

Abstract
Heme oxygenase (HO)-1 is a protective antioxidant enzyme that prevents cardiomyocyte apoptosis, for instance, during progressive cardiomyopathy. Here we identify a fundamental aspect of the HO-1 protection mechanism by demonstrating that HO-1 activity in mouse heart stimulates the bigenomic mitochondrial biogenesis program via induction of NF-E2–related factor (Nrf)2 gene expression and nuclear translocation. Nrf2 upregulates the mRNA, protein, and activity for HO-1 as well as mRNA and protein for nuclear respiratory factor (NRF)-1. Mechanistically, in cardiomyocytes, endogenous carbon monoxide (CO) generated by HO-1 overexpression stimulates superoxide dismutase-2 upregulation and mitochondrial H2O2 production, which activates Akt/PKB. Akt deactivates glycogen synthase kinase-3β, which permits Nrf2 nuclear translocation and occupancy of 4 antioxidant response elements (AREs) in the NRF-1 promoter. The ensuing accumulation of nuclear NRF-1 protein leads to gene activation for mitochondrial biogenesis, which opposes apoptosis and necrosis caused by the cardiotoxic anthracycline chemotherapeutic agent, doxorubicin. In cardiac cells, Akt silencing exacerbates doxorubicin-induced apoptosis, and in vivo CO rescues wild-type but not Akt1−/− mice from doxorubicin cardiomyopathy. These findings consign HO-1/CO signaling through Nrf2 and Akt to the myocardial transcriptional program for mitochondrial biogenesis, provide a rationale for targeted mitochondrial CO therapy, and connect cardiac mitochondrial volume expansion with the inducible network of xenobiotic and antioxidant cellular defenses.
Keywords: mitochondria, heme oxygenase, carbon monoxide, NF-E2–related factor 2, nuclear respiratory factor-1, superoxide dismutase, hydrogen peroxide
Progressive cardiomyopathy attends diverse stresses ranging from aberrant calcium signaling to inflammation to direct cardiomyocyte toxicity.1 The hallmarks of cardiac decompensation, such as apoptosis and myocyte depletion, often imply mitochondrial pathogenesis, particularly after inciting agents such as doxorubicin,2 which inhibits the expression of nuclear and mitochondrial encoded genes involved in mitochondrial biogenesis. This deficit, however, can be averted with low-dose carbon monoxide (CO),2 also a product of endogenous heme oxygenase-1 (HO-1, Hmox1), which catalytically degrades potentially toxic heme to biliverdin.3-5
Among the arsenal of antioxidant enzymes, HO-1 is strategically induced by its heme substrate, but also indirectly by endotoxin, hypoxia, and heavy metals against which it protects.5 HO-1/CO signaling operates in part and in similarity to the nitric oxide (NO) synthases, through heme–protein binding, eg, soluble guanylate cyclase (GC),6,7 and like NO,8,9 CO activates mitochondrial biogenesis. In the heart, however, CO acts independently of endothelial NO synthase on gene transactivation for nuclear respiratory factor (NRF)-1 and -2, as well as the PGC-1α coactivator and mitochondrial transcription factor A (Tfam), necessary for mitochondrial biogenesis.2,10
CO binding to the reduced a3 heme of cytochrome c oxidase also enhances mitochondrial hydrogen peroxide (H2O2) production, which despite its potential toxicity, serves signal transduction11 and contributes to retrograde activation of mitochondrial biogenesis.2,10 CO activates the prosurvival phosphatidylinositol-3 (PI3)-kinase/Akt pathway, and Akt phosphorylates NRF-1, an integral transcription factor for mitochondrial biogenesis,12-14 before it enters the nucleus,15 but how CO activates the transcriptional programming is not known.
HO-1 activation by PI3-kinase/Akt as well as transcriptional regulation of Hmox-1 by NF-E2-related factor (Nrf)216,17 puts HO-1 in a position to exert oxidation-reduction (redox) control over cell processes. A basic leucine zipper transcription factor, Nrf2 is activated by electrophiles and reactive oxygen species, and enhances cell-protective gene expression by interacting with ARE motifs usually located 5′ to the transcription start site (TSS).18-21 Nrf2 is sequestered in the cytoplasm by Kelch-like ECH-associated protein (Keap)1, which represses its transcriptional role,22 but oxidation of cysteine-rich Keap1 frees Nrf2 to enter the nucleus to participate in ARE transactivation.18,23-25 Many electrophiles also enhance Nrf2 gene transcription, and the Nrf2/Keap1 complex is regulated by specific protein kinases24,26 such as glycogen synthase kinase (GSK)3β, which inhibits Nrf2 nuclear translocation.27,28
Here we analyzed the NRF-1 promoter for ARE motifs and examined the hypothesis that endogenous CO stimulates mitochondrial H2O2 production and induces NRF-1 responsive genes via Nrf2 occupancy of NRF-1 promoter sites to increase NRF-1 expression and activity. By implication, HO-1 would serve mitochondrial biogenesis through transcriptional integration with the major antioxidant enzyme defenses. Our findings delineate a new role for HO-1/CO in the coordination of mitochondrial biogenesis as well as a previously unsuspected Nrf2-based redox component for the regulation of mitochondrial mass in the heart.
Materials and Methods
Materials
Antibodies to citrate synthase, GSK3β, Nrf2, Bach1, Bid, Bclxl, and PolRMT were obtained from Santa Cruz and to caspase 3, Akt, Ser473-phospho-Akt, phospho-GSK3β, and phospho-Bad from Cell Signaling. Phospho Ser/Thr/Tyr antibody was from AnaSpec and antitubulin and porin from Sigma. NRF-1 and Tfam antibodies were developed and characterized in laboratory.2,10,15 All secondary and fluorescent antibodies were from Invitrogen. My-Akt expression vector was from Origene; HO-1 and mCAT vectors were developed and characterized in house.2,10 Small interfering (si)RNA oligonucleotides were from Ambion.
