

Abstract
Glypicans are a family of glycosylphosphatidylinositol (GPI)-anchored, membrane-bound heparan sulfate (HS) proteoglycans. Their biological roles are only partly understood, although it is assumed that they modulate the activity of HS-binding growth factors. The involvement of glypicans in developmental morphogenesis and growth regulation has been highlighted by Drosophila mutants and by a human overgrowth syndrome with multiple malformations caused by glypican 3 mutations (Simpson-Golabi-Behmel syndrome). We now report that autosomal-recessive omodysplasia, a genetic condition characterized by short-limbed short stature, craniofacial dysmorphism, and variable developmental delay, maps to chromosome 13 (13q31.1-q32.2) and is caused by point mutations or by larger genomic rearrangements in glypican 6 (GPC6). All mutations cause truncation of the GPC6 protein and abolish both the HS-binding site and the GPI-bearing membrane-associated domain, and thus loss of function is predicted. Expression studies in microdissected mouse growth plate revealed expression of Gpc6 in proliferative chondrocytes. Thus, GPC6 seems to have a previously unsuspected role in endochondral ossification and skeletal growth, and its functional abrogation results in a short-limb phenotype.
Introduction
Heparan sulfate proteoglycans (HSPGs) are involved in various biological processes, such as growth-factor signaling, cell adhesion, intracellular membrane trafficking, and tumor metastasis.1–3 Glypicans are a family of glycosylphosphatidylinositol (GPI)-anchored cell-surface HSPGs sharing a highly conserved three-dimensional structure.4–6 They play key roles in the regulation of growth-factor signaling and morphogen gradients during development.4,7,8 Mutations in dally (division abnormally delayed), an ortholog of mammalian glypicans 3 and 5 (GPC3 [MIM 300037] and GPC5 [MIM 602446]) in Drosophila, implicate glypicans in the control of cell fates and division.9 The only known human disorder caused by mutations in a glypican core protein is Simpson-Golabi-Behmel syndrome (MIM 312870), an X-linked overgrowth/malformation syndrome caused by mutations in GPC310 and occasionally by deletion of clustered GPC3 and GPC4 (MIM 300168).11
Autosomal-recessive omodysplasia (MIM 258315) is a genetic condition characterized by proximally shortened limbs, facial dysmorphism, and severe short stature. The term omodysplasia derives from “omos,” the Greek word for humerus, and was first applied by Maroteaux to a series of patients with syndromal short stature and hypoplastic humeri.12 An autosomal-dominant form involving only the upper limbs was later recognized as a separate disorder13 (MIM 164745) distinct from the recessive form. To date, 22 cases of recessive omodysplasia have been reported.14 Skeletal features comprise proximal limb shortening, distal tapering of long tubular bones, proximal radioulnar diastasis, and anterolateral dislocation of the radial head. Facial features include frontal bossing, a flat nasal bridge, low set ears, a long philtrum, anteverted nostrils, and frontal capillary hemangiomas. Variable findings are cryptorchidism, hernias, congenital heart defects, and cognitive delay.14,15 Adult height ranges between 132 and 144 cm (−7.0 to −5.5 SD).15
We report that omodysplasia maps to chromosome 13 and is caused by homozygosity for null mutations in GPC6 (MIM 604404), which encodes for the latest described human glypican gene.16
Material and Methods
Patients and Samples
We investigated eight patients and two products of conception from five families (Figure 1) and one additional isolated patient (patient 9). All patients except for patient 9 have been previously reported: family 1 is of Gypsy origin17; families 218 and 319 are Arabic-Muslim (Lebanese); family 4, previously reported by Elcioglu et al. (cases 3 and 4)15, is Swedish; and family 5 is Arabic-Muslim.20 Consanguinity was known in families 1, 2, 3, and 5. Patient 9 is an adopted child; neither clinical information nor biological materials were available from the biological parents. Rhizomelic limb shortening was noted at prenatal ultrasound at 24 weeks; she was born at 38 weeks of gestational age with birth weight 3118 g (−0.41 SD), length 41 cm (−4.76 SD), and head circumference 35 cm (+0.44 SD). A heart ultrasound was normal. At age 4 years, she was 84 cm (−4.38 SD) and 16.7 kg (+0.85 SD); she presented gross motor delay but normal cognitive development.
Figure 1.
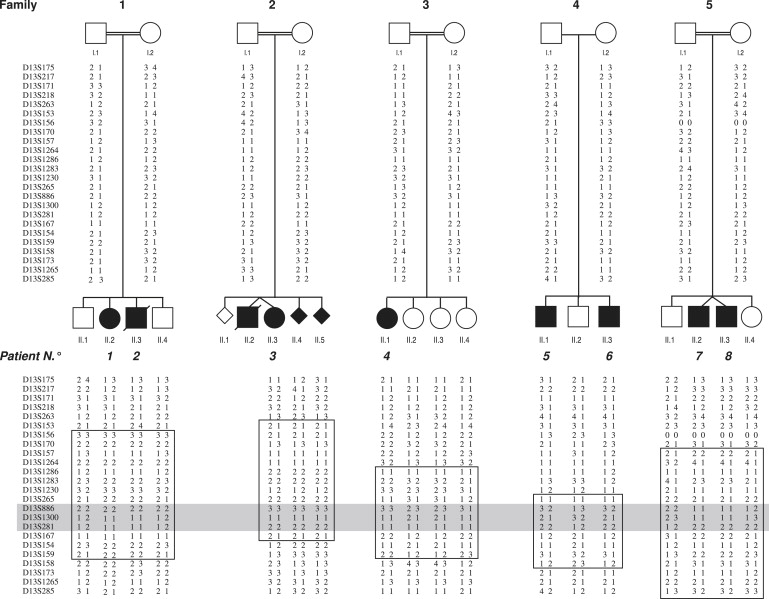
Pedigrees and Haplotypes on Chromosome 13 of the Five Omodysplasia Families
In squares: regions compatible with linkage in single families. In gray: common region of homozygosity in affected individuals of consanguineous families.
All affected individuals fulfilled the clinical and radiological criteria for the diagnosis of omodysplasia. Motor delay was a feature of all patients, but mental retardation was present only in patients 1 and 5. Additional clinical abnormalities were cryptorchidism (patients 2, 5, 6, 7, and 8) and congenital heart defect (patient 2). The radiographic findings for patient 4 (whose clinical history and description were reported in Di Luca and Mitchell19, without radiographic images) and the clinical and radiographic findings for patient 9 (previously undescribed) are reported in Figure 2.
Figure 2.
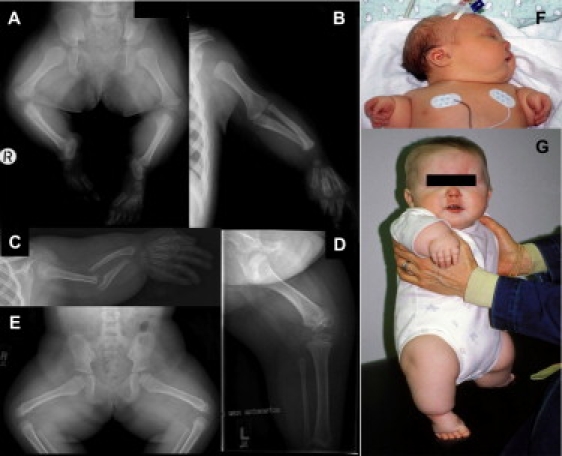
Clinical and Radiographic Features of Omodysplasia
(A and B) Radiographic features of female patient 4, age 16 months. Note shortened femora and humeri with mild tapering and distally flared metaphyses; the tibiae are also short but are less affected. Note also the radioulnar diastasis and relative preservation of the acral skeletal elements.
