

Abstract
Congenital generalized hypertrichosis terminalis (CGHT) is a rare condition characterized by universal excessive growth of pigmented terminal hairs and often accompanied with gingival hyperplasia. In the present study, we describe three Han Chinese families with autosomal-dominant CGHT and a sporadic case with extreme CGHT and gingival hyperplasia. We first did a genome-wide linkage scan in a large four-generation family. Our parametric multipoint linkage analysis revealed a genetic locus for CGHT on chromosome 17q24.2-q24.3. Further two-point linkage and haplotyping with microsatellite markers from the same chromosome region confirmed the genetic mapping and showed in all the families a microdeletion within the critical region that was present in all affected individuals but not in unaffected family members. We then carried out copy-number analysis with the Affymetrix Genome-Wide Human SNP Array 6.0 and detected genomic microdeletions of different sizes and with different breakpoints in the three families. We validated these microdeletions by real-time quantitative PCR and confirmed their perfect cosegregation with the disease phenotype in the three families. In the sporadic case, however, we found a de novo microduplication. Two-color interphase FISH analysis demonstrated that the duplication was inverted. These copy-number variations (CNVs) shared a common genomic region in which CNV is not reported in the public database and was not detected in our 434 unrelated Han Chinese normal controls. Thus, pathogenic copy-number mutations on 17q24.2-q24.3 are responsible for CGHT with or without gingival hyperplasia. Our work identifies CGHT as a genomic disorder.
Main Text
Congenital generalized hypertrichosis (CGH), a condition characterized by excessive hair growth all over the body as compared to the normal of the same age, sex, and race, has attracted a great attention from the scientific community and the general public since the Middle Ages.1–3 It was considered an example of atavism and at one time even thought to be the missing ape-human link required to prove Darwin's theory.3,4 It is now believed that most people with CGH have an unknown genetic defect. CGH represents a group of phenotypically and genetically heterogeneous conditions.1 Nine different genetic syndromes include CGH as a major phenotype.1 The first genetic locus for human hypertrichosis was identified by traditional linkage analysis in a large Mexican family with X-linked CGH (MIM 307150).5 The Ambras type hypertrichosis universalis congenita (MIM 145701) has been shown to be associated with complex rearrangements of chromosome 8.1,6 Congenital generalized hypertrichosis terminalis (CGHT) with gingival hyperplasia (MIM 135400) is a distinct entity with characteristic features including universal overgrowth of fully pigmented terminal hairs, gingival hypertrophy, and a coarse facial appearance.7 Julia Pastrana, known as the world's most famous bearded lady in 1850–1860s, might be the first case reported in medical literature.2,8,9 This rare condition may be inherited as an autosomal-dominant trait.2,9,10 In an Irish family with CGHT, coarse face, and normal gingivae, an autosomal-dominant inheritance has also been observed.11 However, the underlying genetic defects for CGHT with or without gingival hyperplasia have not been discovered.
Genomic disorders refer to the human genetic diseases caused by submicroscopic microdeletion or microduplication of a genomic region that often includes dosage-sensitive gene(s).12 All disease-causing copy-number variations (CNVs), whether they are inherited or de novo, can be collectively classified as copy-number mutation. They may alter gene dosage, interrupt a gene, or exert long-range positional effects on expression pattern of the genes outside the CNV region, thereby producing the highly varied phenotypes seen in genomic disorders.13,14 With the application of modern high-resolution microarray-based technologies, numerous new genomic disorders have been uncovered.14,15
We ascertained three Han Chinese families (SY, GD, and BJ) with autosomal-dominant CGHT and coarse face (Figures 1A–1D and 1F). Families SY, GD, and BJ have ten, nine, and three affected individuals in four, four, and two generations, respectively (Figure 1F). Sixteen affected individuals of the three families were available for physical examination. Universal overgrowth of the terminal hairs and coarse face were the common phenotypic features among the affected individuals of these families (Figures 1A–1D). Most affected individuals showed excessive growth of terminal hairs all over the body as compared to the normal hair pattern and growth in Han Chinese of the same age and sex. Hair overgrowth on the back and limbs was particularly remarkable (Figures 1A and 1C). Affected females in family SY presented with less marked hypertrichosis. All affected individuals showed bushy and dark eyebrows, a broad and flat nose (in affected adults) or a bulbous soft nose (in affected children), large ears with thick, long, and hairy lobes, and thickened lips (Figures 1B and 1D). Approximately half of the affected individuals had a relatively large head (Figure 1C). With the exception of the widely spaced teeth observed in the affected adult individuals of family GD, no affected individual had obvious dental abnormalities. All affected individuals showed normal gingival growth and mental development.