Animals
Studies of mice were approved by the Institutional Animal Care and Use Committee: male 10 to 12 week-old C57BL/6 or Akt1−/− (Jackson) or transgenic mice that express green fluorescent protein (GFP) in mitochondria (mtGFP-tg, gift from Hiroshi Shitara and Hiromichi Yonekawa of Tokyo Metropolitan Institute of Medical Science).29 Akt1−/−/mtGFP-tg hybrids were generated by cross-breeding. Cardiomyopathy was induced by 1 injection of doxorubicin (Dox, Sigma-Aldrich; 15 mg/kg IP).2 At appropriate times, mice were anesthetized, aortas transected and the hearts removed and snap-frozen. For histology and TUNEL staining, hearts were perfusion-fixed with 0.9% NaCl followed by 10% formalin. Serial changes in cardiac function were assessed noninvasively in sedated mice by Doppler ultrasound (Visual Sonics Vevo 770).
Cell Culture
Murine atrial cardiomyocyte HL-1 cells, a generous gift from Dr William C. Claycomb (LSU Medical Center), were cultured in Claycomb medium with 10% FBS, 100 μmol/L norepinephrine, and 4 mmol/L l-glutamine in gelatin/fibronectin-coated flasks or plates. Rat neonatal cardiomyoblasts (H9c2 cells) were cultured as before.2,10 Cells were cultured at 37°C with 5% CO2 and 95% air. Cells were transfected with empty or siRNA vectors using the FuGene HD transfection reagent (Roche) and efficiencies of 65–80% transfection and gene suppression achieved. Dox and dichloromethane (DCM) (CO-releasing molecule) were used as reported.10
Immunomethods
Western blots were performed on cell and heart lysates and nuclear extracts after protein separation by SDS-PAGE. After transfer, primary and secondary antibodies were applied and the signals developed with ECL. Bands were quantified on digitized images from the mid-dynamic range, and data expressed relative to tubulin or β-actin.30 Immunoprecipitation was performed as described.10,15 Cells or tissues were immunolabeled with primary antibodies to HO-1 (1:200), Nrf2 (1:100) or NRF-1 (1:200) and immunofluorescence quantified.2,10
MtDNA Copy Number and Respiratory Proteins
mtDNA was determined by SYBR green quantitative PCR (qPCR). Fluorescence intensities were recorded and analyzed on an ABI Prism 7000 sequence-detector system (Applied Biosystems). MtDNA-encoded cytochrome c oxidase subunit I and NADH dehydrogenase subunit I were quantified by RT-PCR and normalized to nuclear-encoded 18S and/or GAPDH.2,10
Real-Time PCR
qPCR was performed using an ABI PRISM 7000 Sequence Detection System with TaqMan gene expression and premix assays (Applied Biosystems). The primer pairs are shown in Table I in the online data supplement, available at http://circres.ahajournals.org. 18S RNA served as an endogenous control.2,10 Quantification of gene expression was determined using the comparative threshold cycle CT and RQ method.
Bioinformatics
Promoter analysis was performed with consensus sequences for transcription factor binding located with DNASIS (Hitachi Software; Alameda, Calif) and confirmed with MatInspector (Genomatix Software; München, Germany). Putative ARE canonical binding sites of 92% to 100% homology were identified (Ensembl Gene ID ENSMUSG00000058440). NRF-1 promoter regions were analyzed for ARE consensus motifs for the mouse, rat, and human.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed as described.2,31 Heart samples were evaluated for Nrf2 enrichment by quantitative real-time PCR (qPCR) in 20 μL mixtures of SYBR Green master mix and 0.1 μmol/L primer. Nrf2 enrichment was evaluated using primers for regions upstream of the mouse NRF-1 genes (sequences available on request). The relative efficiency of each PCR primer was determined using input DNA and adjusted accordingly. DNA in each ChIP sample was normalized to the corresponding input chromatin (ΔCt) and enrichment defined as change in Ct in treated versus untreated control samples (ΔΔCt), relative to IgG. Exponential ΔΔCt values were converted to linear values (2-ΔΔCt) for graphics.
Statistical Analysis
Grouped data are expressed as the means±SEM for n=4 to 6 replicates. Statistical significance was tested with the unpaired Student’s t test or two-way analysis of variance using commercial software. Differences were considered significant at P<0.05.
Results
Nuclear Nrf2
One hour of CO breathing reinforced HO-1 gene expression and enzyme activity in the mouse heart in vivo (Figure 1A).2 Because Hmox-1 promoter contains multiple ARE motifs, we examined cardiac Nrf2 expression and activation by CO and found increased nuclear Nrf2 localization at 12 hours by confocal microscopy (Figure 1B), confirmed by Western analysis of nuclear protein (Figure 1C). This CO dose produced a 3-fold increase in cardiac Nrf2 mRNA levels by 6 hours (Figure 1D) and protected against Dox cardiotoxicity as shown by stability of ventricular ejection fraction using serial ultrasound measurements (Figure 1E).
Figure 1.

Nuclear Nrf2 accumulation and cardiac protection by CO. CO breathing in mice induces HO-1 mRNA expression and enzyme activity in Wt heart after 24 hours (A). Cardiac Nrf2 nuclear localization by confocal microscopy after CO is enhanced at 12 hours (B), confirmed by nuclear protein Western blot (C). Cardiac Nrf2 mRNA increases 3-fold within 6 hours and remains elevated for 12 hours after CO (D). *P<0.05 compared with preexposure control (n=4). CO given at days 0 and 7 after Dox (E) prevents the decline in right and left ventricular ejection fraction at 14 days by Doppler ultrasound.
