

Summary
Human IgM+IgD+CD27+ B cells have mutated Ig genes and harbor a splenic marginal zone phenotype. The group of R. Küppers has studied the expression of the enzyme activation-induced cytidine deaminase (AID) in human spleen samples by immunocytochemistry and failed to detect a significant AID-expressing subset in the marginal zone region. The consequences on the possible origin of these cells are discussed.
Keywords: Animals; Antigens, CD27; immunology; B-Lymphocyte Subsets; cytology; immunology; Cell Lineage; immunology; Cytidine Deaminase; Cytosine Deaminase; immunology; metabolism; Humans; Immunoglobulins; genetics; Mutation; Spleen; cytology; immunology
Introduction: “Memory” B cell subsets in humans
R. Küppers and colleagues made the seminal observation that blood B cells in humans were very different from mice’s one because they contained 30–40% of memory B cells displaying the CD27 marker at their surface and a mutated Ig receptor [1]. Among these memory B cells, about half of them are switched cells and surprisingly, the other half are IgM-positive cells, distributed in two subsets, IgM+IgD+ and IgM-only (i.e. IgD-negative) B cells. In their discussion these authors proposed, among other hypothesis, that these CD27+ IgM-positive cells could in fact be the equivalent of the mutated IgM+ cells made during the formation of the pre-immune repertoire in sheep.
At first and without any clear explanation for the discrepancy with Küppers’s initial observation, a clear CD27+ IgM-only subset has not been observed by two different groups in either blood or spleen, its distinct presence being mostly linked with pathological situations (immunodeficiency or auto-immunity)[2,3]. In AID-deficient patients for example, in which there is a molecular block within germinal centers (GC) that prevents hypermutation and isotype switch [4], one can detect a clear IgM-only CD27+ B cell subset in blood, suggesting that these cells could be GC precursors of switched cells that would now accumulate and thus appear in the circulation [2].
Asking the question of the origin of IgM+IgD+CD27+ B cells, it was observed that hyper-IgM patients who lack either a functional CD40 or CD40L protein and therefore have no GC and switched cells (similarly to the equivalent experimental K.O. mice) always display IgM+IgD+CD27+ B cells with a mutated Ig receptor in blood [2,5]. The frequency of these cells was often lower than in normal individuals of the same age, while the frequency of somatic mutations was close to normal. Now comes the issue of the interpretation of these results. Where do these mutated IgM+IgD+ B cells come from in hyper-IgM patients? Are they made in cryptic germinal centers, which have not been described but may still exist in these patients, or do they belong to a separate B cell diversification pathway? The first explanation would mean that these cryptic structures would allow the generation and the mutation of the IgM+IgD+ cells specifically but not the formation of any IgM-only and switched cells. Not a totally satisfactory explanation.
No marginal zone (MZ) B cell lineage in humans?
It has been shown thereafter that blood and splenic IgM+IgD+CD27+ B cells, which represent 10 to 30% of total B cells in normal individuals, display a marginal zone phenotype (IgMhigh, IgDlow, CD21+, CD23−, CD1+) and are involved in T-independent responses [2,3]. The marginal zone B cell sub-population was originally described as a separate lineage in rodents, bearing the CD21highCD23−IgM+IgDlow/− CD1+ phenotype, but, in contrast to humans, carrying a germline non-mutated Ig receptor [6,7]. Differentiation of this subpopulation occurs in mice as a cell lineage choice at the transitional B cell stage, resulting in either a follicular or a marginal zone phenotype. This binary cell fate decision is, like many others in development, mediated by the Notch pathway, Notch2 acting via RBP-J, with its function being counteracted by MINT in this specific case [8,9]. Many other factors, involved in particular in cell migration or in BCR signal transduction, have profound impact on the follicular versus marginal zone B cell development [7,10].
