

Abstract
Mitochondrial complex I deficiency is the most prevalent and least understood disorder of the oxidative phosphorylation system. The genetic cause of many cases of isolated complex I deficiency is unknown because of insufficient understanding of the complex I assembly process and the factors involved. We performed homozygosity mapping and gene sequencing to identify the genetic defect in five complex I-deficient patients from three different families. All patients harbored mutations in the NDUFAF3 (C3ORF60) gene, of which the pathogenic nature was assessed by NDUFAF3-GFP baculovirus complementation in fibroblasts. We found that NDUFAF3 is a genuine mitochondrial complex I assembly protein that interacts with complex I subunits. Furthermore, we show that NDUFAF3 tightly interacts with NDUFAF4 (C6ORF66), a protein previously implicated in complex I deficiency. Additional gene conservation analysis links NDUFAF3 to bacterial-membrane-insertion gene cluster SecF/SecD/YajC and to C8ORF38, also implicated in complex I deficiency. These data not only show that NDUFAF3 mutations cause complex I deficiency but also relate different complex I disease genes by the close cooperation of their encoded proteins during the assembly process.
Introduction
Disorders of the oxidative phosphorylation (OXPHOS) system have an incidence of approximately 1:5000 living births, frequently resulting in severe multisystem disease with a consequence of early childhood death.1–3 The molecular cause is improper function and/or assembly of one or more of the five multiprotein enzyme complexes of the OXPHOS system.
The most prevalent OXPHOS disorder is isolated complex I deficiency (MIM 252010). So far, sequencing efforts have attributed about half of the cases to mutations in one of the seven mitochondrial and 38 nuclear genes that encode the structural components of complex I. Frequently, these mutations disturb complex I assembly, an intricate 45-component puzzle.4 At present, we know that complex I assembly involves the formation of multiple assembly intermediates, presumably starting with several highly conserved subunits (NDUFS2, NDUFS3, and NDUFS7).4–9 This de novo assembly is most likely accompanied by subunit exchange to maintain complex I integrity.7 In addition, aided by recent developments in bioinformatics and genetics, the number of putative complex I assembly proteins has grown considerably. Five of these have been implicated in complex I deficiency (NDUFAF1, NDUFAF2, C6ORF66, C8ORF38, and C20ORF7),10–14 and an additional three have been shown to be required for complex I assembly (Ecsit, AIF, and Ind1).15–17
Most likely, many more nonstructural proteins required for proper complex I function, assembly, and stability await discovery. This is illustrated by the fact that only half of the cases of isolated complex I deficiency can be genetically explained.1–3 In a recent paper, Pagliarini and colleagues have used comparative genomics to predict proteins involved in complex I biology and disease, ultimately verified by RNA interference for several candidates and the discovery of two complex I-deficient patients that harbor mutations in candidate gene C8ORF38 (MIM 612392).13 One of the candidate genes picked up in this study is C3ORF60, hereafter referred to as NDUFAF3.
Here, we describe five complex I-deficient patients with mutations in the NDUFAF3 gene, and we perform a detailed study of the requirement of the NDUFAF3 protein for complex I assembly. We show cooperation between NDUFAF3 and previously described chaperone NDUFAF4 (C6ORF66) and reveal gene-order conservation with C8ORF38 and genes involved in the Sec membrane-insertion machinery in bacteria. The discovery of cooperation between proteins encoded by different complex I disease genes is an important step forward in the understanding of the molecular basis of complex I assembly defects.
Subjects and Methods
Case Reports
Family I consisted of consanguineous parents of Muslim origin and their five children. Their second (I-1), third (I-2), and fifth (I-3) children, two females and one male, were born at 34–38 weeks of gestation, and their birth weights were 1490–2560 g. The three newborns presented similarly with severe lactic acidosis (peak level 27 mM, n < 2.2) at 1–3 days of life, which persisted and led to their death at three months of age. Throughout their life, increased muscle tone developed but physical examination was otherwise normal. Specifically, the patients followed moving objects, smiled socially, and had normal brain MRI, EEG, abdominal ultrasound, and echocardiogram results. Treatment with dichloroacetate, carnitine, thiamine, and CoQ10 did not affect the course of the disease.