(C–E) Radiographic features of female patient 9, age 5 years. Note markedly short, “club shaped” humeri; shortened femora; radioulnar diastasis; and relative acral preservation.
(F and G) Clinical features of patient 9 at birth (F) and age 1 year (G). Note posteriorly rotated ears, mild micrognathia, persistent capillary hemangioma, and marked rhizomesomelic limb shortening.
Blood samples were obtained from all individuals except for the first unaffected fetus and the deceased twin in family 2 (Figure 1). Genomic DNA was purified from peripheral blood leukocytes according to standard techniques. Skin biopsies were obtained from the unaffected parents of family 2, the affected child in family 3 (patient 4), and the unaffected father of family 4. Fibroblasts were cultured in Dulbecco's modified Eagle's medium (GIBCO) with 10% fetal calf serum and antibiotics and incubated at 37°C with 5% CO2. Total RNA was extracted from fibroblasts via the RNeasy Mini Kit (QIAGEN) and was kept at −80°C.
Appropriate written informed consent was obtained from all individuals. The study protocol was approved by the Ethical Committee of the University of Lausanne, Switzerland as well as by the National Helsinki committee of Israel.
Linkage Study
We performed a genome-wide linkage scan in all five families by using microsatellite markers (Linkage Mapping Set V2.5; Applied Biosystems) at an average distance of 10 cM. We performed a multipoint linkage analysis with GeneHunter version 2.1_r4_beta. Disease gene frequency was set to 0.001, under the assumption of an autosomal-recessive mode of inheritance with complete penetrance. A unique region of interest was refined with ten extra markers of Genethon and deCODE maps.
Mutation Analysis
GPC5 (MIM 602446) and GPC6 genes were both studied in genomic DNA of affected subjects.
For PCR amplification of GPC6, oligonucleotide primers were designed on the basis of the GPC6 sequence (ENSG00000183098), targeting all nine exons and 3′- and 5′-UTR-regions. All primers were custom synthesized by Microsynth, and their sequences are reported in Table S1. The entire coding sequence (including intron-exon boundaries) and UTR regions were amplified by PCR. Amplicon sizes and fragment-specific annealing temperatures are reported in Table S1. To verify the amplification products, we used a 2% agarose gel electrophoresis. The amplified fragments were purified with the standard protocol of Montage PCRμ96 (Millipore). Amplification products were analyzed by direct sequencing with the fluorescent dideoxy-terminator method according to standard procedures (BigDye Terminator V1.1 Cycle Sequencing Kit, Applied Biosystems) on an automatic sequencer, ABI 3100-Avant (Applied Biosystems).
GPC6 cDNA was amplified by RT-PCR from total RNA extracted from fibroblasts. On the basis of the GenBank sequence of GPC6 mRNA (accession number NM_005708), five overlapping fragments of different sizes were amplified by one-step reverse-transcription PCR (SuperScript One-Step RT-PCR with Platinum Taq, Invitrogen) according to the manufacturer's instructions. The cDNA-specific primers (Microsynth), the length of the amplicons, and the annealing temperature for each PCR are reported in Table S2. The cDNA amplicons were migrated on 3% agarose gel NuSieve GTG Agarose, (BioConcept); when single bands were obtained, PCR products were directly purified according to the standard protocol of Montage PCRμ96 (Millipore); when double bands were visible on gel, each band was excised and purified according to the standard protocol of S.N.A.P. Gel Purification Kit (Invitrogen). Primers used for sequencing were the same as for amplification. To prevent potential degradation of transcripts containing premature termination codons (PTCs) by nonsense-mediated mRNA decay (NMD),21 we treated fibroblast cells (80%–90% confluent) from the parents of family 2, patient 4, the father of family 4, and one control with cycloheximide (CHx) (Sigma-Aldrich). We treated cultured cells by incubating cells that were 80%–90% confluent in a 75 cm2 flask with 100 μg/ml CHx for 6 hr prior to the RNA extraction. After incubation, cells were washed with 5 ml phosphate-buffered saline, the flask was placed on ice, and cells were collected via scraping. The cells were transferred to a 15 ml new tube and centrifuged 10 min at 1500 rpm at room temperature. After careful removal of the supernatant, the tube was put on ice, and RNA isolation was performed according to standard protocols. The protocols for cDNA sequencing were the same as those for genomic DNA.
Comparative Genomic Hybridization Array
Patients 3–9 and parents of patients 3–6 were studied by comparative genomic hybridization array (aCGH) with the Agilent Human Genome CGH Microarray Kit 244KA. The aCGH platform is a 60-mer oligonucleotide-based microarray that allows a genome-wide survey and molecular profiling of genomic aberrations with a resolution of ∼20 kb. Labeling and hybridization were performed according to the protocols provided by Agilent. In brief, after digestion and purification with the QIAprep Spin Miniprep kit (QIAGEN), the Agilent Genomic DNA Labeling Kit Plus was used for labeling test and reference DNAs (1.3 μg) by random priming with either Cy3-dUTP or Cy5-dUTP. After the labeling reaction, the individually labeled test and reference samples were concentrated with Microcon YM-30 filters (Millipore) and then combined. After probe denaturation and pre-annealing with Cot-1 DNA, hybridization was performed at 65°C with rotation for 40 hr. Four steps were done with Agilent Oligo CGH washes: wash buffer 1 at room temperature for 5 min, wash buffer 2 at 37°C for 1 min, acetonitrile rinse at room temperature for 1 min, and 30 s at room temperature in Agilent's Stabilization and Drying Solution. All slides were scanned on an Agilent DNA microarray Scanner. Data were obtained with Agilent Feature extraction software v9.1. Graphical overviews were obtained with the CGH analytics software (v3.4.27) according to hg17 genome assembly (May 2004 release) subsequently translated to the hg18 (March 2006). Copy-number variations were checked in the database of genomic variants; all studied patients have been considered as white because no specific data about copy-number variations is available for Arabic Muslims.
Quantitative Multiplex PCR of Short Fluorescent Fragments
In order to confirm the genomic deletions and duplication detected by aCGH with a different method and in all family members, we used the previously described quantitative multiplex PCR of short fluorescent fragments (QMPSF) method.22–25 We designed oligonucleotide primer pairs for amplification of short exon fluorescent fragments corresponding to the nine GPC6 exons to construct multiplex PCR; their sequences, as well as amplicon sizes, are reported in Table S3. The multiplex reaction also contained an internal control primer pair that amplified a short sequence (237 bp) of the DSCR1 gene (MIM 602917). All forward primers of each pair were also 5′ labeled with 6-FAM fluorochrome.