The Nrf2 response to CO was examined in neonatal rat cardiomyoblasts and beating mouse cardiomyocytes (HL-1 cells) with the same results. At low concentrations of DCM/CO (50 μmol/L), nuclear Nrf2 content increased by more than fivefold (data for HL-1 cells shown in Figure 2A, top). Also in HL-1 cells, DCM/CO disrupted the Nrf2 association with Keap1, an effect not observed in cells transfected with mitochondrial-targeted catalase (mCAT) and indicating an oxidant mechanism (Figure 2A, bottom).
Figure 2.

Nrf2 expression in mouse cardiomyocytes. HL-1 cells exposed to DCM/CO (50 μmol/L) increase nuclear Nrf2 content by >5-fold along with cytoplasmic Keap1 dissociation that is blocked by mitochondrial-targeted catalase (mCAT) (A). B, DCM/CO increases Nrf2, HO-1, and SOD2 mRNA levels by 3- to 4-fold, whereas Nrf2 silencing abrogates the mRNA responses and scrambled siRNA has no effect. *P<0.05 compared with control (n=4). C, DCM/CO increases HO-1 enzyme activity by >10-fold, also abrogated by Nrf2 silencing but not by scrambled siRNA. *P<0.05 (n=4). D, Top, HL-1 cells treated with DCM/CO (50 μmol/L) or overexpressing HO-1 increase Akt phosphorylation and nuclear Nrf2 accumulation. mCAT inhibits HO-1-induced Akt activation and Nrf2 nuclear translocation. D, Bottom, PI3-kinase inhibition shows Akt primarily regulates Nrf2 translocation, not exclusion of the Bach1 nuclear repressor. DCM/CO produces greater Akt activity but less nuclear Bach1, whereas Akt inhibition does not affect nuclear Bach1 but reduces Nrf2 translocation. Bach1 siRNA increases nuclear Nrf2 with stable Akt activity.
DCM/CO increased HO-1 mRNA levels 3-fold and mitochondrial superoxide dismutase (SOD)2 mRNA fourfold, whereas Nrf2 gene silencing abrogated the ability of DCM/CO to increase mRNA levels for both enzymes (Figure 2B). After DCM/CO, HO-1 enzyme activity increased tenfold; once again this was abrogated by Nrf2 silencing (Figure 2C).
Nrf2 nuclear translocation involves not only Keap1 oxidation but Nrf2 phosphorylation. Because CO induces mitochondrial H2O2 generation and Akt activation,10 we compared the CO effects on Akt with Nrf2 nuclear translocation (Figure 2D, top). Treatment of HL-1 cells with DCM/CO or overexpression of active HO-1 provoked strong increases in Akt phosphorylation and Nrf2 nuclear translocation. When mitochondrial H2O2 release is attenuated by cotransfection with mCAT, Akt activation (68%) and Nrf2 nuclear translocation (60%) are attenuated (Figure 2D). To determine whether Akt regulates Nrf2 translocation directly or excludes the Bach1 repressor from the nucleus,32 HL-1 cells were treated with LY29 to inhibit PI3-kinase (Figure 2D, bottom). Here DCM/CO causes half as much nuclear Bach1 accumulation, whereas Akt inhibition has no effect on nuclear Bach1 but Nrf2 translocation is reduced by ≈60%. Bach1 siRNA increased nuclear Nrf2 content by 38% with no change in Akt activity (Figure 2D), implying that CO modulates Nrf2 nuclear translocation via Akt independently of Bach1.
HO-1 Increases Nrf2 Occupancy of NRF-1 Promoter ARE Motifs
CO induces NRF-1 mRNA and protein10; thus to investigate a similar effect of HO-1 on NRF-1 expression, HL-1 cells were transfected with HO-1. By confocal microscopy, active HO-1 compared with empty vector increased NRF-1 expression and nuclear protein accumulation (Figure 3A). Simultaneous cotransfection with HO-1 and active Akt containing an amino-terminal myristoylation signal (myr-Akt) yielded a further 3-fold increase in nuclear NRF-1 protein (Figure 3A and 3B). These increases in nuclear NRF-1 are accompanied by 8- to 10-fold increases in the downstream expression of Tfam (Figure 3B), as well as in mtDNA copy number (Figure 3C).
Figure 3.
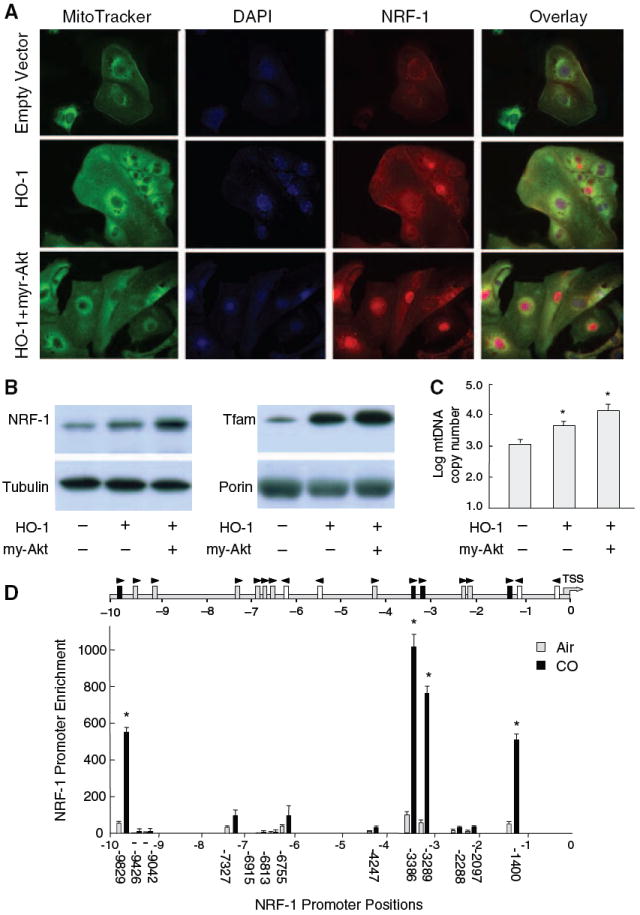
HO-1 increases nuclear NRF-1 and Nrf2 occupancy of NRF-1 ARE promoter motifs. Active HO-1 increases NRF-1 expression and nuclear protein by confocal microscopy in HL-1 cells (A). Cotransfection with HO-1 and active Akt containing an amino-terminal myristoylation signal (myr-Akt) produces a further 3-fold increase in nuclear NRF-1 protein (A and B) accompanied by 8- to 10-fold increases in Tfam expression (B, right) as well as mtDNA copy number (C). A search of the NRF-1 promoter for Nrf2-binding sites 10 kb upstream of the TSS identified multiple ARE motifs investigated by ChIP (D). Arrows indicate orientation of each ARE motif relative to the ARE consensus. Gray boxes are motifs meeting the consensus sequence and black boxes show active motifs. The graph shows DNA enrichment by Nrf2 ChIP at each NRF-1 ARE motif. Values along the abscissa indicate positions of DNA regions amplified by qPCR. Black bars depict significant DNA enrichment after DCM/CO at positions −1400, −3289, 3386, and −9829. Nrf2 binding increased 11 to 13-fold at each site. Values represent the means±SEM of independent experiments performed in triplicate (*P<0.05).