The patient from the second family (II-1) was a male, the fifth child born to first cousins of Arab-Muslim origin. His parents and four older siblings were healthy. He was born at term and had a birth weight of 2900 g. At three weeks of age, he became hypoactive and sucked poorly. Physical examination revealed macrocephaly (head circumference +3 SD), a weak cry, wide anterior fontanelle, and axial hypotonia. Generalized tonic movements were intermittently seen, and fundoscopy revealed bilateral pallor of the optic disc. Plasma lactate was 5.4 mM, and CSF lactate was 6 mM (n < 1.8). The EEG recording showed a burst suppression pattern. At three months of age, there was no eye contact and marked axial hypotonia with brisk tendon reflexes and a lack of sucking were observed. Breathing was irregular, and the patient was mechanically ventilated and fed via a nasogastric tube until his death at four months of age. Muscle biopsy was performed at two months of age.
The patient from the third family (III-1) was a daughter of unrelated parents of Jewish origin. She was admitted at three months of age because of myoclonic seizure disorder. A brain MRI revealed diffuse brain leukomalacia, and an abdominal ultrasound disclosed left hydroureter and hydronephrosis. Skin fibroblasts showed mitochondrial complex I deficiency. She had respiratory failure and was readmitted to the hospital two more times. She died at six months of age.
Enzyme Measurements
For enzymatic assays, mitochondria were isolated from muscle and fibroblasts as described.18,19 Mitochondrial isolation and measurement of rotenone-sensitive NADH-ubiquinone reductase activity were performed as previously described.18,19 Complex I function was assessed by three assays: NADH-ferricyanide reductase, rotenone-sensitive NADH-ubiquinone reductase, and rotenone-sensitive NADH-cytochrome c reductase (complexes I and III). Activities were normalized to citrate synthase, a mitochondrial matrix enzyme20 (patients of families I and II), or to cytochrome c oxidase (patient III-1).
Homozygosity Mapping and Sequencing
All experiments involving DNA of the patients, their relatives, healthy controls, and patient cells were approved by the institutional review boards of the participating centers. Homozygosity mapping of DNA was performed with the Affymetrix Human Mapping 50K Array Xba240 for patients I-1 and I-2 of family 1 and with the GeneChip Human Mapping 250K Nsp Array for family 2, as previously described.21 The NDUFAF3 gene was amplified from genomic DNA in five exons by PCR (primer sequences available upon request). For exon 1, all three alternative spliced exons were analyzed. Fragments included both DNA sequences of the individual exons, the splice donor and splice acceptor sites. The amplimers thus obtained were subjected to DNA sequence analysis as described by Hoefs et al.22
Modeling
Homology modeling was performed with the use of the NMR structure of Rpa2829 protein from Rhodopseudomonas palustris as a template (Protein Data Bank [PDB] file 2FVT, 30% sequence identity). The mitochondrial target signal at the N terminus and the 11 C-terminal residues could not be modeled. A model of the aligned residues was built with the WHAT IF web server for modeling and with the WHAT IF and Yasara Twinset for model optimization and analysis.23 Default parameters were used during the entire modeling and model-optimization procedures.