Figure 1.
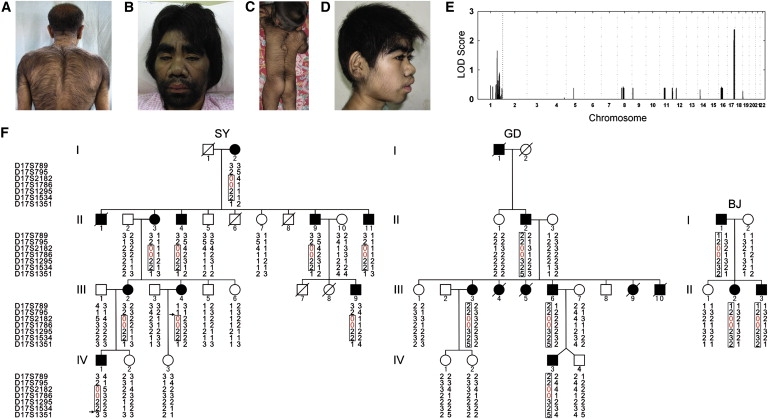
Phenotypes and Genetic Mapping of Congenital Generalized Hypertrichosis Terminalis on Chromosome 17q24.2-q24.3
(A) A 60-year-old male (II-4 of family SY) showing excessive hair growth on his back and shoulders.
(B) A 40-year-old bearded female (III-3 of family GD) showing a coarse face with bushy and dark eyebrows, a broad and flat nose, and thickened lips.
(C) A 3-year-old boy (IV-3 of family GD) showing excessive hair growth on his back and limbs and a relative large head.
(D) A 13-year-old bearded girl (II-2 of family BJ) showing a coarse face with bushy and dark eyebrows, long eyelashes, a bulbous soft nose, large ears with thick and hairy lobes, and thickened lips.
(E) Genome-wide parametric multipoint linkage analysis in family SY yielded a maximum LOD score of 2.37 for SNPs from chromosome 17q24.2-q24.3.
(F) Haplotype analysis of the three families (SY, GD, and BJ) with autosomal-dominant CGHT and coarse face. Recombinations in two affected individuals of SY (III-4 and IV-1), indicated by arrows, define a critical interval between markers D17S795 and D17S1351. The linked haplotype is boxed and the null alleles are indicated in red.
We collected blood samples from participating family members after obtaining informed consent and approval of the Peking Union Medical College Institutional Review Board. Genomic DNA was extracted by standard methods from 19 (9 affected, 6 unaffected, and 4 spouse), 11 (4 affected, 5 unaffected, and 2 spouse) and 5 (3 affected, 1 unaffected, and 1 spouse) members of families SY, GD, and BJ, respectively. DNA samples from the previously described Irish family with autosomal-dominant CGHT and coarse face were also included in linkage analysis.11
To identify candidate loci for CGHT, we first carried out a genome-wide linkage scan in the four-generation family SY (Figure 1F). DNA samples were genotyped on the Affymetrix GeneChip Human Mapping 50K Array Hind 240 containing 57,244 SNPs. Array experiments were carried out according to the protocol of the manufacturer. The Affymetrix GeneChip Operating Software (GCOS) was used in image processing. Genotypes were called with the Affymetrix GeneChip Genotyping Analysis Software (GTYPE 4.0). Parametric multipoint linkage analysis was performed with the dChipLinkage software under the assumptions of autosomal-dominant inheritance with 99% penetrance, a disease allele frequency of 0.1%, and equal SNP allele frequency (50%). Mendelian errors and unlikely genotypes were deleted prior to linkage analysis. Our multipoint linkage analysis produced a maximum logarithm of the odds (LOD) score of 2.37 with markers on chromosome 17q24.2-q24.3 (Figure 1E). To confirm the genome-wide linkage scan and define the critical region, we then typed in the family seven microsatellite markers (D17S789, D17S795, D17S2182, D17S1786, D17S1295, D17S1534, and D17S1351) from the same chromosome region and performed two-point linkage analysis. Two-point LOD scores were calculated with the MLINK program of the LINKAGE package. The parameters used in linkage analysis were autosomal-dominant inheritance, complete penetrance, a mutation rate of zero, equal male-female recombination rate, equal microsatellite-allele frequency, and a disease-allele frequency of 0.01%. PCR primers used for genotyping are given in Table S1 available online. We obtained a maximum LOD score of 3.91 at θ = 0 for two markers (D17S2182 and D17S1786) (Table S4), confirming the genetic linkage. Consistent with the genome scan, haplotype analysis in two recombinants (III-4 and IV-1 of family SY, Figure 1F) placed the critical region to an interval between markers D17S795 and D17S1351, representing a genomic extent of 4 Mb containing nine RefSeq genes. By typing the same microsatellite marker set, we detected haplotype sharing among affected members in families GD and BJ (Figure 1F). Notably, all affected individuals in the three Chinese CGHT families showed a null allele for markers D17S2182 and D17S1786 on the disease haplotypes, suggesting the possibility of a recurrent genomic microdeletion (Figure 1F). Our haplotype analysis with this microsatellite marker set excluded genetic linkage to 17q24.2-q24.3 in the Irish family. Further genome-wide linkage scan in this family yielded a maximum LOD score of 1.77 with SNP markers on a chromosome 6 region (data not shown), pointing to the presence of genetic heterogeneity in autosomal-dominant CGHT with coarse face.
To confirm whether the CGHT phenotype is associated with a recurrent microdeletion on 17q24.2-q24.3, we performed CNV analysis with the Affymetrix Genome-Wide Human SNP Array 6.0 containing more than 906,600 SNPs and more than 946,000 copy-number probes. Genomic DNA samples were genotyped with the SNP Array 6.0 in accordance with the manufacturer's protocols. The Affymetrix Genotyping Console 3.0 software was used for genotype calling, genotyping quality control, and CNV identification. Copy-number-state calls were determined with the Canary algorithm embedded in the Affymetrix Genotyping Console 3.0 package. We analyzed one affected individual per family and detected genomic microdeletions of different sizes and with different breakpoints in all the three familial cases (Figure 2A). To validate these nonrecurrent microdeletions by an alternative method, we also performed multiple qPCR assays to determine the relative copy number (RCN) of the target genomic regions. We designed 11 qPCR assays by using the Primer Express v2.0 software (Applied Biosystems). Information about the primer sequences and amplicon position is given in Table S2. The qPCR was carried out as described previously.16 The quantification of the target sequences was normalized to an assay from chromosome 21, and the RCN was determined on the basis of the comparative ΔΔCT method with a normal control DNA as the calibrator.16 The experiments were repeated three times. A ∼0.5-fold RCN and a ∼1.5-fold RCN were used for deletion and duplication, respectively. We detected an RCN of 0.5 for the corresponding qPCR targets in all analyzed individuals and thus provided independent confirmation of all the three microdeletions (Figure 2B).
Figure 2.
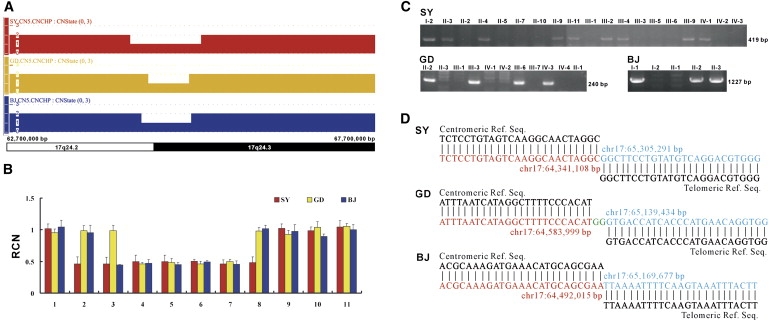
Identification of Inherited Microdeletions on Chromosome 17q24.2-q24.3 in Three CGHT Families
(A) Copy-number state of a 5 Mb genomic region on chromosome 17q24.2-q24.3 in three affected individuals, one per family.
(B) Real-time quantitative PCR (qPCR) assays of 11 amplicons validating the microdeletions. The deletion is seen as a ∼0.5-fold normalized relative copy number (RCN). Error bars represent SD.
(C) Amplification of deletion junctions showing segregation of the microdeletions with the phenotypes in families SY, GD, and BJ. The sizes of long-range PCR fragments are shown.
