPK kinase, MAPK mitogen-activated protein kinase
The different Ptprr transcripts give rise to multiple PTPRR protein isoforms (Fig. 1b). Mutagenesis studies revealed exactly which AUG start codons are being used in the various Ptprr mRNAs [12]. Intriguingly, the second as well as the third start site in the two types of PTPPBSγ mRNAs are preferred by the ribosomes, generating isoforms PTPPBSγ-42 and PTPPBSγ-37 with sizes of 42 and 37 kDa, respectively. PTPBR7 and PTP-SL are 72 and 60 kDa in size, respectively. PTPBR7 is a classical type I transmembrane protein: its functional signal-peptide is cleaved of in the endoplasmic reticulum (ER) and a hydrophobic segment further on in the protein functions as transmembrane anchor. The PTP-SL isoform contains two hydrophobic regions in the N-terminal part, which led originally to the prediction that it also would be a type I transmembrane protein [13]. However, subsequent studies revealed that PTP-SL is lacking a functional signal peptide and thus may be a membrane-associated protein [15]. Only recently, the use of a selective cell permeabilization method in combination with dedicated antisera directed against PTPRR domains allowed the determination of PTP-SL’s exact membrane topology to be a type III transmembrane protein [17]. The two smallest isoforms, PTPPBSγ-42 and PTPPBSγ-37, are located in the cytosol. Interestingly, the two receptor-type isoforms, PTPBR7 and PTP-SL, form homo- and heteromeric complexes, whereas the cytosolic species PTPPBSγ-42 and PTPPBSγ-37 cannot. Furthermore, in line with data for a number of other RPTPs, the enzymatic activity of PTPRR monomers towards an artificial substrate decreased significantly upon multimerization [17]. This opens up the possibility that the activity of the receptor-type PTPRR isoforms can be modulated through ligand binding. Ligand hunting strategies using tagged PTPBR7 ectodomain fusion proteins [18] may serve in the identification of such molecules.
PTPRR Can Bind and Inactivate Mitogen-Activated Protein Kinases
Growth factor binding induces dimerization of their receptor tyrosine kinase (RTK), which results in increased kinase activity and the generation of phosphotyrosine sites on the RTK cytosolic portion that serve as docking sites for interacting proteins [19, 20]. The consequent recruitment, assembly, and phosphorylation of these downstream signaling complexes, in turn, switches on many different signaling pathways, including the intensively studied mitogen-activated protein kinase (MAPK) cascades [20]. MAPK pathways are organized into three layers of consecutive acting kinases that through the dual kinase specificity of MAPK-kinases lead to the phosphorylation of two regulatory residues (pT-X-pY) in MAPKs, serine/threonine kinases that continuously shuttle between the nucleus and the cytoplasm (Fig. 1c). Once activated by dual phosphorylation, MAPKs can phosphorylate a wide range of cytosolic and nuclear substrates [21, 22]. Termination of MAPK signaling thus can be achieved, in principle, by phosphatases that are either serine/threonine-specific (PP phosphatases), tyrosine-specific (classical PTPs), or of the dual-specificity type (DSPs) [23–25]. The subcellular localization and activity of such MAPK phosphatases is highly relevant for proper spatiotemporal control of MAPK activity and thus a key determinant of the final cellular response triggered by ligand-induced activation of RTKs [26, 27].
In the human genome, a subset of 11 DSP genes encode phosphatases that are especially suited to bind, dephosphorylate, and thus inactivate MAPKs, and these are generally referred to as MKPs (acronym for MAP kinase phosphatases) [28]. An additional three genes encode classical, tyrosine-specific PTPs capable of binding and inactivating MAPK family members, among which PTPRR [29]. Both the dual-specific MKPs and the tyrosine-specific MAPK phosphatases (MAPK-PTPs) have in common the presence of a 16-amino acid kinase interaction motif (KIM), located just N-terminal of the phosphatase domain, which is required for the association with MAPKs. All the PTPRR isoforms contain such a KIM (Fig. 1b) which in part determines the binding specificity towards the various MAPKs [30, 31]. Association specificity is also influenced by a small sequence that is located in between the KIM and the PTP domain [32]. As a substrate, the MAPK-PTPs prefer ERK1/2/5 and p38 over JNK [31–33]. Intriguingly, overexpression of MAPK-PTPs demonstrated that they not only are able to inactivate MAPKs but that their binding also prevents the translocation of the MAPKs into the nucleus [31, 34, 35]. Furthermore, upon binding to activated MAPKs, the MAPK-PTP itself first becomes phosphorylated by the MAPK (on a threonine residue just outside the KIM domain) before it can dephosphorylate the protein [30]. Importantly, the interaction between the two proteins is regulated by protein kinase A (PKA). Phosphorylation of a conserved serine residue within the KIM domain, which can be reversed by PP1 [36, 37], abolishes the binding and subsequent dephosphorylation of MAPKs and results in the release of MAPKs to the nucleus [34, 38]. This PKA-mediated regulatory principles also hold for most of the dual-specificity MKPs that contain a KIM-like domain [28].