Complementation
We obtained the NDUFAF3 isoform A open reading frame (ORF) sequence (GI:41327780, without stop codon) flanked by Gateway ATTB sites (Invitrogen) by PCR and cloned this into pDONR201 by using the Gateway BP Clonase II enzyme mix (Invitrogen) to generate the pEntry vectors. GFP-tagged COX8 leader was cloned as described previously.22 These constructs were then used to make baculovirus and perform complementation as described previously.22
RNA Interference
For transfection, HeLa cells were plated in 2 ml of Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) (without antibiotics) in 6-well plates with a cell density of 2.0 × 105 cells per well. The next day, cells were transfected with siRNA duplex in the presence of oligofectamine (7.5 μl) (Invitrogen) and opti-MEM (Invitrogen), for a final concentration of 100 nM siRNA in a total volume of 2.4 ml per well. Control was cyclophilin B (Dharmacon), NDUFAF3 #1 antisense strand: 5′-AUGUAAGUGAAGUCCCUCC dTdT-3′, #2 antisense strand: 5′-AGGAAGUUGAAGGUGGCAC dTdT-3′; NDUFAF4 #1 antisense strand: 5′- UGGAUAGAGACUAAUCUGC dTdT-3′, #2 antisense strand: 5′-AUCUUUGGAAUCAACAUAC dTdT-3′. Cells were incubated at 37°C in a CO2 incubator for 48 hr, after which the cells were transferred to new wells. After 24 hr, these cells were transfected for a second time and incubated for 24 hr before further analysis.
Generation of Inducible Cell Lines and Cell Culture
NDUFS3 (GI:4758787), NDUFAF3 (GI:41327780), and NDUFAF4 (GI:215599255) ORF sequences (without stop codon) flanked by Gateway AttB sites (Invitrogen) were created by PCR, in accordance with the manufacturer's instruction, and cloned into pDONR201 with the use of Gateway BP Clonase II enzyme mix (Invitrogen). Gateway tandem affinity purification (TAP) and green fluorescent protein (GFP) destination vectors are available in our lab (previously described in 5). For inducible NDUFS3-TAP and -GFP, NDUFAF3-TAP and -GFP, and NDUFAF4-TAP and -GFP vectors, the pDONR201 vector containing NDUFS3, NDUFAF3, and NDUFAF4 inserts was recombined with the TAP and GFP destination vectors via the Gateway LR Clonase II Enzyme Mix (Invitrogen). All constructs were transfected into Flp-In T-REx293 cells (Invitrogen) with SuperFect Transfection Reagent (QIAGEN), in accordance with the manufacturer's protocols. HeLa and Human Embryonic Kidney (HEK) 293 cells were cultured in DMEM (Biowhitaker) supplemented with 10% fetal calf serum (v/v) and 1% penicillin and streptomycin (v/v) (GIBCO). The inducible cell lines were treated with 1 μg/ml doxycycline (Sigma Aldrich) for 24 hr for expression of the transgene. For the chloramphenicol-inhibition experiment, a final concentration of 50 μg/ml of chloramphenicol (Sigma Aldrich) was added to the culture medium for 3 days. On the final day, 1 μg/ml doxycycline was added for 24 hr for induction of the transgenes before cell harvest. Patient skin fibroblasts were cultured in medium 199 supplemented with 10% fetal calf serum, bicarbonate, and antibiotics. The cells were cultured until confluency.
Localization Studies
For confocal imaging, HEK293 cells expressing inducible NDUFAF3-GFP were cultured in a Wilco dish (Intracel, Royston, UK), washed with phosphate-buffered saline (PBS), and incubated with 1 μM Mitotracker Red (Invitrogen) for 15 min and with 10 μM Hoechst 3342 (Invitrogen) for 30 min, both at 37°C. Before imaging, the culture medium was replaced by a colorless HEPES-Tris (HT) solution (132 mM NaCl, 4.2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5.5 mM D-glucose, and 10 mM HEPES, pH 7.4) and fluorescence images were taken on a ZEISS LSM510 Meta confocal microscope (Carl Zeiss). Images were acquired at a rate of 10 Hz with the use of a ×63 oil-immersion objective (N.A. 1.4; Carl Zeiss). Zoom factor 2 and pinhole settings were selected for the attainment of an optical section thickness of < 1 μm. Measurements were performed at 20°C in the dark. Confocal images of GFP and MitoTracker Red fluorescence were simultaneously collected with the use of an argon laser (laser power 1%) with a 488 nm dichroic mirror and a 500–530 nm band-pass barrier filter in combination with a helium-neon (HeNe) 1 laser (laser power 43%) with