Fluorescent multiplex PCR products (1.2 μl) were mixed with 18 μl of formamide and 0.3 μl of size-standard 500 LIZ (GeneScan 500 LIZ Size Standard, Applied Biosystems) and then separated on an ABI Prism 310 Genetic Analyzer at 60°C with POP4 polymer (Applied Biosystems). Results were analyzed with Genescan 3.7 Software (Applied Biosystems) after superimposition of fluorescent profiles of patient and control DNA based on the size standard. Profiles were then normalized to DSCR1 peak intensities. Then, DSCR1-normalized peak levels of corresponding amplicons were visually compared. The normal two-copy number of amplified fragments appears as a completely superimposed peak area. Heterozygous exon deletion is indicated by a 2-fold reduction in the height (approximately 50%) of the corresponding peak, whereas homozygous exon deletion appears as the absence of a peak. Heterozygous exon duplication is represented by a one-and-a-half (approximately 30%–50%) peak-size increase.
Sequencing of Genomic Breakpoints
In order to determine the exact length of the deletions and to define their breakpoints, we applied a strategy based on genomic amplification of a series of intronic fragments that contained an overlapping common primer sequence: the sequence of the forward primer of each amplicon is complementary, in the inverted 5′–3′ direction, to the sequence of the reverse primer of the previous amplicon (Figure S1). Primers were designed in the intronic regions delimited by the rearranged aCGH probes and based on the sequence ENSG00000183098 (chromosome 13: 92,677,711–93,853,948). All nucleotide primers (Microsynth) are available upon request. All fragments were amplified in patients and their parents, as well as in two control individuals, by touch-down PCR in a GeneAmp PCR System 9700 (Applied Biosystems). Cycling conditions are available upon request. We used a 2% agarose gel electrophoresis to verify the amplification in patients and controls. For the deletions found in families 2, 3, and 5 and patient 9, a certain number of fragments were contained, at least partially, within the deletion and failed to amplify from DNA of homozygous probands when control DNA amplified correctly. We then amplified a single amplicon containing the breakpoint by using as forward primer the complementary sequence to the reverse primer of the last amplified fragment (5′ to the deletion) and as reverse primer the complementary sequence to the forward primer of the next amplified fragment (3′ to the deletion) (Figure S1). The final amplicons containing the deletion breakpoints (Table S4) were purified by the standard protocol of Montage PCRμ96 (Millipore) and sequenced with the same primers as those used for amplification. The sequencing reaction was carried out as described above for genomic DNA analysis. For family 4, we amplified all fragments by QMPSF in patients and parents to detect the fragments that increased their peak size by one-and-a-half and contained the start and end points of the duplication (Figure S1). For technical reasons, the duplication breakpoints were not directly sequenced. Primer sequences of the amplicons harboring the breakpoint of each rearrangement are reported in Table S4.
Note: Identified sequence variants and mutations were named according to the recommendations of the Human Genome Variation Society. Mutations were described according to the GPC6 cDNA reference sequence NM_005708. For nucleotide numbering, the A of the ATG translation initiation site was used as position +1. For amino acid numbering, the initiation methionine of GPC6 protein was used as position 1. Numbering of the genomic sequence for naming the breakpoints starts with nucleotide +1 of the Ensembl reference sequence ENSG00000183098 (corresponding to the NCBI nucleotide 92677111 on chromosome 13).
Study of Glypican 6 Expression in Mouse Growth Plate
Quantitative Reverse-Transcriptase Polymerase Chain Reaction
RNA from the proliferative, prehypertrophic, and hypertrophic zones was extracted and amplified from femurs of 2-week-old mice as described previously.26 RNA from each maturation zone (100 ng) was reverse transcribed in a 20 μl reaction with random hexamers and the Transcription High-Fidelity cDNA Synthesis Kit (Roche). Quantitative PCR was carried out with the Universal Probe Library oligo set (Roche) and the LightCycler 480 Probes Master Kit (Roche). PCR reactions were performed in 10 μl reactions on a LightCycler 480II real-time PCR instrument (Roche) with 1 ng of cDNA. The data were normalized to the housekeeping gene mitochondrial ATP synthase (Atp5b), which we have shown is expressed at consistent levels throughout the growth plate (data not shown). All samples were run in triplicate, and experiments were repeated on the growth-plate cartilage zones microdissected from two mice.
Immunofluorescence
Tibiae from 2-week-old wild-type mice were harvested immediately after death and dissected and snap frozen in OCT compound in liquid-nitrogen-cooled isopentane and stored at −80°C. Serial 9 μm coronal frozen sections were collected on superfrost glass slides and air dried for 1 hr. The sections were fixed for 10 min in 100% methanol. Immunostaining was performed in phosphate-buffered saline containing 1% bovine serum albumin. The glypican 6 antibody (catalog # AF1053, R&D Systems) was used at 15 μg/ml, and the antibody against collagen X (kindly provided by Dr. R. Wilson) was used at a dilution of 1:1000. Collagen X was used as a marker of hypertrophic zone chondrocytes. Primary antibody binding was detected with secondary antibodies conjugated to Alexa Fluor 488 (donkey anti-goat IgG green) or 594 (donkey anti-rabbit IgG red) (Molecular Probes, Eugene, OR), diluted 1:1000. Slides were mounted in Fluorosave reagent (Calbiochem) and examined on an Olympus IX70 microscope.
Results
Recessive Omodysplasia Maps to Chromosome 13
The initial linkage-mapping screening revealed two regions with a positive LOD score, one on chromosome 18 (18q12.2–q21.2, 7 cM, maximum LOD score 2.31) and the other on chromosome 13 (13q31.1–q32.2, 15.7 cM, LOD score 3.2). However, only the interval on chromosome 13 showed a common region of homozygosity in patients of consanguineous parents and was thus selected for further screening. Fine mapping with ten additional markers confirmed linkage and reduced the region to 4.5 cM flanked by D13S265 (89.3 Mb) and D13S167 (93.8 Mb) with a maximum LOD score Zmax = 4.0 at theta = 0, at the D13S886 locus (Figure 1). According to the National Center for Biotechnology Information website search, this 4.5-Mb-long region contains 15 genes. GPC5 and GPC6 were first considered as possible candidate genes on the basis of their putative function as coreceptors for growth factors involved in cellular growth control and differentiation during development.
Mutations in GPC6 Cause Omodysplasia
Direct sequencing of GPC5 coding region, intron-exon boundaries, and 5′ and 3′ UTR regions did not reveal any mutation in affected individuals.
Molecular studies on GPC6 include genomic direct sequencing, aCGH studies, QMPSF, genomic breakpoint analysis, and cDNA amplification and sequencing. All mutations found are summarized in Figures 3 and 4; the rearranged aCGH probes and the genomic breakpoints of each rearrangement are reported in Table 1.
Figure 3.