Distinct Subcellular Localization for Receptor-Type PTPRR Isoforms
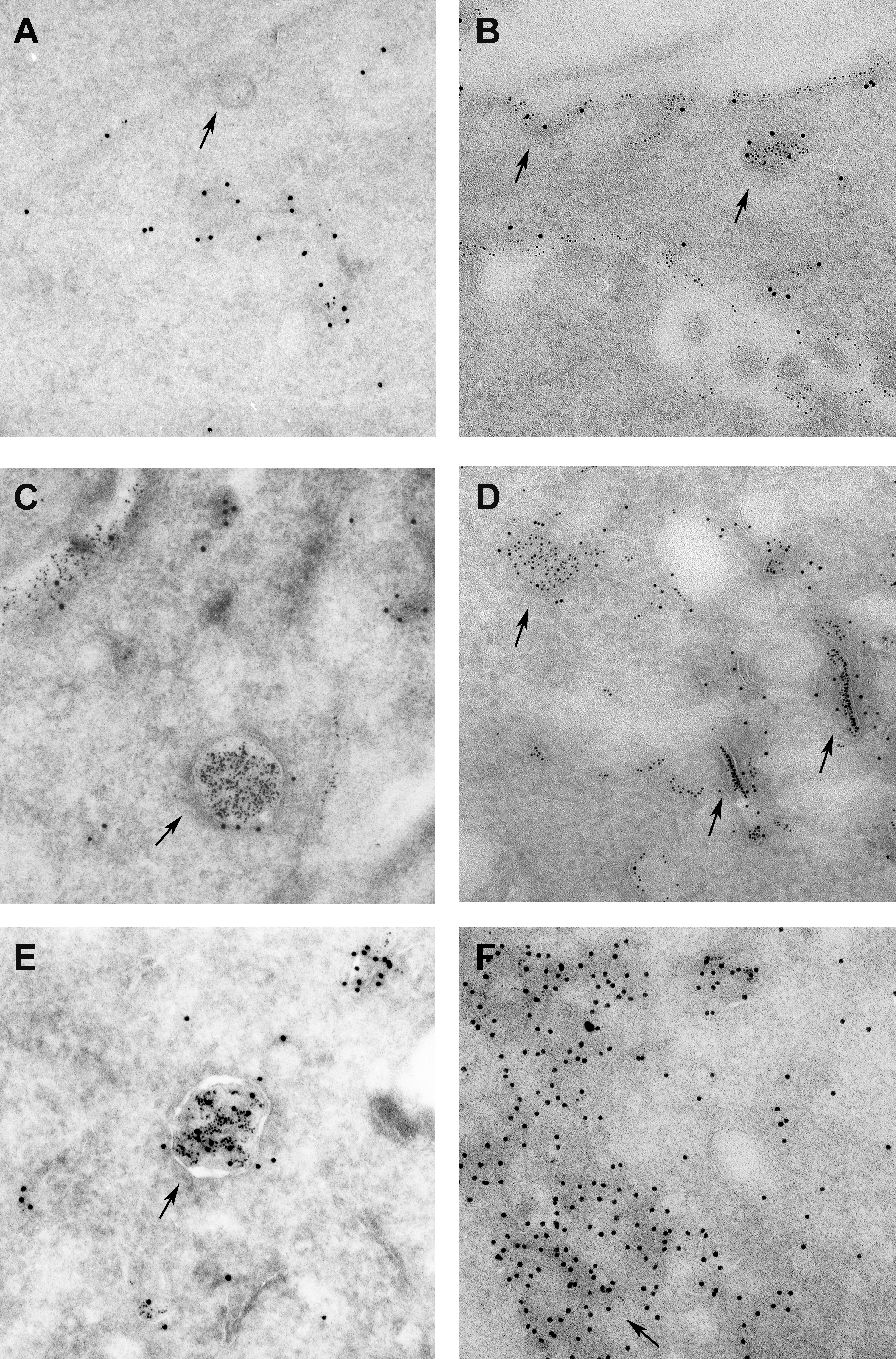
The PTPRR receptor-type isoforms PTPBR7 and PTP-SL share a common membrane topology [17] and both localize at the trans-Golgi network and on endocytic vesicles [15, 39]. However, PTPBR7 is found at the cell surface, whereas PTP-SL is not. Since detailed knowledge on their subcellular localization bears relevance for retrograde signaling cascades by endocytosed growth factor receptors, PTPBR7 and PTP-SL decorated vesicles were classified to different stages of the endocytic pathway (see Electronic Supplementary Material). Endogenous expression of PTPRR proteins in either primary neurons (Electronic Supplementary Material Fig. S1) or PC12 cells [40] is very low but reflects the localization pattern observed in transfected cells [39]. Fluorescent Dextran uptake experiments with mouse Neuro-2a cells transiently expressing EGFP-tagged PTPBR7 (Electronic Supplementary Material Fig. S2) or PTP-SL (Electronic Supplementary Material Fig. S3) corroborated that PTPBR7 and PTP-SL are present on endosomes [39]. Electron microscopic monitoring of timed 5-nm bovine serum albumin gold uptake established that PTP-SL locates predominantly to late endosomes, whereas PTPBR7 is also present at the plasma membrane and endosomes from earlier stages (Electronic Supplementary Material Fig. S4). Co-transfection of Neuro-2a cells with PTPBR7 and PTP-SL expression plasmids confirmed that PTPBR7 and PTP-SL both are present on late endosomal vesicles (Fig. 2). It should be noted, however, that it is unlikely that co-localization of the two isoforms will actually occur in vivo since PTPBR7 transcripts are detectable throughout the developing and adult brain, but in PCs, in fact, PTP-SL transcripts take over during maturation [15]. Single-positive PTPBR7 vesicles are particularly found in the vicinity of the plasma membrane, reminiscent of the early endocytic compartment in which PTP-SL is sparse (Fig. 2a–c). Analysis of the dynamics of the vesicles harboring receptor-type PTPRR isoforms, by time-lapse fluorescence microscopy of PTPBR7-EGFP and PTP-SL-EGFP proteins in living cells, revealed both anterograde and retrograde movement (Electronic Supplementary Material Fig. S5) with an average speed that did not differ significantly between PTPBR7 and PTP-SL positive vesicles. The PTPRR positive vesicle trafficking patterns are reminiscent of movement along microtubule tracks (Electronic Supplementary Material Fig. S6). Together, these findings demonstrate that PTPBR7 is present on vesicles throughout the endocytic pathway from the plasma membrane to the Golgi apparatus. In contrast, PTP-SL is only present on vesicles participating in the later stages of endocytosis, close to the trans-Golgi network, indicative of differential functions of these PTPRR isoforms within the endocytic pathway.