Apart from very young children, all marginal zone B cells in humans (more than 95%) carry a mutated Ig receptor, and there is clearly no individualized unmutated subset among them (the low frequency of germline receptors corresponding to what is expected from a heterogeneous distribution of mutation frequencies). In this issue of Eur. J. Immunol., Willenbrock et al. study the distribution of AID-positive cells in the human spleen [11], since the expression of AID is an absolute requisite for the occurrence of Ig gene hypermutation [4,12]. They found the frequency of AID-positive cells to be very low in the splenic marginal zone, and thus favor the proposition that most marginal zone B cells are in fact derived in humans from a T-dependent response taking place in germinal centers.
One should emphasize that the obvious conclusion of this proposition is that, as opposed to rodents, a distinct marginal zone B cell sub-population does not exist as such in humans. Taking into account the clear anatomical differences between rodent and human spleen, it would suggest that, instead of being a separate B cell lineage, marginal zone B cells arise in humans as a branching point during germinal center differentiation, after some accumulation of somatic mutation but before the onset of isotype switch (fig. 1). We obviously do not favor this interpretation and think, along with others, that, although the splenic marginal zone is a complex lymphoid niche, humans have evolved like rodents a separate sub-population of B cells which reside in this splenic compartment, are pre-activated and respond rapidly to blood-born TI antigens [7,13,14].
Fig. 1. What origin for marginal zone B cells in humans?
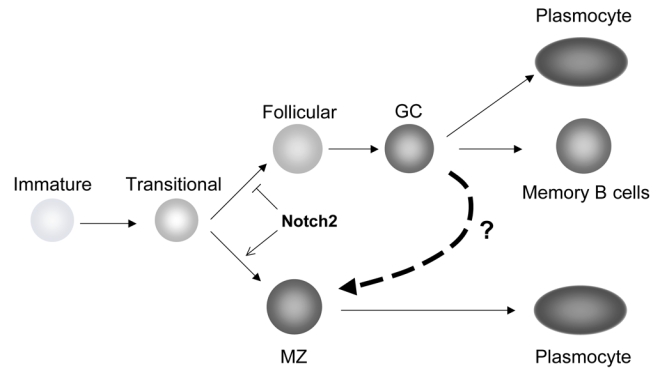
Transitional B cells in the mouse have been described as undergoing a binary cell fate, depending upon Notch2 activation, to become either follicular B cells (than can later be activated in germinal centers, leading again to either memory B cells or plasmocytes) or marginal zone B cells, that can be engaged in rapid plasmocytic differentiation in the response to blood-born antigens [10]. Marginal zone B cells in humans have been proposed to undergo diversification of their Ig genes outside of an immune response [2]. An alternate proposition (marked by the dotted arrow) is that splenic marginal zone B cells in humans are a subset arising during germinal center differentiation, after the onset of somatic mutation and before isotype switch [11].
Other sites for diversification of IgM+IgD+CD27+ B cells?
Hypermutation being the hallmark of a T-dependent germinal center reaction, the confusion arise from the fact that marginal zone B cells in humans carry a mutated Ig receptor and that vaccination against polysaccharidic antigens raise mutated antibodies [15]. However evolution has taught us that hypermutation and gene conversion can be used in several species in a T-independent and external antigen-free mode in order to generate a B cell pre-immune repertoire [16,17]. Moreover these molecular mechanisms that introduce diversity in the three VH/VL complementarity-determining regions (CDRs) can provide these animals with a diversified antibody repertoire with all range of affinities against proteic and polysaccharidic determinants present at the surface of pathogens.
Strikingly in humans this circulating and splenic marginal zone B cell population is already well expanded and mutated at a stage at which T-independent responses against encapsulated bacteria cannot take place (before 2 years), which led us to suggest that these cells could in fact mutate their IgM receptor “à la sheep” while they are generated, before responding to external antigens [2]. The place where this pre-diversification would take place was obviously left open at that time, and if the splenic marginal zone appears as an obvious candidate, marginal zone-like regions in secondary lymphoid organs or gut-associated lymphoid tissues are among the many other possible sites of diversification. Results on the selection of specific clones belonging to this circulating and splenic marginal zone compartment after a T-independent immunization showed by CDR3 spectratyping that the B cells clones involved, that carried an already mutated V gene, seemed to undergo a second round of diversification during the few days of the response, a process which would add one to three mutations on the selected B cell clone [2]. What the results of the paper of Willenbrock et al. [11] tell us is that the short round of hypermutation taking place in marginal zone B cells after a T-independent challenge may only concern a very small number of cells, or that they would emigrate rapidly after stimulation, or even possibly, that the intra-clonal diversification observed represent independent activation of clonally related cells and not reactivation of the hypermutation process during the T-independent immune response.