a 543 nm dichroic mirror and a 560 nm long-pass filter. With the multitrack setting used, Hoechst fluorescence was subsequently imaged with the use of a 405 nm diode laser (laser power 10%) and a 420–480 band-pass barrier filter. Image processing and analysis were performed with Image Pro Plus 5.1 (Media Cybernetics, Bethesda, MD, USA). For biochemical fractionation, HEK293 mitochondria were obtained by pottering as described previously.24 Mitochondria were sonicated on ice for 10 s at 10% amplitude (Branson Digital Sonifier, type W-250D) in isotonic buffer (0.25 M sucrose, 5 mM Tris-HCL, 0.2 mM EDTA, 1 mM PMSF, protease inhibitor mix [Amersham], pH 7.5), followed by a 30 min spindown (10.000 g, 4°C) to pellet unbroken mitochondria. The supernatant was submitted to ultracentrifugation at 100,000 g for 1 hr. The resulting supernatant contains the nonmembrane proteins. The pellet was subsequently resuspended in carbonate buffer (0.1 M Na2CO3, 1 mM PMSF, Protease Inhibitor Mix [Amersham], pH 11.5) for unfolding of the membrane structures and incubated for 30 min on ice. After a second round of ultracentrifugation at 100,000 g for 1 hr, the supernatant containing peripheral membrane proteins was separated from the integral membrane fraction.
Blue-Native- and SDS-PAGE Analysis and CI In-Gel Activity Assay
One-dimensional 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and 5%–15% blue-native (BN)-PAGE was performed as described previously.6,25 Lanes were loaded with 40 (SDS analysis) or 80 (BN analysis) μg of solubilized mitochondrial protein. After electrophoresis, gels were processed further for in-gel complex I activity assay, in-gel fluorescence detection, immunoblotting, or two-dimensional 10% SDS-PAGE as described by Nijtmans et al.25 For blotting, proteins were transferred to a PROTAN nitrocellulose membrane (Schleicher & Schuell).
Antibodies and ECL Detection
Affinity-purified NDUFAF3 antibody was manufactured by Eurogentec with the use of two peptides: no. 1, APRRGHRLSPADDELY; no. 2, RQRGIAVEVQDTPNAC. The NDUFAF4 antibody was kindly provided by H. Lorberboum-Galski, Jerusalem, Israël. The GFP antibody was kindly provided by Frank van Kuppeveld, Nijmegen, The Netherlands. Complex I subunit antibodies were NDUFS3 (Invitrogen), ND1 (a gift from Anne Lombes, Paris, France), NDUFS2 (a gift from Brian Robinson, Montreal, Canada), our NDUFA2 antibody,8 NDUFA5, NDUFA9, and NDUFS3 (Invitrogen). Other antibodies used were raised against SDHA, Core II, COX5A, ATPase α (all Invitrogen), cytochrome c, and mtHSP70 (both Abcam). Secondary antibodies that were used are peroxidase-conjugated anti-mouse or anti-rabbit IgGs (Invitrogen). Signal was generated with ECL Plus (Amersham Biosciences) and exposed to X-ray film (Kodak).
Tandem Affinity Purification and FT-MS Analysis
Tandem affinity purification (TAP) was performed according to the protocol for the InterPlay Mammalian TAP System (Stratagene). Proteins were eluted by incubation in 50 mM NH4HCO3 (pH 8.0) at 95°C for 5 min. Subsequent Fourier transform-mass spectrometry (FT-MS) analysis was performed with a nano-HPLC Agilent 1100 nanoflow system connected to a linear quadrupole ion trap-Fourier transform mass spectrometer (LTQ-FT, Thermo Electron). The eluate was applied to 10% SDS-PAGE for the removal of buffer components from the TAP procedure. Subsequently, the band containing the eluted proteins was excised from the gel, and proteins were in-gel reduced with 10 mM dithiotreitol and alkylated with 50 mM iodoacetamide before in-gel digestion with trypsin. Peptides were extracted from the gel and were purified and desalted with Stage tips.26 Peptide-identification experiments were performed with a nano-HPLC Agilent 1100 nanoflow system connected online to a linear ion trap-Fourier transform (LTQ-FT) (Thermo Electron). Liquid chromatography (LC) and MS settings are further explained in Table S1, available online. Peptides and proteins were identified with the use of the Mascot (Matrix Science) algorithm for search of a local version of the NCBInr database. First-ranked peptides were parsed from Mascot database search html files with MSQuant for the generation of unique first-ranked peptide lists.