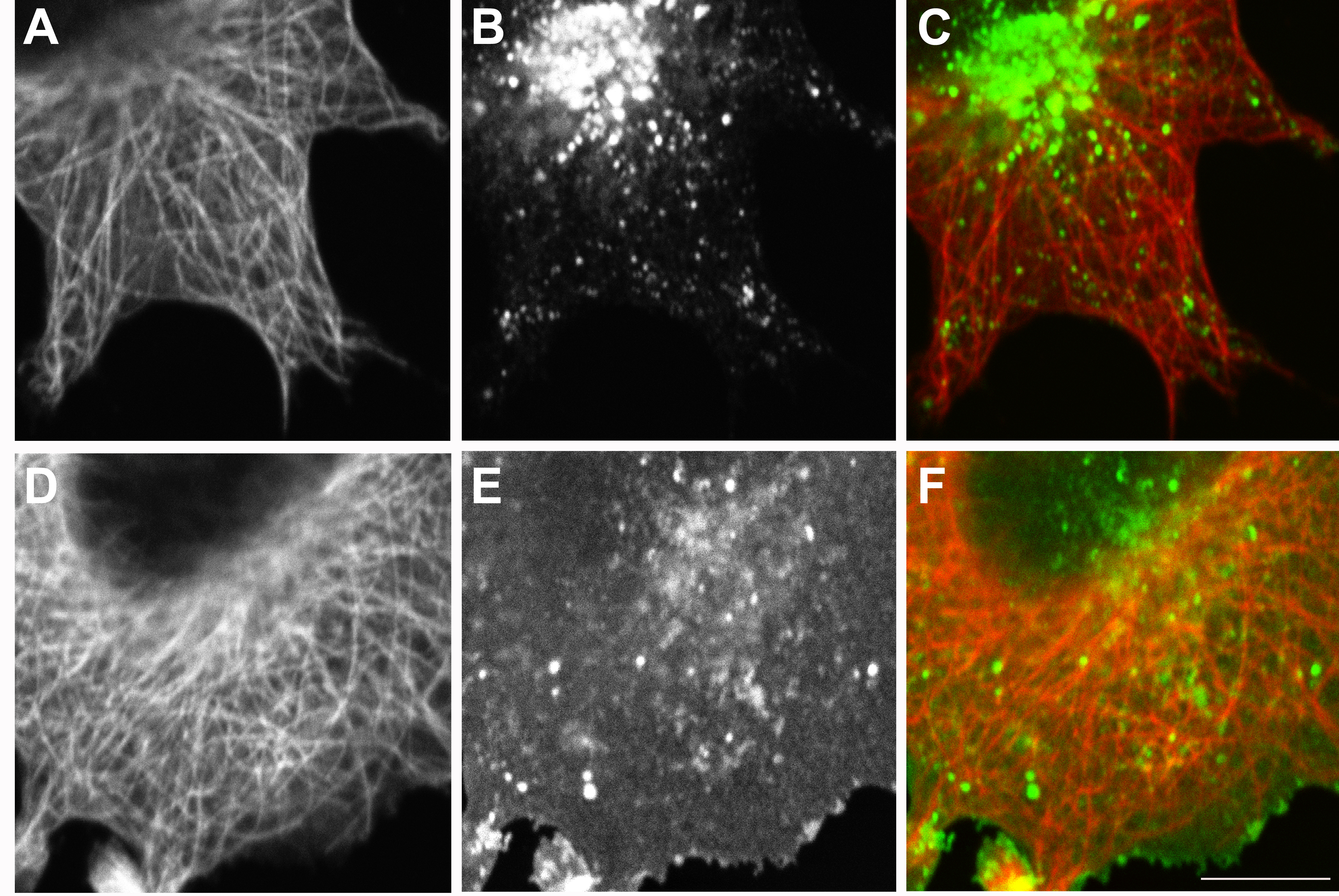
Fig. 2.

PTPBR7 as well as PTP-SL localize in late endocytic vesicles. Neuro-2a cells were co-transfected with pPTP-SL-EGFP and pSG8/PTPBR7-FL-VSV. EGFP fluorescence of PTP-SL-EGFP was recorded directly (b, e) and PTPBR7 proteins were visualized immunohistochemically using the P5D4 antibody against the VSV tag (a, d). Yellow color in c indicates overlap of PTP-SL-EGFP (green) and PTPBR7 (red) signals. After 20 min of Alexa-633-labeled Dextran uptake, triple-labeled vesicles that are positive for PTPBR7 (d), PTP-SL (e), and Dextran (f) can be detected (arrows), corroborating that PTPBR7 and PTP-SL co-localize at vesicles of the late endosomal compartment. Bar indicates 10 μm
Towards the Function of PTPRR Protein Isoforms
It could be that the PTPRR isoforms PTPBR7 and PTP-SL are merely cargo proteins that for example need to be delivered to lysosomes for degradation. In line with this, all PTPRR isoforms are short-lived proteins with half-lives varying from 3 to 5 h [11]. However, PTPRR proteins do not co-localize with lysosomal marker proteins [39] and both anterograde and retrograde movements for PTPRR-positive vesicles are observed (Electronic Supplementary Material Fig. S5), whereas cargo proteins are expected to travel unidirectional and to accumulate at locations within the cell. This leaves two other options, namely that these phosphatases make part of the endocytic transport machinery itself and/or are involved in localized signaling events.