Because HO-1 expression is regulated by Nrf2, we checked for Nrf2-binding sites in the NRF-1 promoter, core ARE motifs conforming to RTGAYnnnGC or its reverse, 10 kb upstream of the TSS.18 Seventeen consensus AREs were identified in the mouse and 13 on the plus-strand close to the NRF-1 TSS were investigated by ChIP analysis for Nrf2 interactions (Figure 3D) by analyzing immunoprecipitated DNA for enrichment by qPCR using primers flanking the ARE motifs. We found baseline Nrf2 occupancy to be minimal in control hearts, but after CO, found significant DNA enrichment at positions −1400, −3289, 3386 and −9829 bp (Figure 3D). The remaining ARE motifs did not respond to CO. These data demonstrate that after CO, Nrf2 regulates NRF-1 promoter activity at multiple sites.
Nrf2/HO-1 Induction of NRF-1 Regulates Mitochondrial Biogenesis and Opposes Cardiomyocyte Apoptosis
Cell protection assessed in rat H9c2 cardiomyoblasts with or without HO-1 overexpression before and after Dox is shown in Figure 4A. To connect nuclear Nrf2 to NRF-1 expression and the HO-1 protective function, we compared Nrf2 and NRF-1 expression before and after Dox as well as without or with HO-1 transfection. Nrf2-dependent NRF-1 responses were evaluated with 2 nuclear-encoded genes upregulated by NRF-1 with which Dox interferes, Tfam and PolRMT, that regulate mitochondrial mRNA levels (Figure 4A). HO-1 transfection doubles mitochondrial Tfam and PolRMT protein and prevents Dox-mediated loss of these proteins (Figure 4A). The HO-1 protective effect is congruent with mitochondrial biogenesis at 24 hours as reflected in greater mitochondrial MTT reduction, increased citrate synthase (CS) expression (Figure 4B), and enhanced mRNA levels for mitochondrial-encoded OXPHOS proteins ND1 and COX subunit I (Figure 4C).
Figure 4.
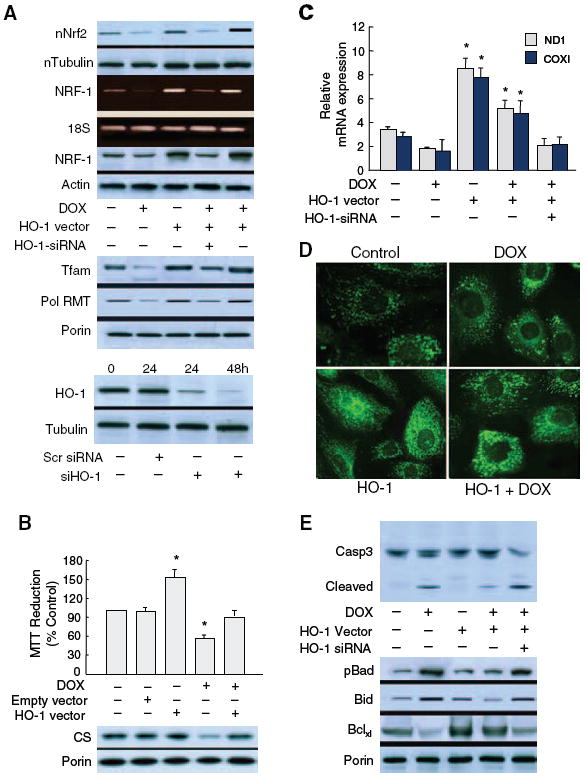
Nrf2/HO-1 induction of NRF-1 activates mitochondrial biogenesis and prevents doxorubicin-induced cardiomyocyte apoptosis. Nuclear Nrf2 and NRF-1 expression were evaluated in H9c2 cardiomyoblasts without and with HO-1 overexpression or silencing compared with readouts of 2 nuclear-encoded downstream genes, Tfam and PolRMT, that regulate mitochondrial mRNA levels and with which Dox interferes (A, top). HO-1 overexpression doubles mitochondrial Tfam and PolRMT protein and attenuates Dox interference (A, middle). A, Bottom, demonstrates effective HO-1 silencing at 24 and 48 hours. Mitochondrial functional protection by HO-1 is reflected by enhanced MTT reduction and citrate synthase (CS) expression (B) and mRNA for mitochondrial-encoded subunits COX I and ND1 (C). Dox-induced changes in Mitotracker green show organelle modification from a fine reticulum to vesicles and aggregates within 24 hours (D, top). In contrast, mitochondrial density in cells overexpressing HO-1 is greater than control, and changes in mitochondrial structure occur sporadically after Dox (D, bottom). E, Dox-induced caspase 3 cleavage; HO-1 overexpression limits and HO-1 silencing exacerbates caspase 3 cleavage. Dox also decreases mitochondrial Bcl-XL and increases mitochondrial phosphorylated Bad (Ser-112) protein, which is counteracted by HO-1 overexpression. HO-1 silencing reverses the effects of HO-1 expression on mitochondrial pBad, Bid, and Bcl-XL.