Gene-Order-Conservation Analysis
To assess the gene-order conservation among bacteria, we used STRING27 in the “protein mode,” using as query the most similar bacterial protein to NDUFAF3: protein amb2521, from the alpha-proteobacterium Magnetospirillum magneticum.
Results
Clinical and Enzymatic Data
We identified five patients from three families that presented with a severe metabolic disorder. Shortly after birth, all affected children from family I presented with lactic acidemia. Besides the children's increased muscle tone, physical examination and radiological and electrophysiological studies were normal. Patient II-1 showed macrocephaly and severe muscle weakness and hypotonia as the most prominent clinical findings. Patient III-1 developed diffuse leukomalacia. Despite various interventions and supplementation, all patients deteriorated and died at 2–6 months of age. Enzymatic analysis of OXPHOS function in fibroblast and muscle revealed a severe isolated complex I deficiency in all investigated patients (Table 1 and Tables S2–S4).
Table 1.
Complex I Activities in Patient Fibroblast and Muscle Mitochondria
| Patient | Base Change | Amino Acid Change | Fibroblasta | Musclea |
|---|---|---|---|---|
| Control | - | - | 217 | 241 |
| I-1 | c.229 G→C | G77R | n.d.b | 23 (32%) |
| I-2 | c.229 G→C | G77R | 16 (39%) | 49 (40%) |
| I-3 | c.229 G→C | G77R | n.d.b | n.d.b |
| II-1 | c.365 G→C | R122P | 5 (18%) | 60 (26%) |
| III-1 | c.2 T→C; c.365 G→C | M1T; R122P | 9 (33%) | n.d.b |
NADH:ubiquinone oxidoreducatase activity in nmol/min/mg (percentage of control, normalized to citrate synthase).
Not determined.
Genetic Analysis and Modeling of NDUFAF3 and the G77R and R122P Substitutions
Multiple genomic stretches of successive homozygous SNPs, longer than 4 Mb, were present in the DNA of the patients from families 1 and 2. One of the regions shared in the patients of family 1 spanned 33.83 Mb (24.23–58.06 Mb, rs10510540– rs6445945) on chromosome 3. Patient II-1 of family 2 also had a large homozygous region on chromosome 3, spanning 26.6 Mb (45.98−72.59 Mb, rs3796376–rs11128275). The SNP haplotype was different for each family, excluding a common founder mutation. The genomic sequence of the four coding exons of NDUFAF3 was determined. The three patients of family 1 were homozygous for c.229 G→C resulting in G77R, the parents and the second twin were heterozygous for the mutation, and the older healthy daughter was normally homozygous. Patient II-1 was homozygous for c.365 G→C resulting in R122P. His parents and two siblings were heterozygous for the mutation, and two other siblings were normally homozygous. Patient III-1 was compound heterozygous for two mutations: c.2 T→C resulting in M1T and c.365 G→C resulting in R122P. Three healthy children in the same family were heterozygous for the c.365 G→C mutation. Each of the described mutations was absent in at least 210 ethnic control alleles. The M1T substitution disrupts the start codon. An overview of the c.229 G→C and c.365 G→C mutations and evolutionary conservation of the substituted amino acids is shown in Figures 1A and 1B, respectively.
Figure 1.