Accurate packaging of cargo and (coated) vesicle formation and membrane fusion in vivo requires tight regulation for which phosphorylation is indeed one of the mechanisms [41]. For example, EGF-induced tyrosine phosphorylation of the β2-subunit of the adapter protein complex AP-2 turned out to be important in regulating EGFR protein turnover [42]. Likewise, the activity of PTP-MEG2 (PTPN9) on secretory vesicle membranes is required to locally bind and dephosphorylate NSF and promote secretory vesicle fusion [43]. PTPRR interacted with the β4-subunit of the AP-4 complex in a yeast two-hybrid system and co-localized with AP-4 at the Golgi apparatus and on endosomes in neuronal cells [39], suggesting a role in AP-4-mediated vesicle sorting. In line with this, co-expression of PTP-SL and β4-adaptin in Neuro-2a cells led to a displacement of PTP-SL from vesicles and the Golgi apparatus and a redistribution throughout the cytoplasm [39].
The alternative option would be a role for PTPRR in spatiotemporal control of growth factor signaling involving receptor internalization [44]. For example, after ligand binding and autophosphorylation at the plasma membrane, the EGF tyrosine kinase receptor is internalized and subsequently dephosphorylated by PTP1B that resides at the ER membrane, eventually resulting in receptor silencing (signal termination) and RTK breakdown in lysosomes [45]. Localized signaling has also been demonstrated for the MAPK scaffold Sef that resides on the Golgi apparatus; it binds ERK—thus blocking nuclear translocation—without inhibiting its activity in the cytoplasm [46]. In view of their KIM domain-mediated association with MAPKs, resulting in MAPK inactivation and prevention of nuclear translocation [30, 31, 33], a comparable role for PTPRR isoforms may be expected. Most of these studies, however, involved (ectopic) overexpression of PTPRR isoforms. The endogenous expression of PTPRR in PC12 cells [16], a paradigm model for the study of spatial and temporal control of MAPK signaling [26, 27], allowed assessment of the impact of PTPRR on MAPK activity under more physiological conditions. Modulation of PTPRR activity in these cells, however, had no effect on the temporal profile of EGF- and NGF-induced ERK1/2 activity [40]. Since in PC12 cells the two other MAPK-PTPs are not expressed, this finding suggests that rather the dual-specificity MKPs serve as key MAPK regulators.
Biological Activities of Human PTPRR Proteins
Information on putative roles of PTPRR isoforms in health and disease is limited. The human PTPRR gene has been excluded as a candidate for a type 2 diabetes locus on chromosome 12q15 [47], but appeared implicated in the pathogenesis of inv(12)-type leukemia [48]. Exon 4 of the TEL gene was found fused to exon 7 of the PTPRR gene, resulting in a TEL/PTPRR chimeric gene. This fusion gene generates ten different transcripts of which only one encodes a chimeric product consisting of the TEL transcriptional repressor fused to a large part of the PTPRR phosphatase domain. This TEL-PTPRR fusion protein, however, lacks catalytic activity, indicating that it is most likely through interference with the functioning of wild-type TEL that this chimera causes leukemia [48]. Another study addressed a potential role for the cytosolic PTPPBSγ isoforms in cartilage formation [49]. Using an embryonic mandibular explant culture model system and antisense oligodeoxynucleotides, evidence was provided that PTPPBSγ may regulate chondroblast proliferation in early skeletal development.
The advent of microarray expression analyses has generated both confusing and surprising data on the PTPRR gene. While analyzing the impact of CIITA, a master transcriptional regulator of MHC class II genes, on global mRNA levels, it was noted that PTPRR transcripts are down-regulated upon overexpression of this transcription factor in a human B cell line and in IFN-γ-induced fibroblasts [50]. Subsequent assessment of the role of CIITA in repressing PTPRR expression using CIITA overexpressing and knockout mice, however, demonstrated that significant PTPRR expression is restricted to the brain and that its transcript levels are not influenced by CIITA [51]. In line with this, no derepression of gene Ptprr was observed in B cells or fibroblasts from CIITA-deficient mice. Another microarray-based study reported PTPRR expression in endometrial tissue and real-time RT-PCR data showed that median gene expression for PTPRR increased 47-fold from the proliferative to the secretory phase in the menstrual cycle [52]. A further 2.4-fold up-regulation was noted in the endometrium of women with endometriosis compared to controls, which led the authors to speculate that PTPRR overexpression may be a predisposing factor in the etiology of endometriosis by preventing normal differentiation of endometrial cellular function [52]. And finally, gene expression profiling of postmortem orbitofrontal cortex in violent suicide victims revealed that PTPRR expression is 1.5-fold higher in suicide tissue as compared to control samples [53]. This raised interest because also significantly decreased levels of MAPK1 and MAPK3 and increased amounts of MAP kinase phosphatase 2 (MKP2) have been detected in the prefrontal cortex and hippocampus of depressed suicide subjects [54]. Whether and how these alterations in the cerebral signaling circuitry might be part of the pathophysiology of suicidal behavior will of course require much more study.