(D) Sequence analysis of the deletion junctions. The colored sequences represent the deletion junctions. The precise positions of the proximal and distal breakpoints are indicated in red and blue, respectively.
To amplify the deletion junctions, we designed long-range PCR primers from the sequences flanking the breakpoints on the basis of the SNP Array 6.0 genomic coordinates. Information about the primer sequences is given in Table S3. With use of the long-range PCR assays, we detected the corresponding microdeletions in all affected individuals of the three study families, but not in the unaffected family members (Figure 2C). Sequence analysis of the PCR fragments revealed the breakpoint positions and defined the precise sizes of the three nonrecurrent microdeletions to be 964,182 bp, 555,432 bp, and 677,661 bp in affected cases of families SY, GD, and BJ, respectively (Figure 2D). For affected cases of families SY, GD, and BJ, the proximal and distal breakpoints were located in unique and L2 repeat, L1PA16 repeat and L1MC4 repeat, and two unique sequences, respectively. Microhomology between the proximal and distal breakpoints was not present (Figure 2D). Thus, the microdeletions in our three CGHT families most likely occurred through a nonhomologous end-joining mechanism.
We also recruited a 31-year-old Chinese man (KK) with extreme CGHT, coarse face, and gingival hyperplasia (Figure 3A), a phenotype closely resembling that of Julia Pastrana. KK was recorded as one of the world's hairiest men and has 96% of his body covered with thick, jet-black, and long (>5 cm) terminal hairs. He has a large head completely out of proportion to the rest of his body (Figure 3A). Oral examination revealed an abnormal dental occlusion and a delayed tooth eruption. He had surgical treatment for deformed nose and hyperplastic gingivae. His parents were apparently normal. We extracted genomic DNA from KK and his parents. Our high-resolution CNV screening with the SNP Array 6.0 revealed a genomic microduplication on chromosome 17q24.2-q24.3 that is 1,429,960 bp in size (CN_761503 to SNP_A-8412453) (Figure 3B). This microduplication was only present in KK and not in his parents (Figure 3B), indicating that the rearrangement is de novo in origin. We also validated the microduplication with the multiple qPCR assays (Figure 3C) and confirmed its de novo nature by an assay of the amplicon 7 (Figure 3D).
Figure 3.
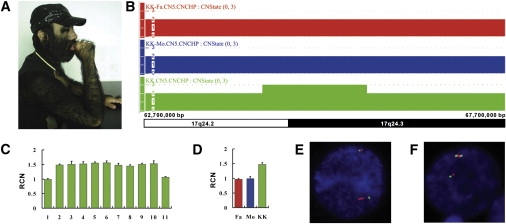
Identification of a De Novo Inverted Microduplication on Chromosome 17q24.2-q24.3 in KK
(A) Photograph of KK showing extreme hair overgrowth.
(B) Copy-number state of a 5 Mb genomic region on chromosome 17q24.2-q24.3 showing the presence of a microduplication in KK (green) but not in his parents (blue and red).
(C) Eleven qPCR assays validating the microduplication in KK. The duplication is seen as a ∼1.5-fold RCN. Error bars represent SD.
(D) The qPCR assay of the amplicon 7 confirming the de novo nature of the microduplication in KK. Error bars represent SD.
(E) Two-color FISH of a representative interphase nucleus showing a “one fused red - one normal green” signal pattern. BAC probes are CTD-2331H1 (red), the most proximal fully duplicated BAC clone, and CTD-2309E11 (green).
(F) Two-color FISH of a representative interphase nucleus showing a green-fused red-green signal order. BAC probes are CTD-2331H1 (red) and RP11-648L7 (green), the central duplicated BAC clone.
We used two-color interphase fluorescence in situ hybridization (FISH) to determine the orientation of the microduplication found in KK. The bacterial artificial chromosome (BAC) clones CTD-2331H1, RP11-648L7, and CTD-2309E11 were selected to cover the ABCA8 (MIM 612505), MAP2K6 (MIM 601254), and SOX9 (MIM 608160) genes, respectively. The CTD-2331H1 BAC DNA was labeled with SpectrumGreen-dUTP (Abbott Molecular Inc.) and the RP11-648L7 and CTD-2309E11 BAC DNAs were individually labeled with Cy3-dUTP (GE Healthcare Life Sciences). The interphase nuclei were cohybridized with a pair of SpectrumGreen-dUTP-labeled and Cy3-dUTP-labeled probes, counterstained with DAPI, and visualized by fluorescent microscopy. Our interphase FISH analysis for KK with the most proximal fully duplicated BAC clone CTD-2331H1 as the test probe (red) and the CTD-2309E11 BAC clone as an intrachromosomal reference probe (green) showed a fused red signal in most nuclei (Figure 3E). When we used the CTD-2331H1 clone (red) and the central duplicated BAC clone RP11-648L7 (green), 69 of the 106 counted interphase nuclei revealed a green-fused red-green signal order (Figure 3F). These two-color FISH signal patterns are consistent with an inverted duplication (Figure 4).