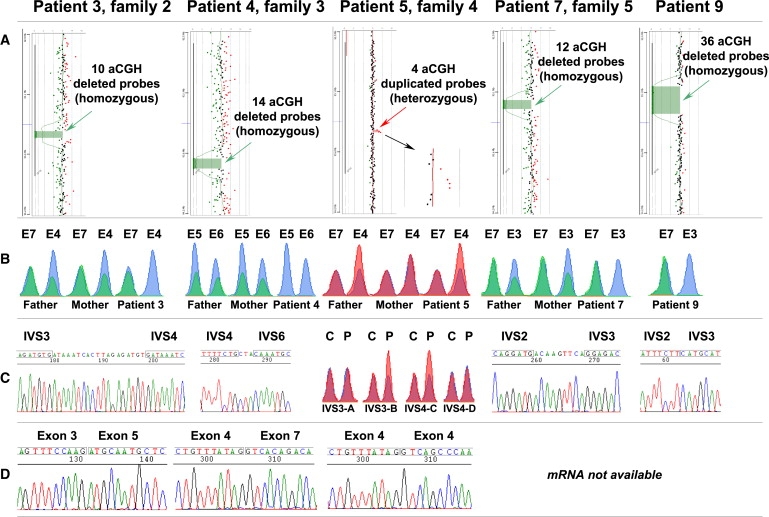
GPC6 Mutations Detected with Different Methods in Families 2, 3, 4, and 5 and in Patient 9
(A) aCGH results of patients 3, 4, 7, and 9 (homozygous deletions) and patient 5 (heterozygous duplication).
(B) QMPSF results for one proband and parents of families 2, 3, 4, and 5 (data of other siblings are not shown) and of patient 9. The peaks obtained for the different exons in control DNA appear in blue, whereas peaks amplified from patients' and parents' DNA appear in green (families 2, 3, and 5 and patient 9) or red (family 4). In family 2, the peak of exon 4 (E4) shows a 2-fold intensity reduction in the parents compared to the control (heterozygous deletion), and it is absent in the proband (homozygous deletion). In family 3, the peaks corresponding to exons 5 and 6 (E5 and E6) show a 2-fold intensity reduction in the parents compared to the control (heterozygous deletion) and are absent in the proband (homozygous deletion); the peak of a control exon is not shown for space reasons. In family 4, the peak of exon 4 (E4) shows a 2-fold intensity increase in the father and the proband (heterozygous duplication) and overlaps with the control in the mother. In family 5, the peak corresponding to exon 3 shows a 2-fold intensity reduction in the parents compared to the control (heterozygous deletion) and is absent in the proband (homozygous deletion). In patient 9, the peak of exon 3 (E3) is absent (homozygous deletion), whereas the peak of the control exon (E7) overlaps with the control peak.
(C) Breakpoint sequencing results: in family 2, IVS3 and IVS4 are truncated by a genomic deletion encompassing exon 4; 19 bp are inserted in the breakpoint between the two intronic sequences. In family 3, IVS4 and IVS6 are truncated by a genomic deletion encompassing exons 5 and 6; 3 bp are inserted in the breakpoint between the two intronic sequences. In family 5, IVS2 and IVS3 are truncated by a genomic deletion encompassing exon 3; 9 bp are inserted in the breakpoint between the two intronic sequences. In patient 9, IVS2 and IVS3 are truncated by a genomic deletion encompassing exon 3. Primers and genomic location of the amplicons shown here are reported in Table S4. In family 4, the borders of the genomic duplication were mapped by QMPSF: fragments IVS3-A and IVS4-D are located 5′ and 3′ of the duplication, respectively; IVS3-B and IVS4-C present enhanced amplification, indicating that at least one primer is located within the rearrangement (see also Table S4).
(D) cDNA sequencing results: exon 4 is missing in family 2; exons 5 and 6 are missing in family 3; exon 4 occurs twice in family 4.
Figure 4.
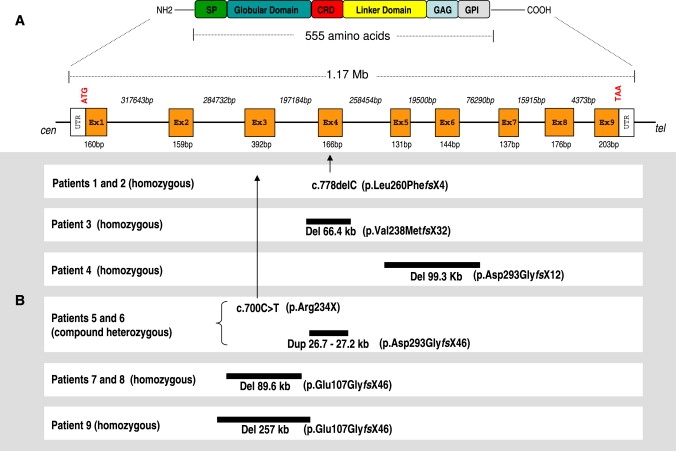
Summary of GPC6 Mutations Found in the Studied Patients with Respect to the Gene and Protein Structure
(A) Genomic and protein structure of GPC6 gene. The proposed protein domains are shared by all glypicans. SP—signal peptide; CRD—cysteine-rich domain; GAG—heparan-sulfate (HS) glycosylation site; GPI—glycophosphatidyl-inositol binding site, which anchors glypican to the plasma membrane.
(B) Schematic representation of genomic mutations found in omodysplasia patients.
Table 1.
Genomic Rearrangements Causing Omodysplasia: aCGH Probes Involved in Genomic Rearrangements, Effect on GPC6 Gene, and Localization of Genomic Breakpoints
| Patient (Family) | aCGH probesa (5′ to the Rearrangement) |
aCGH Probea (3′ to the Rearrangement) |
|||||
|---|---|---|---|---|---|---|---|
| Last Normally Detected | First within Rearrangement | Rearrangement | Effect on GPC6 Gene | Last within Rearrangement | First Normally Detected | Genomic Breakpointb | |
| 3 (2) | A_16_P19915562 | A_16_P19915577 | 10 probes deleted (66492 bp) | Deletion of exon 4 | A_16_P19915750 | A_16_P40055309 | g.770152_836646 del66495 |
| 4 (3) | A_16_P40055823 | A_16_P19916323 | 14 probes deleted (99317 bp) | Deletion of exons 5 and 6 | A_16_P40056128 | A_16_P19916617 | g.1026129_1125444 del99316 |
| 5 and 6 (4) | A_16_P19915593 | A_16_P19915610 | 4 probes duplicated (26-27 kb) | Duplication of exon 4 | A_16_P02838707 | A_16_P19915685 | Between 784663 and 811908 |
| 7 and 8 (5) | A_16_P02838121 | A_16_P19914778 | 12 probes deleted (89632 bp) | Deletion of exon 3 | A_16_P19915025 | A_16_P19915064 | g.514476_604107 del89632 |
| 9 | A_16_P02837882 | A_16_P02837902 | 36 probes deleted (257017 bp) | Deletion of exon 3 | A_16_P19915147 | A_16_P19915180 | g.376472_6633489 del257018 |
Agilent Human Genome CGH Microarray Kit 244KA.
Numbering of the genomic sequence for naming the breakpoints starts with nucleotide +1 of the Ensembl reference sequence ENSG00000183098 (corresponding to the NCBI nucleotide 92677111 on chromosome 13).
In family 1, the affected individuals (patients 1 and 2) were homozygous for a single base deletion (c.778 delC) in exon 4; this deletion is predicted to result in a frameshift starting at codon 260 and leading to a PTC (p.Leu260PhefsX4). Parents and unaffected siblings were heterozygous for the mutation.