PTPRR-Deficient Mice Display Motor Coordination Defects
As an alternative approach to gain insights in the physiological role of PTPRR isoforms, PTPRR-deficient mice have been generated [55]. Knockout animals appeared healthy and were fertile, and inheritance of the mutant allele followed Mendelian rules. The high expression levels of PTPRR isoforms in the hippocampal area and especially in the cerebellar Purkinje cells led to the assessment of mouse basic behavior and locomotion. Nocturnal activity of PTPRR-deficient mice was significantly reduced as compared to wild-type control mice. In addition, PTPRR-deficient mice displayed significant defects in their fine motor coordination and balance skills that are reminiscent of a mild ataxia [55]. Since MAPK signaling pathways are implicated in various neuropathological states [56] and PTPRR can interact with several MAPKs, it should perhaps not come as a surprise that ERK1/2 phosphorylation levels were indeed found to be significantly elevated in PTPRR-deficient brains. However, brain morphology, Purkinje cell number, or dendritic branching was not affected [55].
The majority of ataxia mouse models display histological phenomena (i.e., loss of cerebellar granule cells or PCs) that explain cerebellar dysfunction [57]. Only a few mouse models exist that display ataxic behavior in combination with normal cerebellar morphology. These include junctophilin 3 [58], carbonic anhydrase-related protein VIII (Car8) [59], calbindin [60], and calretinin [61] knockout animals. Collectively, these models point to alterations in Ca2+ homeostasis related to Purkinje cell physiology as causative for the ataxia. Junctophilins contribute to the formation of junctional membrane complexes, by spanning the endoplasmic/sarcoplasmic reticulum membrane and interacting with the plasma membrane, that function as structural platforms for Ca2+-mediated crosstalk between cell surface and intracellular channels. Car8 has no carbonic anhydrase enzymatic activity but was identified as a binding protein for the IP3 receptor expressed in cerebellar PCs [62]. Thus, it is conceivable that Car8 plays a role in fine-tuning of the intracellular calcium level in the PC dendrites to optimize parallel fiber innervation and proper synaptic formation and/or maintenance in the cerebellum. Calbindin and calretinin both are calcium-binding proteins that turned out to be crucial in the shaping of intraneuronal Ca2+ fluxes like PC firings. In the cerebellar cortex of alert mice, calbindin or calretinin deficiency resulted in high-frequency oscillations that emerged from interplay between synchronized PC assemblies and coupled interneurons in the cerebellar molecular layer [61].
Calcium, Phosphorylation, and Motor Coordination
Also the work on human disorders demonstrates that aberrant cerebellar Ca2+ signaling may result in cerebellar ataxia. For example episodic ataxia 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) result from mutations in the pore-forming subunit of P/Q-type voltage-gated Ca2+ channels [57, 63, 64]. EA2 manifests as an episodic, fully reversible ataxia that only after decades may lead to persistent ataxia and mild cerebellar degeneration. Several other ataxia syndromes are directly linked to alterations in the neuronal phosphoproteome [63, 64] that ultimately may have bearing for cell membrane receptors and ion channels [65, 66]. In SCA12, for example, a repeat expansion in the regulatory subunit PR55/Bβ alters phosphatase PP2A expression and, consequently, the phosphorylation status of target proteins (including MAPKs). And SCA14 mutations create a PKCγ superkinase that, in response to Ca2+ influx, readily phosphorylates brain glutamate receptors [65]. Thus, some SCA mutations apparently alter the phospho-status of neuronal Ca2+ channels and receptors and change their membrane localization, activity, and hence functioning in synaptic connectivity and plasticity [67]. The notion that MAPK signaling is a key process in synaptic plasticity [68] and that aberrant ERK1/2 activity has been noted in several neuropathologies [56, 69] nicely merges with the above.
Continued excessive stimulation of Ca2+-permeable glutamate receptors or voltage-dependent Ca2+ channels (VDCCs) is expected to result in neuronal excitotoxicity. The chronic Ca2+ overshoot and concomitant inappropriate activation of Ca2+-dependent processes in cells may then result in production of free radicals, altered mitochondrial function, and ultimately cell death. Apparently, Purkinje cells and cerebellar granule cells are quite vulnerable to such cell stress conditions, but it is believed that excitotoxicity may also contribute to other neurodegenerative illnesses like Alzheimer’s, Parkinson’s, and Huntington’s disease [70].
Concluding remarks
The picture emerges that aberrancies in cellular phosphorylation-dependent processes can deregulate, at the transcriptional and/or the posttranslational level, the calcium-ion-dependent communication circuitry of cerebellar neurons, thereby ultimately causing ataxias. In Fig. 3, we provide a simplified model in which the various lines of evidence are integrated. Activated growth factor receptors recruit distinct adaptor proteins and start up various signaling cascades, such as the PI3K, PLCγ, and MAPK pathways. Transient ERK activation via Ras will affect cytosolic targets that may well include transmembrane proteins regulating cellular Ca2+ concentration (e.g., glutamate-gated ion channels and VDCCs) and membrane potential (e.g., Kv4.2). Following receptor endocytosis, Rap1-mediated sustained activation of ERK will lead to alterations in gene expression. PTPRR may indirectly influence such transcriptional events by inactivating ERK, but could also, much like the proteins mutated in SCA12 and SCA14 patients, directly dephosphorylate and thus regulate relevant cytosolic proteins or even ion channels and receptors. The net effect on Ca2+ levels thus may resemble that caused by EA2 mutations in VDCCs.
Fig. 3.