Figure 4.
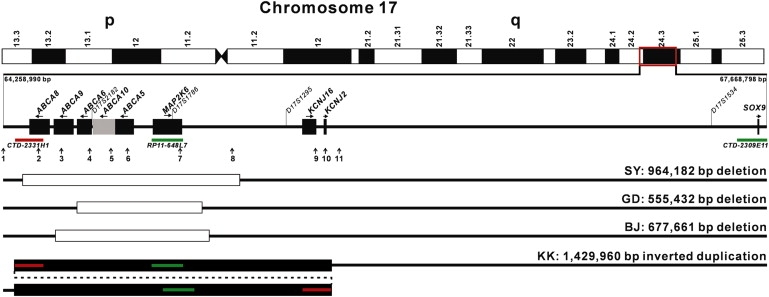
Schematic Diagram of the 17q24.2-q24.3 Region with a Summary of the Copy-Number Mutations Identified in the Present Study
Positions of the RefSeq genes, microsatellite markers, BAC clones, and qPCR amplicons (vertical arrows 1–11) are shown. Open bars represent the sizes of the three microdeletions in affected individuals of families SY, GD, and BJ. Solid bars display the inverted microduplication in KK. Color bars indicate the positions and sizes of the BAC clones used in the two-color interphase FISH.
The four CNVs identified in our familial and sporadic CGHT cases shared a common overlapping genomic region that contains four genes, ABCA5 (MIM 612503), ABCA6 (MIM 612504), ABCA10 (MIM 612508), and MAP2K6 (Figure 4). CNV of this genomic region was not detected in 434 unrelated Han Chinese normal controls. We isolated human hair follicles from a scalp skin tissue donated by a healthy individual after a cosmetic surgery and extracted total RNA. With use of RT-PCR, we could detect mRNA expression of all four genes (Figure S1). Information about the primer sequences is given in Table S5.
The CGHT-coarse face-gingival hyperplasia syndrome was first reported in the years before the discovery of Mendel's laws of heredity.8,9 However, not much has changed since then and the rare condition is still on the list of Mendelian phenotypes for which the underlying molecular basis remains unknown. We have identified a genetic locus for CGHT and found four overlapping 17q24.2-q24.3 CNVs (Figure 4), including three nonrecurrent microdeletions that show perfect cosegregation with the disease phenotype in three Chinese families with CGHT and coarse face, and one de novo inverted microduplication in a sporadic case with extreme CGHT, coarse face, and gingival hyperplasia. CNV of the common minimal region delineated by these four genomic rearrangements is not reported in the public Database of Genomic Variants and was not detected in our unrelated normal controls. Together, our results strongly support a causal relationship between the CNVs on 17q24.2-q24.3 and the CGHT with coarse face or with coarse face and gingival hyperplasia. Our finding of the copy-number mutations on the same chromosome region also suggested that the autosomal-dominant CGHT with coarse face could represent a variant of the CGHT-coarse face-gingival hyperplasia syndrome. Our work has established this rare syndrome as a genomic disorder.