In family 2, genomic PCR reactions with DNA of affected subjects (patient 3 and two products of conception) failed to amplify exon 4, despite good amplification of DNA from the parents and control DNA. The rest of the genomic sequence in the probands and the whole coding region (including exon 4) in the parents were identical to the reference sequence. These results, confirmed in a second PCR amplification, suggested a possible genomic deletion around exon 4. The aCGH study revealed a homozygous genomic deletion in the affected living child (patient 3) (Figure 3A) and heterozygosity for the same deletion in the parents. The deletion was confirmed by QMPSF (Figure 3B) in all family members: the peak corresponding to exon 4 shows a heterozygous deletion in the parents and a homozygous deletion in affected individuals (patient 3 and two products of conception). All the other GPC6 exons showed a normal profile (data not shown), confirming that the boundaries of the deletion in this family were within IVS3 and IVS4. The genomic breakpoint of the deletion is shown in Table 1. Moreover, in all subjects bearing the mutation, an additional 19 base pairs were found at the junction of the breakpoints (Figure 3C); this sequence was not present in the published genomic sequence of IVS3 and IVS4. RT-PCR amplification from fibroblast total RNA of both heterozygous parents resulted in two products, one corresponding to the wild-type GPC6 sequence (579 bp) and the other to a shorter fragment (413 bp) (Figure S2A) missing exon 4 (c.712_877 del) (Figure 3D). The relative abundances of wild-type and mutant mRNAs in heterozygous carriers (Figure S2A) as well as the enhancement (Figure S2B) of mutant mRNA amplification by CHx treatment, which prevents mRNA degradation during translation, are concordant with the mutant mRNA's being subject to NMD. The deleted sequence predicts an out-of-frame translation with PTC (p.Val238MetfsX32).
In family 3, PCR reactions with genomic DNA of the affected child (patient 4) failed to amplify exons 5 and 6 in two independent PCRs, despite good amplification of DNA from other family members and control DNA. The rest of the genomic sequence in these individuals and the whole coding region (including exons 5 and 6) in unaffected family members were identical to the reference sequence. These results, confirmed in a second PCR amplification, suggested a possible genomic deletion around exons 5 and 6. The aCGH study revealed a genomic homozygous deletion in the affected child (Figure 3A) and a heterozygous deletion in the parents. QMPSF analysis was performed in all family members: the results showed that parents and unaffected siblings were heterozygous for the deletion of exons 5 and 6 and that the proband was homozygous for the same deletion (Figure 3B). All other GPC6 exons showed a normal profile in all individuals (data not shown), confirming that the boundaries of the deletion in this family were within IVS4 and IVS6. The genomic breakpoint of the deletion is shown in Table 1. As for family 2, three additional base pairs not present in the published genomic sequence were found in all subjects at the breakpoint junction (Figure 3C). RT-PCR amplification from fibroblast total RNA of the affected patient resulted in one main product (660 bp in Figure S2C) corresponding to GPC6 cDNA sequence missing exons 5 and 6 (c.878_1152 del) (Figure 3D). Inhibition of NMD by CHx treatment enhanced the abundance of the mutant transcript (660 bp) relative to the control mRNA (935 bp) (Figure S2D). The deleted sequence predicts a shift to an out-of-frame translation (p.Asp293GlyfsX12). RT-PCR also revealed a second amplification byproduct of 494 bp (Figure S2C), the sequence of which missed exons 4, 5, and 6. The deletion of the three exons predicts an in-frame, but shorter (408 amino acids), translation product, which lacks most of the cysteine-rich domain essential for the globular conformation of the GPC6 core protein. Given the proven presence of exon 4 in the genomic sequence (seen by QMPSF and sequencing of the breakpoints in both the affected patient and heterozygote parents) and the lack of enhancement by NMD inhibition (Figure S2D), this cDNA may derive from a less efficient splicing from exon 3 to exon 7.
In family 4, the affected individuals (patients 5 and 6) and their mother had a heterozygous single base substitution (c.700C > T) leading to a nonsense mutation (p.Arg234X) in exon 3. No mutation was found at the genomic level by direct sequencing of the other exons and intron-exon boundaries (all well amplified) in affected and nonaffected family members. The aCGH study revealed a genomic heterozygous duplication in the affected children (Figure 3A) and in the father. QMPSF analysis confirmed a heterozygous duplication of exon 4 (Figure 3B) in the father and affected children; this duplication was not present in the mother or unaffected child. All the other GPC6 exons showed a normal profile in all individuals (data not shown), confirming that the boundaries of the duplication in this family were within IVS3 and IVS4. Although the exact boundaries of the duplication were not identified, the start and end of the duplication were localized within the fragments, showing an increase in signal intensity (approximately 30%–50%) (Figure 3C and Table S1). RT-PCR from total RNA extracted from fibroblasts of the father showed a single amplification product (579 bp) (Figure S2A) whose sequence corresponded to the wild-type allele. RT-PCR performed after treatment of fibroblast with CHx allowed the identification of a second product (745 bp) corresponding to the mutant allele (Figure S2B). Direct sequencing showed, as expected, full exon 4 duplication (c.712_877 dup) (Figure 3D). This duplication leads to an out-of-frame translation product at the duplication junction, and the new reading frame terminates with PTC (p.Asp293GlyfsX46).
In family 5, PCR reactions with DNA of the affected twins (patients 7 and 8) failed to amplify exon 3 in two independent PCRs, despite good amplification of DNA from other family members and control DNA, suggesting a possible genomic deletion containing exon 3. The rest of the genomic sequence in these individuals and the whole coding region (including exon 3) in unaffected family members were identical to the reference sequence. The aCGH study performed in one of the affected twins revealed a genomic homozygous deletion (Figure 3A). QMPSF analysis (Figure 3B) was performed in all family members, confirming the heterozygous deletion in the parents and one of the unaffected siblings (II.4 in Figure 1). The other unaffected sibling (II.1 in Figure 1) had a normal profile for exon 3 and thus inherited the two wild-type alleles. All peaks corresponding to the other exons were normal in all individuals (data not shown), confirming that the boundaries of the deletion in this family were located within IVS2 and IVS3. The genomic breakpoint of the deletion is shown in Table 1. As for families 2 and 3, nine additional bp (not present in the published genomic sequence) were found in all subjects at the breakpoint junction (Figure 3C). The deletion predicts an out-of-frame translation product leading to PTC (p.Glu107GlyfsX46).
In patient 9, genomic PCR reactions failed to amplify exon 3 in two independent PCRs, despite good amplification in controls, suggesting a possible deletion encompassing exon 3. The aCGH study revealed a genomic homozygous deletion (Figure 3A). The deletion was confirmed by QMPSF (Figure 3B). All other peaks of GPC6 exons amplified normally (data not shown), confirming that the boundaries of the deletion were located within IVS2 and IVS3. The genomic breakpoints of the deletion are shown in Table 1, and the sequence is shown in Figure 3C. The deletion predicts an out-of-frame translation product (p.Glu107GlyfsX46).
Glypican 6 Expression Study in Mouse Growth Plate
To determine whether glypican 6 may have a role during endochondral ossification, we examined the relative mRNA expression levels within mouse growth plates.
Quantitative RT-PCR performed with total RNA obtained from micro-dissected cartilage growth-plate subzones showed that Gpc6 message was expressed most highly in the proliferative zone and decreased dramatically in the prehypertrophic and hypertrophic zones (Figure 5E). The level of expression in the hypertrophic zone was decreased by more than 50-fold in comparison to the proliferative zone. This was confirmed at the protein level; immunofluorescence demonstrated Gpc6 expression by proliferative-zone chondrocytes (Figure 5D). The localization of collagen X (Col10a1) expression to the hypertrophic zone of the growth plate (Figures 5B and 5E) was used for confirming the accuracy of the cartilage microdissection. The expression patterns in the growth-plate cartilage of other members of the glypican family were also determined by quantitative RT-PCR (Figure S3). These glypicans are expressed throughout the growth plate and do not show the same pattern of differential expression during cartilage hypertrophy.