Dox toxicity is characterized by loss of mitochondrial number and hence Mitotracker signal intensity within 24 hours (Figure 4D). Mitochondrial structure is modified from its normal fine reticulum to vesicles and aggregates (Figure 4D, top images). In contrast, mitochondrial density in HO-1 overexpressing cardiomyocytes is greater than control, and the same Dox dose induces only sporadic mitochondrial structural changes (Figure 4D, bottom images). Dox causes cardiomyocyte apoptosis, confirmed by caspase-3 cleavage, whereas HO-1 overexpression prevents and HO-1 silencing exacerbates caspase-3 cleavage (Figure 4E, top gel). HO-1 overexpression promotes mitochondrial antiapoptotic protein expression, especially mitochondrial Bcl-XL (2.2-fold), compared with control cells (Figure 4E, bottom). HO-1 overexpression also decreases DOX-induced mitochondrial phosphorylated Bad (Ser-112) by 6-fold and Bid by 3-fold, whereas siHO-1 increases total mitochondrial pBad and Bid (Figure 4E). Thus HO-1 facilitates Bcl-XL and opposes Bid and Bad expression and/or mitochondrial translocation, which contributes to an antiapoptotic mitochondrial phenotype.
HO-1 Regulation of Cardiomyocyte Survival Through Akt
Cardiac protection by HO-1/CO in conjunction with Akt stimulates NRF-1 nuclear translocation to activate mitochondrial biogenesis. A role for Akt in HO-1/CO mitochondrial protection against Dox was tested in Akt1-deficient mice crossed with reporter mice expressing mitochondrial-targeted GFP. Cardiac pathology in the GFP reporter mice along with hematoxylin/eosin and TUNEL stains is shown in Figure 5. Akt−/−/GFP mice exposed to Dox show massive mitochondrial damage and cell death compared with wild-type (Wt) mice (Figure 5A) and interval CO treatment, which fully rescues mitochondrial damage and prevents necrosis in the Wt, is not effective in Akt−/− mice (Figure 5A). In addition, CO ameliorates Dox-induced apoptosis in Wt but not in Akt−/− mice (Figure 5B). Dox also introduces a spike in myocardial fibrosis, which CO prevents in Wt but not Akt−/− mice (not shown). Quantification of the mitochondrial reporter signal is shown in supplemental Figure I.
Figure 5.
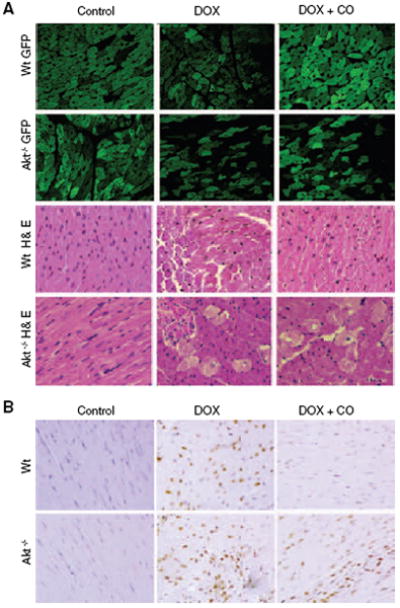
HO-1/CO, mitochondrial biogenesis, and cardiomyocyte survival. Mitochondrial damage and cell loss after Dox is more severe in Akt1−/− mice expressing mitochondrial GFP (Akt1−/−/GFP) than in Wt mice (A), and CO protects mitochondria and rescues cells in Wt but not Akt1−/− mice. Left ventricular myocardial sections stained with hematoxylin/eosin show interstitial edema, focal necrosis, and myofibrillar degeneration 7 days after Dox. Damage is more extensive in Akt−/− than Wt mice and ameliorated by CO in Wt but not Akt1−/− mice. B, TUNEL staining of heart sections from Wt and Akt1−/− mice before and 7 days after Dox. Large numbers of TUNEL-stained nuclei are noted after Dox, and CO protection is limited to the Wt. Supplemental Figure I shows the quantification of cardiac mitochondrial fluorescence in the GFP mice.
To further understand how Akt protects mitochondria, we examined GSK3β, which is negatively regulated by Akt phosphorylation of Ser-9 in the pseudosubstrate domain.33 A computer-assisted analysis of Nrf2 and Bach1 protein revealed no Akt but several canonical GSK3β motifs. Immunoprecipitation of Bach1 and Nrf2 and immunoblotting with anti–phospho-serine/threonine indicated that CO decreases phosphorylation of both Nrf2 and the suppressor, Bach1 (Figure 6A). In comparison, CO increases Akt and GSK3β protein phosphorylation in Wt but not in Akt1−/− mice (Figure 6B). The parsimonious reason is that Akt−/− mice are unable to phosphorylate and inactivate GSK3β during HO-1 activation. Moreover, nuclear translocation of NRF-1 induced by CO in Wt mice is abrogated in Akt−/− mice (Figure 6C). Collectively, these findings indicate that Akt critically offsets the GSK3β effect on nuclear exclusion of Nrf2 as well as facilitates NRF-1 nucleoprotein accumulation.
Figure 6.