The minimal disease genomic region of the four copy-number mutations contains three genes of the ATP-binding cassette transporter subfamily A, ABCA5, ABCA6, and ABCA10, which functions to maintain lipid homeostasis.17 In a large-scale survey of CNVs in normal Han Chinese population, Lin and colleagues found that 4 of the 300 individuals had a 203 kb deletion affecting these three genes.18 These make MAP2K6, the gene producing the human mitogen-activated protein kinase kinase 6, the leading candidate from the overlapping region. Activating mutations in components of the RAS-MAPK pathway can result in a group of phenotypically overlapping genetic syndromes with several dysmorphic features common to the patients reported herein.19 Costello syndrome (MIM 218040), for example, is caused by mutations in HRAS (MIM 190020), an important gene involved in the RAS-MAPK pathway, and often shows coarse face with thick lips, large-lobed ears, gingival hypertrophy, and relative macrocephaly.19,20 Moreover, it has been demonstrated that a gain-of-function mutation in SOS1 (MIM 182530) activates the RAS-MAPK pathway and thus cause gingival overgrowth in hereditary gingival fibromatosis (MIM 135300).21,22 Notably, a hallmark of the cardiofaciocutaneous syndrome (MIM 115150) caused by mutations in BRAF (MIM 164757), KRAS (MIM 190070), MAP2K1 (MIM 176872), or MAP2K2 (MIM 601263) is sparse and curly hair but not the thick and long hair seen in CGHT.23 In a 12-year-old girl with a de novo microdeletion of 17q24.2-q24.3 encompassing MAP2K6, neither CGH nor coarse face was observed.24 Additionally, knockout mice for Map2k6 lack abnormal hair growth.25,26 These facts might argue against the phenotypic contribution of the MAP2K6 gene in our three CGHT families.
Altered expression of the dosage-sensitive genes caused by copy-number mutations has traditionally been viewed as the general pathogenic mechanism for genomic disorders.13,14 Recently, it has been shown that some copy-number mutations can have a position effect, by disrupting regulatory elements or changing local chromatin structure, on expression of the key developmental regulator genes flanking the deleted or duplicated regions.14 Congenital hypertrichosis has been documented in several cases with chromosome 17q trisomy, suggesting that hair overgrowth can result from increased function of the gene(s) on chromosome 17q.27 Of note are two studies that have identified Sox9, a SRY-related HMG-box transcription factor, as an essential regulator for hair follicle stem cells.28,29 Mouse skin inactivated for the Sox9 gene develops alopecia.28 Conceivably, enhanced expression of Sox9 might lead to hair overgrowth. The above-mentioned microdeletion in the girl without CGH was ∼2.3 Mb (chr17:63,260,444-65,593,531), containing 18 RefSeq genes, and had the distal breakpoint within KCNJ16 (MIM 605722).24 Moreover, a reciprocal translocation t(3;17)(p14.3;q24.3) has been reported in a sporadic case with Zimmermann-Laband syndrome (MIM 135500) presenting with hypertrichosis, coarse face, and gingival hyperplasia.30 The breakpoint at 17q24.3 was defined at the telomeric side of KCNJ2 (MIM 600681).31 The human SOX9 locus is located 2,578,690 bp and 1,940,977 bp apart from the MAP2K6 and KCNJ2 loci, respectively (Figure 4). Therefore, the copy-number mutations described in the present study might exert a long-range position effect by altering the local chromatin architecture, resulting in an increased expression of SOX9 in hair follicle stem cells and thus causing a universal hair overgrowth. Constitutive activation of Map2k6 in mouse chondrocytes has been shown to increase the transcriptional activity of Sox9.32 This observation might provide a molecular clue to understanding the pathogenesis of the extreme hypertrichosis in KK. Clearly, further studies are needed to elucidate the molecular mechanisms by which the copy-number mutations on 17q24.2-q24.3 produce the phenotypes.