Schematic overview of growth factor-induced and Ca2+-releasing signaling pathways in neuronal cells. Growth factor (GF) binding to receptors (RTK) may activate PI3K, PLCγ, and MAPK signaling cascades. Transient ERK activation via Ras will affect cytosolic targets that may extend to transmembrane proteins involved in Ca2+ or glutamate signaling. RTK endocytosis may lead to Rap1-mediated sustained activation of ERK and altered transcription. PTPRR targets are depicted in blue. Proteins mutated in SCA6, SCA12, and SCA14 patients are in reddish purple. Proteins that are down-regulated in SCA1 mouse models and connect to IP3 receptor-regulated Ca2+ stores are in orange. Ca2+-modulating proteins (CaBPs) and PTPRR, for which deficiency in mice led to ataxic behavior but not neurodegeneration, are in green
Many dominant ataxias (i.e., SCA1) are caused by an expanding polyglutamine sequence in the responsible protein [63, 64]. In a mouse model for SCA1, the metabotropic glutamate receptor and its auxiliary proteins that connect to IP3 receptor-regulated intracellular Ca2+ stores were all found to be down-regulated [71], providing yet another link to aberrant calcium handling in ataxias. Further support comes from the mouse models that are deficient in Ca2+ binding or Ca2+ modulatory proteins [58–61], which all display ataxic behavior without neurodegeneration. Since PTPRR-deficient mice combine a lack of apparent brain morphological abnormalities with mild ataxic symptoms and hyperactivity of ERK1/2 MAP kinases, further studies towards the underlying mechanism will have bearing for the field of human cerebellar ataxia-type locomotive disorders. This may yield novel candidate SCA genes for the many hereditary cerebellar ataxias that still await disease gene identification and may provide clues to symptomatic or neuroprotective strategies that can be applied in early stages of degenerative ataxias.
Electronic Supplementary Material
Below is the link to the electronic supplementary material
(DOC 42.5 kb)
Below is the image is a link to a high resolution version
{kind=link}
(JPG 5.44 MB)
{kind=link}
(JPG 8.54 MB)
{kind=link}
(JPG 7.16 MB)
{kind=link}
(JPG 9.47 MB)
{kind=link}
(JPG 4.69 MB)
{kind=link}
(JPG 8.65 MB)
Acknowledgments
We thank Bas Wanschers, Frank de Lange, Mietske Wijers, Huib Croes, Jan Kuiper, and Rinske van de Vorstenbosch for technical assistance and Frank Böhmer (Jena, Germany) for generously providing the pNRTIS-21 construct.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s12311-008-0088-y) contains supplementary material, which is available to authorized users.
References
- 1.Andersen JN et al (2004) A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J 18:8–30 [DOI] [PubMed]
- 2.Alonso A et al (2004) Protein tyrosine phosphatases in the human genome. Cell 117:699–711 [DOI] [PubMed]
- 3.Pulido R, Hooft van Huijsduijnen R (2008) Protein tyrosine phosphatases: dual-specificity phosphatases in health and disease. FEBS J 275:848–866 [DOI] [PubMed]
- 4.Hendriks WJAJ, Elson A, Harroch S, Stoker AW (2008) Protein tyrosine phosphatases: functional inferences from mouse models and human diseases. FEBS J 275:816–830 [DOI] [PubMed]
- 5.Tabernero L, Aricescu A, Jones E, Szedlacsek S (2008) Protein tyrosine phosphatases: structure–function relationships. FEBS J 275:867–882 [DOI] [PubMed]
- 6.den Hertog J, Östman A, Böhmer F-D (2008) Protein tyrosine phosphatases: regulatory mechanisms. FEBS J 275:831–847 [DOI] [PubMed]
- 7.Augustine KA et al (2000) Protein tyrosine phosphatase (PC12, BR7, SL) family: expression characterization in the adult human and mouse. Anat Rec 258:221–234 [DOI] [PubMed]
- 8.Watanabe Y et al (1998) Cloning of PCPTP1-Ce encoding protein tyrosine phosphatase from the rat cerebellum and its restricted expression in Purkinje cells. Mol Brain Res 58:83–94 [DOI] [PubMed]
- 9.Shiozuka K, Watanabe Y, Ikeda T, Hashimoto S, Kawashima H (1995) Cloning and expression of PCPTP1 encoding protein tyrosine phosphatase. Gene 162:279–284 [DOI] [PubMed]
- 10.Karim FD, Rubin GM (1999) PTP-ER, a novel tyrosine phosphatase, functions downstream of Ras1 to downregulate MAP kinase during Drosophila eye development. Mol Cell 3:741–750 [DOI] [PubMed]
- 11.Dilaver G et al (2007) Proteolytic processing of the receptor-type protein tyrosine phosphatase PTPBR7. FEBS J 274:96–108 [DOI] [PubMed]
- 12.Chirivi RGS et al (2004) Characterization of multiple transcripts and isoforms derived from the mouse protein tyrosine phosphatase gene Ptprr. Genes Cells 9:919–933 [DOI] [PubMed]
- 13.Hendriks W, Schepens J, Brugman C, Zeeuwen P, Wieringa B (1995) A novel receptor-type protein tyrosine phosphatase with a single catalytic domain is specifically expressed in mouse brain. Biochem J 305:499–504 [DOI] [PMC free article] [PubMed]
- 14.Ogata M, Sawada M, Fujino Y, Hamaoka T (1995) cDna cloning and characterization of a novel receptor-type protein tyrosine phosphatase expressed predominantly in the brain. J Biol Chem 270:2337–2343 [DOI] [PubMed]