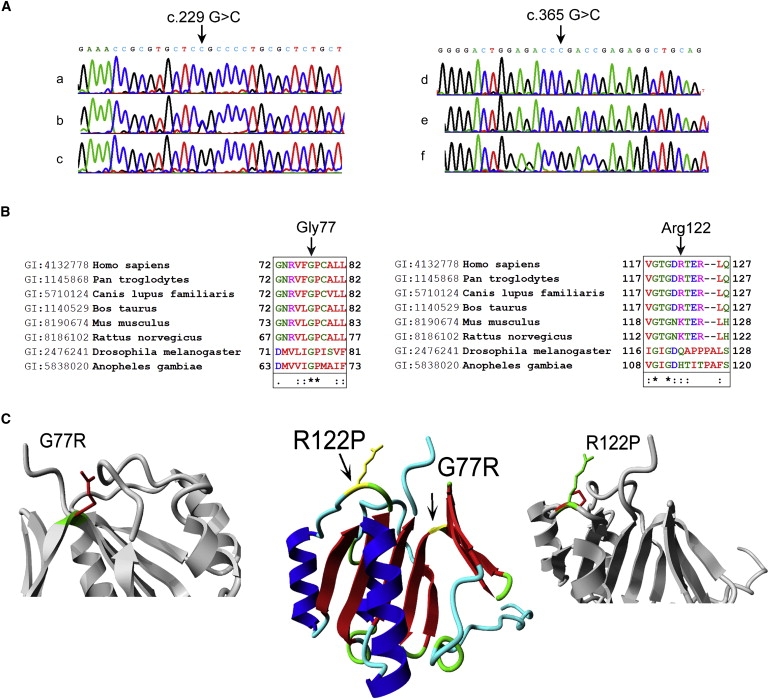
Genetic and Molecular Characterization of the c.229G→C and c.365 G→C Mutations
(A) DNA samples of (a) a patient from family 1, the c.229 G→C mutation indicated by an arrow, (b) an obligate heterozygote from family 1, (c) a control, (d) a patient from family 2, the c.365 G→C mutation indicated by an arrow, (e) an obligate heterozygote from family 2, and (f) a control.
(B) Conservation of the altered amino acids is shown via clustalW alignments. Asterisks (∗) indicate identical amino acids, colons (:) indicate conserved substitutions, and periods (.) indicate semiconserved substitutions.
(C) Model of NDUFAF3 and the two mutations found in the patients. Middle panel: NDUFAF3 is shown in ribbon visualization and colored by secondary structure element (red = beta strand, blue = alpha helix, green = turn, cyan = coil). The positions of the two mutations found in patients are indicated by the arrows and colored yellow. Side panels: the R122P and G77R mutations shown in more detail. The protein is gray. The side chains of both the wild-type residue (green) and the mutant residue (red) are shown. Both mutations result in modifications at the same side of the protein.
A three-dimensional model of NDUFAF3 was made for a better understanding of the structural consequences of the mutations. A BLAST search with the NDUFAF3 sequence against all proteins in the PDB resulted in a protein with unknown function (PDB code 2FVT) that shares 30% sequence identity over a stretch of 129 amino acids. This was used as a template for modeling of the NDUFAF3 protein (Figure 1C, positions of the mutations indicated). The R122P mutation is located at the C-terminal side of the protein and is predicted to change the local structure of the loop, which might influence interactions or the stability of the protein. Gly77 is conserved between the 2FVT template and the NDUFAF3 model and has backbone-torsion angles that are very unfavorable for other residue types (Figure S2). The G77R mutation is therefore energetically unfavorable and likely to disrupt the structure. The model of the NDUFAF3 structure exposes a large hydrophobic domain of about 600 squared Ångstrøms (Figure S3), which is commonly observed for protein-protein interactions.28,29 The homology model and all modeling details are available online.
Requirement of NDUFAF3 for Complex I Assembly and Activity
The specific complex I deficiency was confirmed by SDS- and BN-PAGE analysis for patient II-1 and patient I-2 fibroblasts (Figures 2A and 2B). Protein levels of subunits NDUFS3 and NDUFA9 were decreased (Figure 2A), and the amount and in-gel activity of complex I were severely impaired (Figure 2B, CI WB and CI IGA).