Acknowledgments
We would like to thank all the family members for their participation in the study. This work was mainly supported by the National Natural Science Foundation of China (30730097 and 30721063) and the Ministry of Science and Technology of China (2005DKA21302) (to X.Z.). X.Z. is a Chang Jiang Scholar of Genetic Medicine supported by the Ministry of Education of China. X.Z., M.S., and G.H. are very grateful to K.-L. Sun for his encouragement as a mentor. This paper is also dedicated to Victor A. McKusick. The He Laboratory is supported by the National 863 Program of China (2006AA02A407). The W.H.I.M. group is supported by grants from The Dystrophic Epidermolysis Bullosa Research Association, Pachyonychia Congenita Project, The British Skin Foundation, The National Eczema Society, and The Medical Research Council (reference number G0700314).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Garcia-Cruz D., Figuera L.E., Cantu J.M. Inherited hypertrichosis. Clin. Genet. 2002;61:321–329. doi: 10.1034/j.1399-0004.2002.610501.x. [DOI] [PubMed] [Google Scholar]
- 2.Bondeson J., Miles A.E.W. Julia Pastrana, the nondescript: an example of congenital, generalized hypertrichosis terminalis with gingival hyperplasia. Am. J. Med. Genet. 1993;47:198–212. doi: 10.1002/ajmg.1320470213. [DOI] [PubMed] [Google Scholar]
- 3.Bergman J. Darwin's ape-men and the exploitation of deformed humans. Technical Journal. 2002;16:116–122. [Google Scholar]
- 4.Hall B.K. Atavisms and atavistic mutations. Nat. Genet. 1995;10:126–127. doi: 10.1038/ng0695-126. [DOI] [PubMed] [Google Scholar]
- 5.Figuera L.E., Pandolfo M., Dunne P.W., Cantú J.M., Patel P.I. Mapping of the congenital generalized hypertrichosis locus to chromosome Xq24-q27.1. Nat. Genet. 1995;10:202–207. doi: 10.1038/ng0695-202. [DOI] [PubMed] [Google Scholar]
- 6.Fantauzzo K.A., Tadin-Strapps M., You Y., Mentzer S.E., Baumeister F.A., Cianfarani S., Van Maldergem L., Warburton D., Sundberg J.P., Christiano A.M. A position effect on TRPS1 is associated with Ambras syndrome in humans and the Koala phenotype in mice. Hum. Mol. Genet. 2008;17:3539–3551. doi: 10.1093/hmg/ddn247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Canún S., Guevara-Sanginés E.G., Elvira-Morales A., Sierra-Romero Mdel C., Rodríguez-Asbun H. Hypertrichosis terminalis, gingival hyperplasia, and a characteristic face: a new distinct entity. Am. J. Med. Genet. A. 2003;116A:278–283. doi: 10.1002/ajmg.a.10883. [DOI] [PubMed] [Google Scholar]
- 8.Laurence J.Z. A short account of the bearded and hairy female. Lancet. 1857;2:48. [Google Scholar]
- 9.Sokolov J. Julia Pastrana and her child. Lancet. 1862;1:467–469. [Google Scholar]
- 10.Mangino M., Pizzuti A., Dallapiccola B., Bonfante A., Saccilotto D., Cucchiara E. Hereditary gingival fibromatosis (HGF) with hypertrichosis is unlinked to the HGF1 and HGF2 loci. Am. J. Med. Genet. A. 2003;116A:312–314. doi: 10.1002/ajmg.a.10850. [DOI] [PubMed] [Google Scholar]
- 11.Irvine A.D., Dolan O.M., Hadden D.R., Stewart F.J., Bingham E.A., Nevin N.C. An autosomal dominant syndrome of acromegaloid facial appearance and generalised hypertrichosis terminalis. J. Med. Genet. 1996;33:972–974. doi: 10.1136/jmg.33.11.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lupski J.R. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 13.Lupski J.R., Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1:e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharp A.J. Emerging themes and new challenges in defining the role of structural variation in human disease. Hum. Mutat. 2009;30:135–144. doi: 10.1002/humu.20843. [DOI] [PubMed] [Google Scholar]
- 15.Lupski J.R. Genomic rearrangements and sporadic disease. Nat. Genet. 2007;39:S43–S47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 16.Sun M., Ma F., Zeng X., Liu Q., Zhao X.L., Wu F.X., Wu G.P., Zhang Z.F., Gu B., Zhao Y.F. Triphalangeal thumb-polysyndactyly syndrome and syndactyly type IV are caused by genomic duplications involving the long range, limb-specific SHH enhancer. J. Med. Genet. 2008;45:589–595. doi: 10.1136/jmg.2008.057646. [DOI] [PubMed] [Google Scholar]