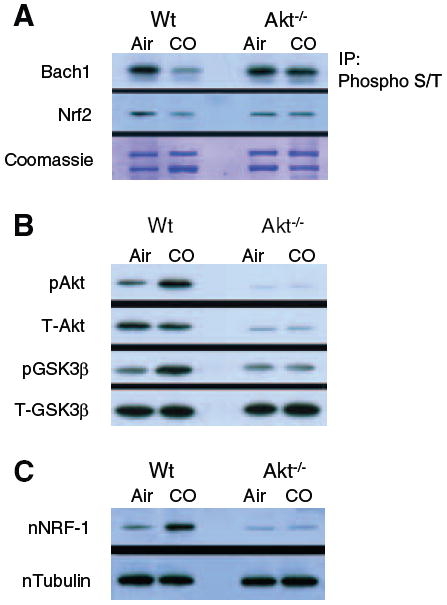
CO and the Akt-GSK3β relationship. In immunoprecipitation studies (A), Nrf2 serine/threonine phosphorylation, as well as Bach1 phosphorylation, decreases after CO (Nrf2 and Bach1 motifs for GSK3β are inhibited by Akt). In contrast to Wt mice, neither Akt nor GSK3β is phosphorylated in Akt1−/− mice after CO (B). Also, nuclear translocation of NRF-1 induced by CO in Wt mice is absent in Akt1−/− mice (C).
Discussion
The demonstration that HO-1 participates in cardiac mitochondrial biogenesis through the Nrf2 transcription factor, which confers resistance against electrophiles and xenobiotics, discloses several novel aspects of cell survival regulation with broad implications. Although we had previously implicated HO-1 in mitochondrial biogenesis through Akt1 activation,10 it was not known that the pathway involves Nrf2 expression, downstream GSK3β blockade, and Nrf2 nuclear translocation leading to Nrf2-dependent activation of NRF-1 transcription. The findings thus establish HO-1/CO, by sequentially activating these 2 transcription factors, as a remarkable component of a prosurvival program of mitochondrial biogenesis linked to the cellular antioxidant defenses.7,34
The main antioxidant function of HO-1 has been thought to derive from the catalytic conversion of pro-oxidant heme to biliverdin, as well as from the induction of iron sequestration proteins.4 A role for endogenous CO has been more difficult to elucidate because CO inhibits respiration, generates reactive oxygen species, and causes apoptosis, yet recapitulates certain protective effects of HO-1,35 and as a pro-oxidant, induces antioxidant enzymes, including HO-1.16,17,36 Although formation of the ferrous carbonyl may prevent indiscriminate redox cycling, loss of heme reactions interferes with cellular functions, and mitochondrial CO binding to the reduced a3 heme of cytochrome c oxidase intensifies the Complex III H2O2 leak rate.37
The pro-oxidant chemistry of CO and regulation of HO-1 by Nrf2,16,21 a critical transcription factor for protection against electrophiles, inflammation, and chemical toxicity,38 offered the possibility that Nrf2 might connect mitochondrial biogenesis with the antioxidant and xenobiotic defenses through a CO-based redox mechanism. Nrf2 protects against toxicants and carcinogens in part by activating phase II detoxifying genes,38-40 and it mediates CO induction of the rate-limiting enzyme for glutathione biosynthesis,17 but the findings here are the first evidence of its role in mitochondrial biogenesis.
To explore nuclear regulation of mitochondrial genes by HO-1, we used CO to boost cardiac HO-1 activity, which also increased Nrf2 mRNA and Nrf2 nuclear protein levels. This HO-1 response, together with SOD2 induction was confirmed by gene silencing to depend in cardiomyocytes on Nrf2 activation by CO. Furthermore, cells transfected with mitochondrial-targeted catalase indicate that Nrf2/Akt activation is contingent on mitochondrial H2O2 production. Classic oxidant activation of Akt entails reciprocal phosphatase inactivation, eg, PTEN,41 which operates after CO binding to cytochrome c oxidase.10 Akt also deactivates GSK3β, which is thereby unable to prevent Nrf2 nuclear translocation.27
In cardiomyocytes, HO-1 overexpression causes NRF-1, a key transcription factor required for mitochondrial biogenesis, to undergo nuclear translocation, and leads to mitochondrial importation of Tfam, the mtDNA transcription factor, and a log increase in mtDNA copy number. HO-1/CO regulates NRF-1 nuclear translocation through phosphorylation by Akt15,10,42 with concurrent NRF-1 gene transcription under Nrf2 control.
The mouse NRF-1 proximal 5′UTR contains multiple ARE motifs, and after CO, 4 plus-strand sites are occupied by Nrf2. The Nrf2 affinities conform to the consensus ARE core motif; however, the short sequence is not the sole determinant of transcription factor binding. Nucleotides flanking the ARE core, as well as motif multiplicity and interactions with small Maf proteins,43 contribute to differential recognition. Although extended ARE flanking sequences are vital for Nrf2 recognition,44-46 the extended motif requirements are not well defined. Although Nrf2 does bind individual motifs, ARE multiplicity enhances Nrf2 promoter-binding, and in silico analysis of the proximal 10kb of the NRF-1 5′UTR also reveals multiple ARE motifs in the rat and human.
HO-1/CO-mediated Nrf2 expression/translocation coupled to mitochondrial biogenesis through Nrf2-Akt activation of HO-1 and NRF-1, opposes the toxicity of the chemotherapeutic, doxorubicin, in vitro and in vivo. Despite its antineoplastic efficacy, 30% of patients who receive doxorubicin develop dilated cardiomyopathy47 which has been linked to oxidative mitochondrial damage, impaired NO signaling, mitochondrial biogenesis and respiratory gene suppression, and widespread intrinsic apoptosis.2,48,49 Mechanistically, Akt1−/− mice are especially susceptible to cardiac necrosis and apoptosis, but unlike Wt mice, cannot be rescued by CO, which no longer activates NRF-1 and Tfam for mitochondrial biogenesis.10 This finding means that HO-1, in conjunction with other regulators of myocardial mitochondrial capacity, is fundamentally tied to the support of energy metabolism and the prevention of cell death.