How much AID does it take to perform hypermutation?
A last point of discussion concerns the sensitivity of AID detection assay. The IgM+IgD+CD27+ shows an Ig gene mutation frequency significantly lower than switched B cells (by a factor of 2, approximately [2]). Moreover, the actual accumulation of mutation could be a much slower process than the one occurring in a few weeks in germinal centers. The obvious question then is: what is the amount of AID necessary to induce somatic mutation? Most AID proteins are present in the cytoplasm of mutating B cells, as a result of a constant nucleo-cytoplasmic shuttling in which the nuclear export dominates [18]. Only a few AID molecules have the opportunity to assemble in a nuclear mutasome at any time point and reach their Ig gene target. It is moreover totally unknown whether the quantitative requirements for AID to perform hypermutation versus isotype switch differ, and to what specific processes the widely different intensities in AID staining of germinal center B cells correspond. It seems therefore somewhat premature to conclude that marginal zone B cells are devoid of any AID activity. Accordingly, semi-quantitative PCR of RNA extracted from various B cell subsets show a 20-fold lower expression in splenic IgM+IgD+CD27+ B cells compared to switched cells (after a double cell sorting), whereas splenic naive B cells were negative, thus arguing against a trivial contamination in the purification procedure (our unpublished data). As AID is also required to induce class switching, these data raise only the question of the sensitivity of the in situ detection, but cannot lead to the formal conclusion that hypermutation is occurring in marginal zone B cells.
In conclusion, if one assumes that, in spite of the lack of detection of AID expression in the splenic marginal zone reported here by Willenbrock et al., a separate marginal zone B cell subset with mutated Ig genes do exist in humans, the question is now to unravel how and where this population is constantly renewed, pre-activated and pre-diversified. Self and commensal bacterial antigens may be necessary to develop these cells but also CD40L triggering by accessory cells such as NKT that can recognize self-lipids presented by CD1 on MZ B cells [19,20]. In such a scheme pre-activation could not occur correctly in hyper-IgM patients lacking the CD40-CD40L interactions, thus explaining the lower development of these cells. At the effector stage, when marginal zone B cells respond to TI antigens in the spleen, switch and differentiate into plasma cells here again they would need non cognate T-cell mediated CD40L signaling (explaining why no switching occurs in T-independent responses in hyper-IgM patients), together with strong Ig cross-linking and TLR signaling, that would drive them rapidly out of the MZ microenvironment.