Figure 5.
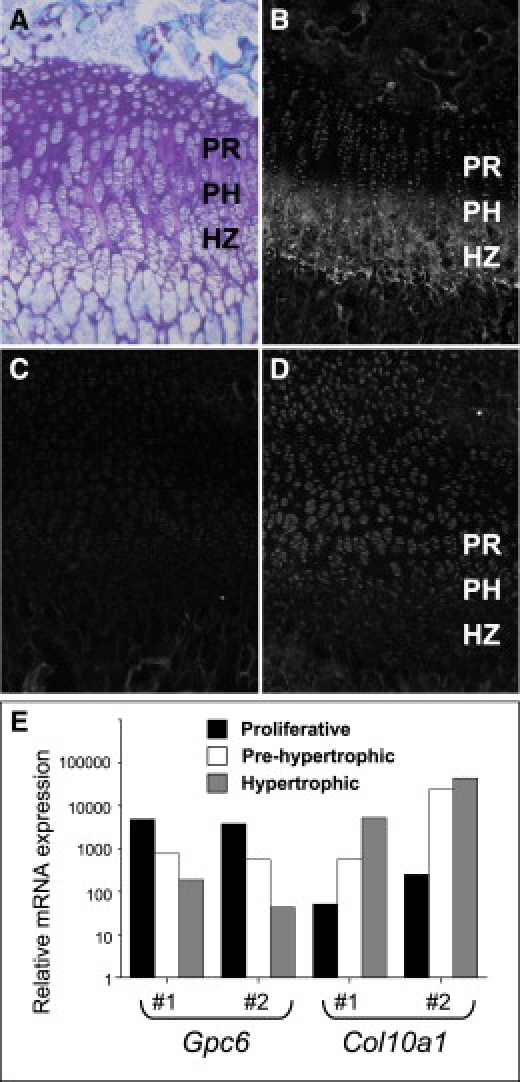
Analysis of GPC6 Protein and mRNA in the Mouse Growth Plate
(A) Toluidine-blue-stained section of growth-plate cartilage from a 2-week-old mouse, showing the organization of the chondrocytes in the proliferative (PR), pre-hypertrophic (PH), and hypertrophic zones (HZ). Immunostaining shows localization of collagen X (B) to the PH and HZ and a gradient of glypican 6 expression (D) from the PR and PH zones to little or no expression in the HZ. (C) Non-immune serum control. (E) Quantitative RT-PCR assay of the mRNA expression levels of Gpc6 and Col10a1 in microdissected cartilage zones from two biological replicates (#1 and #2). Gpc6 and Col10a1 expression was normalized to Atp5b mRNA by a comparative CT method.
Discussion
We report that omodysplasia is caused by recessive loss-of-function mutations in a glypican HSPG gene, GPC6. This finding confirms the role of glypicans in growth control during development, as suggested by the Drosophila mutants dally and dly9,27 and by the association of Simpson-Golabi-Behmel syndrome with GPC3 mutations.10 Furthermore, our data highlight the role of GPC6 in skeletal limb growth.
All mutations found in the individuals affected by omodysplasia predict absence of a functional protein. The relative abundance of wild-type and mutant mRNA in fibroblasts of heterozygous carriers in families 2 and 4 was concordant with the mutant mRNA's being subject to NMD. When escaping NMD and translated, all mutations are expected to disrupt the three-dimensional protein structure and often to abolish multiple highly preserved cysteine residues.28 All predicted mutant proteins would be truncated and thereby lose both the GPI and the HS binding sites (Figure 4), essential for the putative functions of GPC6.28,29 We show that omodysplasia can be caused by both point mutations, leading to stop codon mutations, and/or larger genomic rearrangements akin to genomic disorders30; these rearrangements do not appear to be mediated by low copy repeats because none of these were found at the deletion/duplication junctions or in general within the GPC6 gene. The only other known human disorder caused by mutations in a glypican gene is Simpson-Golabi-Behmel syndrome; in this disorder, loss-of-function mutations have been found in GPC3, and large deletions are responsible for a significant proportion of cases.10,31,32 Similarly to GPC3 rearrangements, GPC6 mutations are found in the entire coding region without any mutational hotspot and include one or more exons. It is noteworthy that genomic rearrangements are frequent in both glypican genes, perhaps because they cover large genomic regions.
Because omodysplasia results from recessive loss-of-function mutations in GPC6, hypomorphic or dominant mutations in the same gene might result in other skeletal phenotypes. Very recently, GPC5 haploinsufficiency has been hypothesized as the molecular cause of upper limb anomalies and growth retardation in 13q deletion syndrome because of its expression in the developing limb bud.33 GPC5 is colocalized with GPC6 on 13q31.2–q31.3, and the two genes are clustered, similarly to GPC3 and GPC4 on chromosome X, suggesting that these members of the glypican family share an evolutionary relationship2 that might reflect a common function.16,34 However, given that intact GPC5 does not compensate for loss of GPC6 in omodysplasia patients and that Gpc5 expression in the mouse growth plate is not significant in comparison to that of other glypicans (Figure S3), their functional relationship is not supported by our data.
GPC6 is one of the six members of the GPI-linked glypican subfamily of heparan sulfate proteoglycans (HSPGs),16,28,29 widely expressed during vertebrate development.29 As for other HSPGs,1 it has been proposed that glypican genes in Drosophila share partially redundant functions.9 GPC4 is the glypican most closely related to GPC6, with which it is 64% identical.16,28,29 Despite strong similarity and coexpression in the growth plate (Figure S3), there appears to be no complementation between these two molecules because mutations in GPC6 are sufficient to cause omodysplasia.
The skeletal phenotype was homogeneous in our patients because only typical patients were selected for the study. No genotype-phenotype correlation could be found between mutations in specific domains and extra-skeletal manifestations. Studies in mouse embryos showed that GPC6 expression is highly distinctive in dental mesenchyme, metanephric cap mesenchyme, intestinal mesenchyme, and blood vessels (dorsal aorta), suggesting a specific function in the development of these organs.29 However, a dysfunction in one of these tissues would not directly explain the extra-skeletal manifestations, such as cryptorchidism or congenital heart defects, that are reported in omodysplasia. In the central nervous system, GPC6 colocalizes with GPC4 particularly in the ventricular zones,29 but it is expressed at a much lower level than GPC4.35 Mental retardation is not a constant feature of omodysplasia and was present in only two patients of our series. The presence of mental retardation as a potential additional effect of consanguinity seems to be excluded because only one of these two patients was born from consanguineous parents, whereas the other one was one of the two affected siblings of nonconsanguineous family 4.