Figure 7 summarizes how HO-1/CO acts on Nrf2-dependent gene expression to drive mitochondrial biogenesis. Endogenous CO enhances mitochondrial H2O2 production, which activates Akt, alleviates cytoplasmic Nrf2 inhibition, and promotes Nrf2 gene expression. Akt phosphorylates and deactivates GSK3β, releasing Nrf2 from inhibition. Nuclear Nrf2 occupies AREs in the HO-1, SOD2, and NRF-1 gene promoters, and facilitates the transcription and translation of all 3 proteins, whereas Akt phosphorylation of NRF-1 facilitates its translocation and activation of downstream genes required for mitochondrial biogenesis.15 SOD2 scavenges mitochondrial superoxide and in its regulatory capacity10,11,50 matches mitochondrial H2O2 to the rate of heme turnover by HO-1. And notably, functional NRF-1 binding sites are found in the promoters of more than 100 genes involved in regulating the mitochondrial transcriptosome, OXPHOS, heme biosynthesis, protein importation, and mitochondrial protein synthesis.13,14
Figure 7.
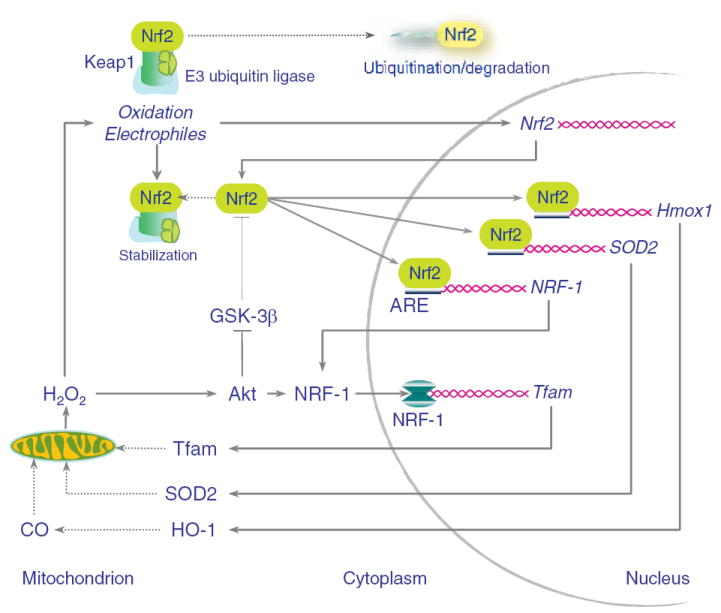
Working diagram of the HO-1/CO/Nrf2 pathway involved in the regulation of mitochondrial biogenesis. Nrf2 constitutively docks with Keap1, which sequesters it in the cytoplasm, allowing it to undergo ubiquitination and degradation (upper left). HO-1/CO increases mitochondrial H2O2 (lower left), which stabilizes Keap1 and results in Nrf2 gene expression and nuclear translocation. Nuclear Nrf2 undergoes binding to AREs in the Hmox1, SOD2, and NRF-1 promoters (far right). HO-1 and SOD2 amplify the mitochondrial H2O2 signal, whereas NRF-1 entry into the nucleus drives transcription for Tfam, PolRMT, and other genes of mitochondrial biogenesis.
In conclusion, HO-1/CO promotes NRF-1 gene expression via Nrf2, which couples mitochondrial biogenesis to the expression of phase II detoxifying and antioxidant enzyme defenses through the cis-acting ARE. In this role, HO-1/CO critically protects against doxorubicin-mediated mitochondrial damage and cardiomyocyte death, and by implication, pulse CO should have a place in targeted mitochondrial therapy. The most important new idea is that mitochondrial biogenesis as a prosurvival factor is integrally connected by redox control to the cellular defenses against xenobiotic toxicity and oxidative stress.
Supplementary Material
Acknowledgments
We thank Craig Marshall, Susan Fields, John Patterson and Lynn Tatro for technical assistance.
Sources of Funding This work was funded by National Heart, Lung, and Blood Institute/NIH grant R01 HL096979 (to C.A.P.) and the GEMI Fund (to H.B.S.).
Footnotes
Disclosures None.
References
- 1.Ashrafian H, Watkins H. Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications cardiomyopathies: therapeutics based on molecular phenotype. J Am Coll Cardiol. 2007;49:1251–1264. doi: 10.1016/j.jacc.2006.10.073. [DOI] [PubMed] [Google Scholar]
- 2.Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest. 2007;117:3730–3741. doi: 10.1172/JCI32967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 4.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 5.Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation. 2008;117:231–241. doi: 10.1161/CIRCULATIONAHA.107.698316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furchgott RF, Jothianandan D. Endothelium-dependent and -independent vasodilation involving cyclic GMP: relaxation induced by nitric oxide, carbon monoxide and light. Blood Vessels. 1991;28:52–61. doi: 10.1159/000158843. [DOI] [PubMed] [Google Scholar]
- 7.Maines MD. The heme oxygenase system: update 2005. Antioxid Redox Signal. 2005;7:1761–1766. doi: 10.1089/ars.2005.7.1761. [DOI] [PubMed] [Google Scholar]
- 8.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valano A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 9.Nisoli E, Carruba MO. Nitric oxide and mitochondrial biogenesis. J Cell Sci. 2006;119:2855–2862. doi: 10.1242/jcs.03062. [DOI] [PubMed] [Google Scholar]
- 10.Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. A new activating role for CO in cardiac mitochondrial biogenesis. J Cell Sci. 2007;120:299–308. doi: 10.1242/jcs.03318. [DOI] [PubMed] [Google Scholar]
- 11.Connor KM, Subbaram S, Regan KJ, Nelson KK, Mazurkiewicz JE, Barthlomew PJ, Aplin AE, Tai YT, Aquirre-Ghiso J, Flores SC, Melendez JA. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J Biol Chem. 2005;280:16916–16924. doi: 10.1074/jbc.M410690200. [DOI] [PubMed] [Google Scholar]
- 12.Scarpulla RC. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene. 2002;286:81–89. doi: 10.1016/s0378-1119(01)00809-5. [DOI] [PubMed] [Google Scholar]
- 13.Scarpulla RC. Transcriptional paradigms in Mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 14.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 15.Piantadosi CA, Suliman HB. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J Biol Chem. 2006;281:324–333. doi: 10.1074/jbc.M508805200. [DOI] [PubMed] [Google Scholar]
- 16.Lee BS, Heo J, Kim YM, Shim SM, Pae HO, Kim YM, Chung HT. Carbon monoxide mediates heme oxygenase 1 induction via Nrf2 activation in hepatoma cells. Biochem Biophys Res Commun. 2006;343:965–972. doi: 10.1016/j.bbrc.2006.03.058. [DOI] [PubMed] [Google Scholar]