- 15.van den Maagdenberg AMJM et al (1999) The mouse Ptprr gene encodes two protein tyrosine phosphatases, PTP-SL and PTPBR7, that display distinct patterns of expression during neuronal development. Eur J Neurosci 11:3832–3844 [DOI] [PubMed]
- 16.Sharma E, Lombroso PJ (1995) A neuronal protein tyrosine phosphatase induced by nerve growth factor. J Biol Chem 270:49–53 [DOI] [PubMed]
- 17.Noordman YE et al (2008) Multimerisation of receptor-type protein tyrosine phosphatases PTPBR7 and PTP-SL attenuates enzymatic activity. Biochim Biophys Acta 1783:275–286 [DOI] [PubMed]
- 18.Stoker A (2005) Methods for identifying extracellular ligands of RPTPs. Methods 35:80–89 [DOI] [PubMed]
- 19.Heldin CH (1995) Dimerization of cell surface receptors in signal transduction. Cell 80:213–223 [DOI] [PubMed]
- 20.Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225 [DOI] [PubMed]
- 21.Krens SF, Spaink HP, Snaar-Jagalska BE (2006) Functions of the MAPK family in vertebrate development. FEBS Lett 580:4984–4990 [DOI] [PubMed]
- 22.Pearson G et al (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22:153–183 [DOI] [PubMed]
- 23.Yao Z, Seger R (2004) The molecular mechanism of MAPK/ERK inactivation. Curr Genomics 5:1–9 [DOI]
- 24.Theodosiou A, Ashworth A (2002) MAP kinase phosphatases. Genome Biol 3:REVIEWS3009 [DOI] [PMC free article] [PubMed]
- 25.Keyse SM (2000) Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol 12:186–192 [DOI] [PubMed]
- 26.Vaudry D, Stork PJ, Lazarovici P, Eiden LE (2002) Signaling pathways for PC12 cell differentiation: making the right connections. Science 296:1648–1649 [DOI] [PubMed]
- 27.Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80:179–185 [DOI] [PubMed]
- 28.Farooq A, Zhou MM (2004) Structure and regulation of MAPK phosphatases. Cell Signal 16:769–779 [DOI] [PubMed]
- 29.Barr AJ, Knapp S (2006) MAPK-specific tyrosine phosphatases: new targets for drug discovery? Trends Pharmacol Sci 27:525–530 [DOI] [PubMed]
- 30.Pulido R, Zúñiga A, Ullrich A (1998) PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J 17:7337–7350 [DOI] [PMC free article] [PubMed]
- 31.Zúñiga A, Torres J, Ubeda J, Pulido R (1999) Interaction of mitogen-activated protein kinases with the kinase interaction motif of the tyrosine phosphatase PTP-SL provides substrate specificity and retains ERK2 in the cytoplasm. J Biol Chem 274:21900–21907 [DOI] [PubMed]
- 32.Muñoz JJ, Tárrega C, Blanco-Aparicio C, Pulido R (2003) Differential interaction of the tyrosine phosphatases PTP-SL, STEP and HePTP with the mitogen-activated protein kinases ERK1/2 and p38alpha is determined by a kinase specificity sequence and influenced by reducing agents. Biochem J 372:193–201 [DOI] [PMC free article] [PubMed]
- 33.Buschbeck M, Eickhoff J, Sommer MN, Ullrich A (2002) Phosphotyrosine-specific phosphatase PTP-SL regulates the ERK5 signaling pathway. J Biol Chem 33:29503–29509 [DOI] [PubMed]
- 34.Blanco-Aparicio C, Torres J, Pulido R (1999) A novel regulatory mechanism of MAP kinases activation and nuclear translocation mediated by PKA and the tyrosine phosphatase PTP-SL. J Cell Biol 147:1129–1136 [DOI] [PMC free article] [PubMed]
- 35.Paul S et al (2007) The striatal-enriched protein tyrosine phosphatase gates long-term potentiation and fear memory in the lateral amygdala. Biol Psychiatry 61:1049–1061 [DOI] [PMC free article] [PubMed]
- 36.Nika K et al (2004) Haematopoietic protein tyrosine phosphatase (HePTP) phosphorylation by cAMP-dependent protein kinase in T-cells: dynamics and subcellular location. Biochem J 378:335–342 [DOI] [PMC free article] [PubMed]
- 37.Valjent E et al (2005) Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci USA 102:491–496 [DOI] [PMC free article] [PubMed]
- 38.Saxena M, Williams S, Tasken K, Mustelin T (1999) Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase. Nat Cell Biol 1:305–311 [DOI] [PubMed]
- 39.Dilaver G et al (2003) Colocalisation of the protein tyrosine phosphatases PTP-SL and PTPBR7 with β4-adaptin in neuronal cells. Histochem Cell Biol 119:1–13 [DOI] [PubMed]
- 40.Noordman YE, Jansen PAM, Hendriks WJAJ (2006) Tyrosine-specific MAPK phosphatases and the control of ERK signaling in PC12 cells. J Mol Signal 1:4 [DOI] [PMC free article] [PubMed]
- 41.Korolchuk V, Banting G (2003) Kinases in clathrin-mediated endocytosis. Biochem Soc 31:857–860 [DOI] [PubMed]
- 42.Huang F, Jiang X, Sorkin A (2003) Tyrosine phosphorylation of the β2 subunit of clathrin adaptor complex AP-2 reveals the role of a dileucine motif in the epidermal growth factor receptor trafficking. J Biol Chem 278:43411–43417 [DOI] [PubMed]
- 43.Huynh H et al (2004) Control of vesicle fusion by a tyrosine phosphatase. Nat Cell Biol 6:831–839 [DOI] [PubMed]
- 44.Gonzalez-Gaitan (2003) Signal dispersal and transduction through the endocytic pathway. Nat Rev Mol Cell Biol 4:213–224 [DOI] [PubMed]