Immunofluorescence on cartilage of the mouse growth plate showed that Gpc6 is expressed in the proliferative zone (Figure 5D). Quantitative PCR of RNA isolated from microdissected growth-plate cartilage confirmed that Gpc6 is expressed more than 50 times more in the proliferative zone than in the hypertrophic zone (Figure 5E). Although the mechanism by which GPC6 absence causes omodysplasia is unknown, these expression data correlate with the morphologic findings in the human omodysplasia growth plate, where the proliferative zone appears expanded compared to controls, as if a functional deficiency in these physeal cells were ineffectively compensated by an increased number of small chondrocytes.36 The differential expression of Gpc6 mRNA in the mouse growth plate was not observed with other glypicans (Figure S3), suggesting a specific, nonredundant functional role for Gpc6 in proliferative chondrocytes. Although animal models for GPC6 functional deficiency are lacking, the omodysplasia phenotype, together with the expression data reported above, suggest that GPC6 plays a role in endochondral ossification and long-bone elongation.
HSPGs are essential for regulation of Indian hedgehog (IHH [MIM 600726]), fibroblast growth factor, and bone morphogenetic protein and for Wnt signaling, all key players in endochondral ossification.8 GPC6 is critical to modulating the response of the growth plate to thyroid hormones in mice37, and similar to the growth plates of patients with omodysplasia36, growth plates of mice harboring homozygous hypomorphic alleles of Ext1 (a HS-polymerizing enzyme) show broadening of the proliferative zone as a result of altered diffusion of the Ihh gradient.38 Although these data do not provide the mechanism of action of glypicans in the growth plate, they show a morphologic correlation between IHH signaling and omodysplasia physeal abnormalities. At the cell surface, glypicans promote the association of growth factors with their receptors.34 Once cleaved by Notum and released in the extracellular matrix, GPC6 core protein may be involved in further modulation of signaling molecules.39 We postulate that the GPC6 mutations in our patients abrogate the function of this HSPG in the growth plate and cause altered growth-factor signaling and morphogen gradients leading to failure of proliferative chondrocyte terminal differentiation and long-bone growth retardation.
Supplemental Data
Supplemental Data include three figures and four tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
Ensembl, http://www.ensembl.org/index.html
UCSC Genome Browser, http://genome.cse.ucsc.edu/index.html
ExPASy, http://www.expasy.org/uniprot/
Database of Genomic Variants, http://projects.tcag.ca/variation/cgi-bin/gbrowse/hg17/ and
Human Genome Variation Society, http://www.hgvs.org/
Centre d'Etude du Polymorphisme Humain (CEPH), http://www.cephb.fr/fr/cephdb/browser.php
Human Genome Variation Society recommendations, http://www.hgvs.org/mutnomen/
LimmaGUI, http://bioinf.wehi.edu.au/limma/
Acknowledgments
We are grateful to the affected individuals and their families for their cooperation. We thank Carole Chiesa for excellent technical assistance, Trevor Cameron for cartilage RNA isolation, and Peter Farlie for help with immunohistochemistry. This work was supported by the Swiss National Research Foundation, grant no. 320000-116506, to L.B. and by an Australian Research Council Discovery grant to J.B.
References
- 1.Kirkpatrick C.A., Knox S.M., Staatz W.D., Fox B., Lercher D.M., Selleck S.B. The function of a Drosophila glypican does not depend entirely on heparan sulfate modification. Dev. Biol. 2006;300:570–582. doi: 10.1016/j.ydbio.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Filmus J. Glypicans in growth control and cancer. Glycobiology. 2001;11:19R–23R. doi: 10.1093/glycob/11.3.19r. [DOI] [PubMed] [Google Scholar]
- 3.Nakato H., Kimata K. Heparan sulfate fine structure and specificity of proteoglycan functions. Biochim. Biophys. Acta. 2002;1573:312–318. doi: 10.1016/s0304-4165(02)00398-7. [DOI] [PubMed] [Google Scholar]
- 4.Filmus J., Selleck S.B. Glypicans: proteoglycans with a surprise. J. Clin. Invest. 2001;108:497–501. doi: 10.1172/JCI13712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filmus J. The contribution of in vivo manipulation of gene expression to the understanding of the function of glypicans. Glycoconj. J. 2002;19:319–323. doi: 10.1023/A:1025312819804. [DOI] [PubMed] [Google Scholar]
- 6.Fransson L.A. Glypicans. Int. J. Biochem. Cell Biol. 2003;35:125–129. doi: 10.1016/s1357-2725(02)00095-x. [DOI] [PubMed] [Google Scholar]
- 7.Hufnagel L., Kreuger J., Cohen S.M., Shraiman B.I. On the role of glypicans in the process of morphogen gradient formation. Dev. Biol. 2006;300:512–522. doi: 10.1016/j.ydbio.2006.08.076. [DOI] [PubMed] [Google Scholar]
- 8.Bishop J.R., Schuksz M., Esko J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 9.Nakato H., Fox B., Selleck S.B. Dally, a Drosophila member of the glypican family of integral membrane proteoglycans, affects cell cycle progression and morphogenesis via a Cyclin A-mediated process. J. Cell Sci. 2002;115:123–130. doi: 10.1242/jcs.115.1.123. [DOI] [PubMed] [Google Scholar]
- 10.Pilia G., Hughes-Benzie R.M., MacKenzie A., Baybayan P., Chen E.Y., Huber R., Neri G., Cao A., Forabosco A., Schlessinger D. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat. Genet. 1996;12:241–247. doi: 10.1038/ng0396-241. [DOI] [PubMed] [Google Scholar]
- 11.Veugelers M., Vermeesch J., Watanabe K., Yamaguchi Y., Marynen P., David G. GPC4, the gene for human K-glypican, flanks GPC3 on xq26: deletion of the GPC3–GPC4 gene cluster in one family with Simpson-Golabi-Behmel syndrome. Genomics. 1998;53:1–11. doi: 10.1006/geno.1998.5465. [DOI] [PubMed] [Google Scholar]
- 12.Maroteaux P., Sauvegrain J., Chrispin A., Farriaux J.P. Omodysplasia. Am. J. Med. Genet. 1989;32:371–375. doi: 10.1002/ajmg.1320320321. [DOI] [PubMed] [Google Scholar]
- 13.Borochowitz Z., Barak M., Hershkowitz S. Nosology of omodysplasia. Am. J. Med. Genet. 1995;58:377–378. doi: 10.1002/ajmg.1320580416. [DOI] [PubMed] [Google Scholar]
- 14.Albano L.M., Oliveira L.A., Bertola D.R., Mazzu J.F., Kim C.A. Omodysplasia: The first reported Brazilian case. Clinics. 2007;62:531–534. doi: 10.1590/s1807-59322007000400023. [DOI] [PubMed] [Google Scholar]
- 15.Elcioglu N.H., Gustavson K.H., Wilkie A.O., Yuksel-Apak M., Spranger J.W. Recessive omodysplasia: Five new cases and review of the literature. Pediatr. Radiol. 2004;34:75–82. doi: 10.1007/s00247-003-1064-9. [DOI] [PubMed] [Google Scholar]