- 17.Kaminski W.E., Piehler A., Wenzel J.J. ABC A-subfamily transporters: Structure, function and disease. Biochim. Biophys. Acta. 2006;1762:510–524. doi: 10.1016/j.bbadis.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 18.Lin C.H., Li L.H., Ho S.F., Chuang T.P., Wu J.Y., Chen Y.T., Fann C.S. A large-scale survey of genetic copy number variations among Han Chinese residing in Taiwan. BMC Genet. 2008;9:92. doi: 10.1186/1471-2156-9-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denayer E., de Ravel T., Legius E. Clinical and molecular aspects of RAS related disorders. J. Med. Genet. 2008;45:695–703. doi: 10.1136/jmg.2007.055772. [DOI] [PubMed] [Google Scholar]
- 20.Digilio M.C., Sarkozy A., Capolino R., Chiarini Testa M.B., Esposito G., de Zorzi A., Cutrera R., Marino B., Dallapiccola B. Costello syndrome: Clinical diagnosis in the first year of life. Eur. J. Pediatr. 2008;167:621–628. doi: 10.1007/s00431-007-0558-0. [DOI] [PubMed] [Google Scholar]
- 21.Hart T.C., Zhang Y., Gorry M.C., Hart P.S., Cooper M., Marazita M.L., Marks J.M., Cortelli J.R., Pallos D. A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. Am. J. Hum. Genet. 2002;70:943–954. doi: 10.1086/339689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jang S.I., Lee E.J., Hart P.S., Ramaswami M., Pallos D., Hart T.C. Germ line gain of function with SOS1 mutation in hereditary gingival fibromatosis. J. Biol. Chem. 2007;282:20245–20255. doi: 10.1074/jbc.M701609200. [DOI] [PubMed] [Google Scholar]
- 23.Roberts A., Allanson J., Jadico S.K., Kavamura M.I., Noonan J., Opitz J.M., Young T., Neri G. The cardiofaciocutaneous syndrome. J. Med. Genet. 2006;43:833–842. doi: 10.1136/jmg.2006.042796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blyth M., Huang S., Maloney V., Crolla J.A., Temple I.K. A 2.3Mb deletion of 17q24.2-q24.3 associated with ‘Carney Complex plus’. Eur. J. Med. Genet. 2008;51:672–678. doi: 10.1016/j.ejmg.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka N., Kamanaka M., Enslen H., Dong C., Wysk M., Davis R.J., Flavell R.A. Differential involvement of p38 mitogen-activated protein kinase kinases MKK3 and MKK6 in T-cell apoptosis. EMBO Rep. 2002;3:785–791. doi: 10.1093/embo-reports/kvf153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki H., Wu J., Hossain K., Ohhata T., Du J., Akhand A.A., Hayakawa A., Kimura H., Hagiwara M., Nakashima I. Involvement of MKK6 in TCRαβintCD69lo: A target population for apoptotic cell death in thymocytes. FASEB J. 2003;17:1538–1540. doi: 10.1096/fj.02-0869fje. [DOI] [PubMed] [Google Scholar]
- 27.Sarri C., Gyftodimou J., Avramopoulos D., Grigoriadou M., Pedersen W., Pandelia E., Pangalos C., Abazis D., Kitsos G., Vassilopoulos D. Partial trisomy 17q22-qter and partial monosomy Xq27-qter in a girl with a de novo unbalanced translocation due to a postzygotic error: case report and review of the literature on partial trisomy 17qter. Am. J. Med. Genet. 1997;70:87–94. doi: 10.1002/(sici)1096-8628(19970502)70:1<87::aid-ajmg16>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 28.Vidal V.P., Chaboissier M.C., Lützkendorf S., Cotsarelis G., Mill P., Hui C.C., Ortonne N., Ortonne J.P., Schedl A. Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr. Biol. 2005;15:1340–1351. doi: 10.1016/j.cub.2005.06.064. [DOI] [PubMed] [Google Scholar]
- 29.Nowak J.A., Polak L., Pasolli H.A., Fuchs E. Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell. 2008;3:33–43. doi: 10.1016/j.stem.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim H.G., Higgins A.W., Herrick S.R., Kishikawa S., Nicholson L., Kutsche K., Ligon A.H., Harris D.J., Macdonald M.E., Bruns G.A. Candidate loci for Zimmermann-Laband syndrome at 3p14.3. Am. J. Med. Genet. A. 2007;143:107–111. doi: 10.1002/ajmg.a.31544. [DOI] [PubMed] [Google Scholar]
- 31.Abo-Dalo B., Kim H.G., Roes M., Stefanova M., Higgins A., Shen Y., Mundlos S., Quade B.J., Gusella J.F., Kutsche K. Extensive molecular genetic analysis of the 3p14.3 region in patients with Zimmermann-Laband syndrome. Am. J. Med. Genet. A. 2007;143A:2668–2674. doi: 10.1002/ajmg.a.32034. [DOI] [PubMed] [Google Scholar]
- 32.Zhang R., Murakami S., Coustry F., Wang Y., de Crombrugghe B. Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation. Proc. Natl. Acad. Sci. USA. 2006;103:365–370. doi: 10.1073/pnas.0507979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.