- 17.Li MH, Jang JH, Na HK, Cha YN, Surh YJ. Carbon monoxide produced by heme oxygenase-1 in response to nitrosative stress induces expression of glutamate-cysteine ligase in PC12 cells via activation of phosphatidylinositol 3-kinase and Nrf2 signaling. J Biol Chem. 2007;282:28577–28586. doi: 10.1074/jbc.M701916200. [DOI] [PubMed] [Google Scholar]
- 18.Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci U S A. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci U S A. 2000;97:12475–12480. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006;25:3605–3617. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol. 2007;36:166–174. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 22.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabavashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 25.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–44682. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- 27.Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–14851. doi: 10.1074/jbc.M513737200. [DOI] [PubMed] [Google Scholar]
- 28.Jain AK, Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282:16502–16510. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]
- 29.Shitara H, Kaneda H, Sato A, Iwasaki K, Hayashi J, Taya C, Yonekawa H. Non-invasive visualization of sperm mitochondria behavior in transgenic mice with introduced green fluorescent protein (GFP) FEBS Lett. 2001;500:7–11. doi: 10.1016/s0014-5793(01)02574-1. [DOI] [PubMed] [Google Scholar]
- 30.Suliman HB, Welty-Wolf KE, Carraway MS, Schwartz DA, Hollingsworth JW, Piantadosi CA. Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated E. coli. FASEB J. 2005;19:1531–1533. doi: 10.1096/fj.04-3500fje. [DOI] [PubMed] [Google Scholar]
- 31.Piantadosi CA, Suliman HB. Transcriptional regulation of SDHa flavoprotein by nuclear respiratory factor-1 prevents pseudo-hypoxia in aerobic cardiac cells. J Biol Chem. 2008;283:10967–10977. doi: 10.1074/jbc.M709741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohira M, Seki N, Nagase T, Ishikawa K, Nomura N, Ohara O. Characterization of a human homolog (BACH1) of the mouse Bach1 gene encoding a BTB-basic leucine zipper transcription factor and its mapping to chromosome 21q22.1. Genomics. 1998;47:300–306. doi: 10.1006/geno.1997.5080. [DOI] [PubMed] [Google Scholar]
- 33.Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol. 2005;17:150–157. doi: 10.1016/j.ceb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 34.Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, Figdor CG. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol Rev. 2003;55:551–571. doi: 10.1124/pr.55.3.5. [DOI] [PubMed] [Google Scholar]
- 35.Piantadosi CA. Biological chemistry of carbon monoxide. Antioxid Redox Signal. 2002;4:259–270. doi: 10.1089/152308602753666316. [DOI] [PubMed] [Google Scholar]
- 36.Cronje FJ, Carraway MS, Freiberger JJ, Suliman HB, Piantadosi CA. Carbon monoxide actuates O(2)-limited heme degradation in the rat brain. Free Radic Biol Med. 2004;37:1802–1812. doi: 10.1016/j.freeradbiomed.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 37.Piantadosi CA, Carraway MS, Suliman HB. Carbon monoxide, oxidative stress, and mitochondrial permeability pore transition. Free Radic Biol Med. 2006;40:1332–1339. doi: 10.1016/j.freeradbiomed.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 38.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem. 1990;265:14648–14653. [PubMed] [Google Scholar]
- 40.Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol. 2003;23:8786–8794. doi: 10.1128/MCB.23.23.8786-8794.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22:5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Shan P, Alam J, Fu XY, Lee PJ. Carbon monoxide differentially modulates STAT1 and STAT3 and inhibits apoptosis via a phosphatidylinositol 3-kinase/Akt and p38 kinase-dependent STAT3 pathway during anoxia-reoxygenation injury. J Biol Chem. 2005;280:8714–8721. doi: 10.1074/jbc.M408092200. [DOI] [PubMed] [Google Scholar]
- 43.Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol. 2005;25:8044–8051. doi: 10.1128/MCB.25.18.8044-8051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- 45.Erickson AM, Nevarea Z, Gipp JJ, Mulcahy RT. Identification of a variant antioxidant response element in the promoter of the human glutamate-cysteine ligase modifier subunit gene. Revision of the ARE consensus sequence. J Biol Chem. 2002;277:30730–30737. doi: 10.1074/jbc.M205225200. [DOI] [PubMed] [Google Scholar]
- 46.Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J. 2003;374:337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97:2869–2879. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- 48.Zhou S, Starkov A, Froberg MK, Leino RL, Wallace KB. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001;61:771–777. [PubMed] [Google Scholar]
- 49.Cole MP, Chaiswing L, Oberley TD, Edelmann SE, Piascik MT, Lin SM, Kiningham KK, St Clair DK. The protective roles of nitric oxide and superoxide dismutase in adriamycin-induced cardiotoxicity. Cardiovasc Res. 2006;69:186–197. doi: 10.1016/j.cardiores.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 50.Zhang HJ, Zhao W, Venkataraman S, Robbins ME, Buettner GR, Kregel KC, Oberley LW. Activation of matrix metalloproteinase-2 by overexpression of manganese superoxide dismutase in human breast cancer MCF-7 cells involves reactive oxygen species. J Biol Chem. 2002;277:20919–20926. doi: 10.1074/jbc.M109801200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.