- 16.Paine-Saunders S., Viviano B.L., Saunders S. GPC6, a novel member of the glypican gene family, encodes a product structurally related to GPC4 and is colocalized with GPC5 on human chromosome 13. Genomics. 1999;57:455–458. doi: 10.1006/geno.1999.5793. [DOI] [PubMed] [Google Scholar]
- 17.Baxova A., Maroteaux P., Barosova J., Netriova I. Parental consanguinity in two sibs with omodysplasia. Am. J. Med. Genet. 1994;49:263–265. doi: 10.1002/ajmg.1320490303. [DOI] [PubMed] [Google Scholar]
- 18.Tan T.Y., McGillivray G., Kornman L., Fink A.M., Superti-Furga A., Bonafe L., Francis D.I., Savarirayan R. Autosomal recessive omodysplasia: Early prenatal diagnosis and a possible clue to the gene location. Am. J. Med. Genet. A. 2005;135:324–327. doi: 10.1002/ajmg.a.30754. [DOI] [PubMed] [Google Scholar]
- 19.Di Luca B.J., Mitchell A. Anaesthesia in a child with autosomal recessive omodysplasia. Anaesth. Intensive Care. 2001;29:71–73. doi: 10.1177/0310057X0102900115. [DOI] [PubMed] [Google Scholar]
- 20.Borochowitz Z., Barak M., Hershkowitz S. Familial congenital micromelic dysplasia with dislocation of radius and distinct face: A new skeletal dysplasia syndrome. Am. J. Med. Genet. 1991;39:91–96. doi: 10.1002/ajmg.1320390120. [DOI] [PubMed] [Google Scholar]
- 21.Furniss D., Critchley P., Giele H., Wilkie A.O. Nonsense-mediated decay and the molecular pathogenesis of mutations in SALL1 and GLI3. Am. J. Med. Genet. A. 2007;143A:3150–3160. doi: 10.1002/ajmg.a.32097. [DOI] [PubMed] [Google Scholar]
- 22.Charbonnier F., Raux G., Wang Q., Drouot N., Cordier F., Limacher J.M., Saurin J.C., Puisieux A., Olschwang S., Frebourg T. Detection of exon deletions and duplications of the mismatch repair genes in hereditary nonpolyposis colorectal cancer families using multiplex polymerase chain reaction of short fluorescent fragments. Cancer Res. 2000;60:2760–2763. [PubMed] [Google Scholar]
- 23.Charbonnier F., Olschwang S., Wang Q., Boisson C., Martin C., Buisine M.P., Puisieux A., Frebourg T. MSH2 in contrast to MLH1 and MSH6 is frequently inactivated by exonic and promoter rearrangements in hereditary nonpolyposis colorectal cancer. Cancer Res. 2002;62:848–853. [PubMed] [Google Scholar]
- 24.Bendavid C., Dubourg C., Gicquel I., Pasquier L., Saugier-Veber P., Durou M.R., Jaillard S., Frebourg T., Haddad B.R., Henry C. Molecular evaluation of foetuses with holoprosencephaly shows high incidence of microdeletions in the HPE genes. Hum. Genet. 2006;119:1–8. doi: 10.1007/s00439-005-0097-6. [DOI] [PubMed] [Google Scholar]
- 25.Niel F., Martin J., Dastot-Le Moal F., Costes B., Boissier B., Delattre V., Goossens M., Girodon E. Rapid detection of CFTR gene rearrangements impacts on genetic counselling in cystic fibrosis. J. Med. Genet. 2004;41:e118. doi: 10.1136/jmg.2004.022400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Little C.B., Mittaz L., Belluoccio D., Rogerson F.M., Campbell I.K., Meeker C.T., Bateman J.F., Pritchard M.A., Fosang A.J. ADAMTS-1-knockout mice do not exhibit abnormalities in aggrecan turnover in vitro or in vivo. Arthritis Rheum. 2005;52:1461–1472. doi: 10.1002/art.21022. [DOI] [PubMed] [Google Scholar]
- 27.Khare N., Baumgartner S. Dally-like protein, a new Drosophila glypican with expression overlapping with wingless. Mech. Dev. 2000;99:199–202. doi: 10.1016/s0925-4773(00)00502-5. [DOI] [PubMed] [Google Scholar]
- 28.Filmus J., Capurro M., Rast J. Glypicans. Genome Biol. 2008;9:224. doi: 10.1186/gb-2008-9-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veugelers M., De Cat B., Ceulemans H., Bruystens A.M., Coomans C., Durr J., Vermeesch J., Marynen P., David G. Glypican-6, a new member of the glypican family of cell surface heparan sulfate proteoglycans. J. Biol. Chem. 1999;274:26968–26977. doi: 10.1074/jbc.274.38.26968. [DOI] [PubMed] [Google Scholar]
- 30.Gu W., Zhang F., Lupski J.R. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veugelers M., Cat B.D., Muyldermans S.Y., Reekmans G., Delande N., Frints S., Legius E., Fryns J.P., Schrander-Stumpel C., Weidle B. Mutational analysis of the GPC3/GPC4 glypican gene cluster on Xq26 in patients with Simpson-Golabi-Behmel syndrome: Identification of loss-of-function mutations in the GPC3 gene. Hum. Mol. Genet. 2000;9:1321–1328. doi: 10.1093/hmg/9.9.1321. [DOI] [PubMed] [Google Scholar]
- 32.Sakazume S., Okamoto N., Yamamoto T., Kurosawa K., Numabe H., Ohashi Y., Kako Y., Nagai T., Ohashi H. GPC3 mutations in seven patients with Simpson-Golabi-Behmel syndrome. Am. J. Med. Genet. A. 2007;143A:1703–1707. doi: 10.1002/ajmg.a.31822. [DOI] [PubMed] [Google Scholar]
- 33.Quelin C., Bendavid C., Dubourg C., de la Rochebrochard C., Lucas J., Henry C., Jaillard S., Loget P., Loeuillet L., Lacombe D. Twelve new patients with 13q deletion syndrome: Genotype-phenotype analyses in progress. Eur. J. Med. Genet. 2009;52:41–46. doi: 10.1016/j.ejmg.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 34.De Cat B., David G. Developmental roles of the glypicans. Semin. Cell Dev. Biol. 2001;12:117–125. doi: 10.1006/scdb.2000.0240. [DOI] [PubMed] [Google Scholar]
- 35.Ford-Perriss M., Turner K., Guimond S., Apedaile A., Haubeck H.D., Turnbull J., Murphy M. Localisation of specific heparan sulfate proteoglycans during the proliferative phase of brain development. Dev. Dyn. 2003;227:170–184. doi: 10.1002/dvdy.10298. [DOI] [PubMed] [Google Scholar]
- 36.Borochowitz Z., Sabo E., Misselevitch I., Boss J.H. Autosomal-recessive omodysplasia: Prenatal diagnosis and histomorphometric assessment of the physeal plates of the long bones. Am. J. Med. Genet. 1998;76:238–244. doi: 10.1002/(sici)1096-8628(19980319)76:3<238::aid-ajmg7>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 37.Bassett J.H., Swinhoe R., Chassande O., Samarut J., Williams G.R. Thyroid hormone regulates heparan sulfate proteoglycan expression in the growth plate. Endocrinology. 2006;147:295–305. doi: 10.1210/en.2005-0485. [DOI] [PubMed] [Google Scholar]
- 38.Koziel L., Kunath M., Kelly O.G., Vortkamp A. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev. Cell. 2004;6:801–813. doi: 10.1016/j.devcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Traister A., Shi W., Filmus J. Mammalian Notum induces the release of glypicans and other GPI-anchored proteins from the cell surface. Biochem. J. 2008;410:503–511. doi: 10.1042/BJ20070511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.