- 45.Haj FG, Verveer PJ, Squire A, Neel BG, Bastiaens PIH (2002) Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum. Science 295:1708–1711 [DOI] [PubMed]
- 46.Torii S, Kusakabe M, Yamamoto T, Maekawa M, Nishida E (2004) Sef is a spatial regulator for Ras/MAP kinase signaling. Dev Cell 7:33–44 [DOI] [PubMed]
- 47.Bektas A, Hughes JN, Warram JH, Krolewski AS, Doria A (2001) Type 2 diabetes locus on 12q15. Further mapping and mutation screening of two candidate genes. Diabetes 50:204–208 [DOI] [PubMed]
- 48.Nakamura F, Nakamura Y, Maki K, Sato Y, Mitani K (2005) Cloning and characterization of the novel chimeric gene TEL/PTPRR in acute myelogenous leukemia with inv(12)(p13q13). Cancer Res 65:6612–6621 [DOI] [PubMed]
- 49.Augustine KA et al (2000) Evidence that the protein tyrosine phosphatase (PC12, BR7, SL) gamma (−) isoform modulates chondrogenic patterning and growth. Int J Dev Biol 44:361–371 [PubMed]
- 50.Nagarajan UM, Bushey A, Boss JM (2002) Modulation of gene expression by the MHC class II transactivator. J Immunol 169:5078–5088 [DOI] [PubMed]
- 51.Otten LA et al (2006) Revisiting the specificity of the MHC class II transactivator CIITA in vivo. Eur J Immunol 36:1548–1558 [DOI] [PubMed]
- 52.Sherwin JR, Sharkey AM, Mihalyi A, Simsa P, Catalano RD, D’Hooghe TM (2008) Global gene analysis of late secretory phase, eutopic endometrium does not provide the basis for a minimally invasive test of endometriosis. Hum Reprod 23:1063–1068 [DOI] [PubMed]
- 53.Thalmeier A et al (2008) Gene expression profiling of post-mortem orbitofrontal cortex in violent suicide victims. Int J Neuropsychopharmacol 11:217–228 [DOI] [PubMed]
- 54.Dwivedi Y, Rizavi HS, Roberts RC, Conley RC, Tamminga CA, Pandey GN (2001) Reduced activation and expression of ERK1/2 MAP kinase in the post-mortem brain of depressed suicide subjects. J Neurochem 77:916–928 [DOI] [PubMed]
- 55.Chirivi RG, Noordman YE, van der Zee CE, Hendriks WJ (2007) Altered MAP kinase phosphorylation and impaired motor coordination in PTPRR deficient mice. J Neurochem 101:829–840 [DOI] [PubMed]
- 56.Colucci-D’Amato L, Perrone-Capano C, di Porzio U (2003) Chronic activation of ERK and neurodegenerative diseases. Bioessays 25:1085–1095 [DOI] [PubMed]
- 57.Pietrobon D (2005) Function and dysfunction of synaptic calcium channels: insights from mouse models. Curr Opin Neurobiol 15:257–265 [DOI] [PubMed]
- 58.Nishi M et al (2002) Motor discoordination in mutant mice lacking junctophilin type 3. Biochem Biophys Res Commun 292:318–324 [DOI] [PubMed]
- 59.Jiao Y et al (2005) Carbonic anhydrase-related protein VIII deficiency is associated with a distinctive lifelong gait disorder in waddles mice. Genetics 171:1239–1246 [DOI] [PMC free article] [PubMed]
- 60.Farre-Castany MA et al (2007) Differences in locomotor behavior revealed in mice deficient for the calcium-binding proteins parvalbumin, calbindin D-28 k or both. Behav Brain Res 178:250–261 [DOI] [PubMed]
- 61.Cheron G, Gall D, Servais L, Dan B, Maex R, Schiffmann SN (2004) Inactivation of calcium-binding protein genes induces 160 Hz oscillations in the cerebellar cortex of alert mice. J Neurosci 24:434–441 [DOI] [PMC free article] [PubMed]
- 62.Hirota J, Ando H, Hamada K, Mikoshiba K (2003) Carbonic anhydrase-related protein is a novel binding protein for inositol 1,4,5-trisphosphate receptor type 1. Biochem J 372:435–441 [DOI] [PMC free article] [PubMed]
- 63.Dueñas AM, Goold R, Giunti P (2006) Molecular pathogenesis of spinocerebellar ataxias. Brain 129:1357–1370 [DOI] [PubMed]
- 64.van de Warrenburg BP, Sinke RJ, Kremer B (2005) Recent advances in hereditary spinocerebellar ataxias. J Neuropathol Exp Neurol 64:171–180 [DOI] [PubMed]
- 65.Verbeek DS, Knight MA, Harmison GG, Fischbeck KH, Howell BW (2005) Protein kinase C gamma mutations in spinocerebellar ataxia 14 increase kinase activity and alter membrane targeting. Brain 128:436–442 [DOI] [PubMed]
- 66.van de Warrenburg BP et al (2003) Identification of a novel SCA14 mutation in a Dutch autosomal dominant cerebellar ataxia family. Neurology 61:1760–1765 [DOI] [PubMed]
- 67.Nakanishi S (2005) Synaptic mechanisms of the cerebellar cortical network. Trends Neurosci 28:93–100 [DOI] [PubMed]
- 68.Thomas GM, Huganir RL (2004) MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5:173–183 [DOI] [PubMed]
- 69.Krab LC, Goorden SM, Elgersma Y (2008) Oncogenes on my mind: ERK and MTOR signaling in cognitive diseases. Trends Genet 24:498–510 [DOI] [PubMed]
- 70.Gee CE, Mansuy IM (2005) Protein phosphatases and their potential implications in neuroprotective processes. Cell Mol Life Sci 62:1120–1130 [DOI] [PubMed]
- 71.Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY, Orr HT (2004) Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet 13:2535–2543 [DOI] [PubMed]
- 72.Lein ES et al (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material
(DOC 42.5 kb)
Below is the image is a link to a high resolution version
(JPG 5.44 MB)
(JPG 8.54 MB)
(JPG 7.16 MB)
(JPG 9.47 MB)
(JPG 4.69 MB)
(JPG 8.65 MB)