Abbreviations
- TI
T-independant
References
- 1.Klein U, Rajewsky K, Küppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998;188:1679–1689. doi: 10.1084/jem.188.9.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weller S, Braun MC, Tan BK, Rosenwald A, Cordier C, Conley ME, Plebani A, Kumararatne DS, Bonnet D, Tournilhac O, Tchernia G, Steiniger B, Staudt LM, Casanova JL, Reynaud CA, Weill J-C. Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a pre-diversified immunoglobulin repertoire. Blood. 2004;104:3647–3654. doi: 10.1182/blood-2004-01-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kruetzmann S, Rosado MM, Weber H, Germing U, Tournilhac O, Peter HH, Berner R, Peters A, Boehm T, Plebani A, Quinti I, Carsetti R. Human IgM memory B cells controlling Streptococcus pneumoniae infections are generated in the spleen. J Exp Med. 2003;197:939–945. doi: 10.1084/jem.20022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 5.Weller S, Faili A, Garcia C, Braun MC, Le Deist F, de Saint Basile G, Hermine O, Fischer A, Reynaud CA, Weill JC. CD40-CD40L independent Ig gene hypermutation suggests a second B cell diversification pathway in humans. Proc Natl Acad Sci USA. 2001;98:1166–1170. doi: 10.1073/pnas.98.3.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumararatne DS, Bazin H, MacLennan IC. Marginal zones: the major B cell compartment of rat spleens. Eur J Immunol. 1981;11:858–864. doi: 10.1002/eji.1830111103. [DOI] [PubMed] [Google Scholar]
- 7.Martin F, Kearney JF. Marginal-zone B cells. Nat Rev Immunol. 2002;2:323– 335. doi: 10.1038/nri799. [DOI] [PubMed] [Google Scholar]
- 8.Saito T, Chiba S, Ichikawa M, Kunisato A, Asai T, Shimizu K, Yamaguchi T, Yamamoto G, Seo S, Kumano K, Nakagami-Yamaguchi E, Hamada Y, Aizawa S, Hirai H. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity. 2003;18:675–685. doi: 10.1016/s1074-7613(03)00111-0. [DOI] [PubMed] [Google Scholar]
- 9.Kuroda K, Han H, Tani S, Tanigaki K, Tun T, Furukawa T, Taniguchi Y, Kurooka H, Hamada Y, Toyokuni S, Honjo T. Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity. 2003;18:301–312. doi: 10.1016/s1074-7613(03)00029-3. [DOI] [PubMed] [Google Scholar]
- 10.Pillai S, Cariappa A, Moran ST. Marginal zone B cells. Annu Rev Immunol. 2005;23:161–196. doi: 10.1146/annurev.immunol.23.021704.115728. [DOI] [PubMed] [Google Scholar]
- 11.Willenbrock K, Jungnickel B, Hansmann ML, Küppers R. Human splenic marginal zone B cells lack expression of activation-induced cytidine deaminase (AID) Eur J Immunol. 2005 doi: 10.1002/eji.200535134. this issue. [DOI] [PubMed] [Google Scholar]
- 12.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 13.Carsetti R, Rosado MM, Wardemann H. Peripheral development of B cells in mouse and man. Immunol Rev. 2004;197:179–191. doi: 10.1111/j.0105-2896.2004.0109.x. [DOI] [PubMed] [Google Scholar]
- 14.Spencer J, Perry ME, Dunn-Walters DK. Human marginal-zone B cells. Immunol Today. 1998;19:421–426. doi: 10.1016/s0167-5699(98)01308-5. [DOI] [PubMed] [Google Scholar]
- 15.Lucas AH, Reason DC. Polysaccharide vaccines as probes of antibody repertoires in man. Immunol Rev. 1999;171:89–104. doi: 10.1111/j.1600-065x.1999.tb01343.x. [DOI] [PubMed] [Google Scholar]
- 16.Reynaud CA, Garcia C, Hein WR, Weill JC. Hypermutation generating the sheep immunoglobulin repertoire is an antigen-independent process. Cell. 1995;80:115–125. doi: 10.1016/0092-8674(95)90456-5. [DOI] [PubMed] [Google Scholar]
- 17.Weill JC, Weller S, Reynaud CA. A bird’s eye view on human B cells. Semin Immunol. 2004;16:277–281. doi: 10.1016/j.smim.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Ito S, Nagaoka H, Shinkura R, Begum N, Muramatsu M, Nakata M, Honjo T. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc Natl Acad Sci USA. 2004;101:1975–1980. doi: 10.1073/pnas.0307335101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, Zhou D, Saint-Mezard P, Wang V, Gao Y, Yin N, Hoebe K, Schneewind O, Walker D, Beutler B, Teyton L, Savage PB, Bendelac A. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- 20.De Libero G, Moran AP, Gober HJ, Rossy E, Shamshiev A, Chelnokova O, Mazorra Z, Vendetti S, Sacchi A, Prendergast MM, Sansano S, Tonevitsky A, Landmann R, Mori L. Bacterial infections promote T cell recognition of self-glycolipids. Immunity. 2005;22:763–772. doi: 10.1016/j.immuni.2005.04.013. [DOI] [PubMed] [Google Scholar]