Figure 2.

Complementation of the Complex I Deficiency in Fibroblasts from Patients I-2 and II-1
NI: not infected. Cox8: infected with GFP-tagged COX8 leader sequence. AF3: NDUFAF3-GFP infected.
(A) SDS-PAGE immunodetections of GFP, NDUFS3, and NDUFA9.
(B) Complex I in-gel activity (CI IGA) and BN-PAGE immunodetections of complex I (ND1), complex V (ATPase α), complex III (core II), complex IV (COX5A), and complex II (SDHA).
(C) Enzymatic activities of complexes I (assayed as NADH-ubiquinone reductase), II, and IV after normalization to citrate synthase activity and untreated control (source data in Table S2).
(D) Two-dimensional BN-SDS immunodetections of NDUFS2 and ND1 in control and patient II-1 fibroblasts, before and after complementation (Compl.).
(E and F) RNA interference-mediated knockdowns of NDUFAF3 and NDUFAF4 in HeLa cells. N: not treated. M: mock transfected. #1, #2: targets 1 and 2. (E) SDS-PAGE immunodetection of NDUFAF3, NDUFAF4, complex I subunits NDUFS3 and ND1, and control CII-70 kDa, after each knockdown.
(F) BN-PAGE analysis of complex I in-gel activity (IGA) and immunodetection of complex I subunit NDUFA9, complex II-70 kDa, and complex III core II subunits.
The fibroblast cell lines were complemented with the use of a NDUFAF3-GFP-expressing baculovirus system. Figure 2A shows that the GFP-tagged COX8 leader (negative control) and NDUFAF3 proteins were present after infection and that the NDUFA9 protein level appears slightly increased after infection with NDUFAF3-GFP. BN analysis in Figure 2B shows the restoration of complex I amount and activity, which could be confirmed by enzymatic assay (Figure 2C and Table S2). Two-dimensional BN- and SDS-PAGE immunodetection (Figure 2D) reveals no accumulation of assembly intermediates for patient II-1 as assayed for peripheral (NDUFS2)- and membrane (ND1)-arm subunits of complex I. NDUFAF3-GFP baculovirus infection of a patient fibroblast cell line harboring the NDUFS7 (MIM 601825) V122M mutation30 did not restore complex I amount and in-gel activity, further supporting the specificity of the NDUFAF3 complementation (Figure S1).
To verify the requirement of NDUFAF3 for complex I assembly, we performed RNA interference in HeLa cells, using two targets (Figure 2E). In addition to the expected decrease of NDUFAF3 (Figure 2E) and complex I amount and activity (Figure 2F) observed, we observed a strong reduction of the NDUFAF4 protein (Figure 2E). NDUFAF4 (C6ORF66) is a previously described complex I assembly protein.12 To investigate a putative interdependence, we performed RNA interference for NDUFAF4, which also resulted in decrease in NDUFAF3 protein (Figure 2E) and complex I amount and activity (Figure 2F). As controls, complexes II and III remained unaffected by both knockdowns.
Mitochondrial Localization and Associations
The human NDUFAF3 gene encodes two isoforms, A and B, which share a “domain of unknown function” (DUF498). In addition, the longer A isoform harbors an in silico predicted N-terminal mitochondrial-targeting sequence of about 35 amino acids (MitoprotII, TargetP, and Predotar). Sequence data of the human NDUFAF3 variants and interspecies homology of the A isoform are summarized in Table S5. To verify the predicted mitochondrial targeting of isoform A, we performed confocal microscopy for analysis of GFP-tagged NDUFAF3 isoform A (Figure 3A) and subcellular fractionation experiments for analysis of the wild-type protein (Figure 3B). Both analyses demonstrate that NDUFAF3 is predominantly (if not exclusively) localized to the mitochondrion. In the submitochondrial fractionation (Figure 3B), NDUFAF3 remained in the pellet after carbonate extraction. This suggests a strong interaction with the mitochondrial membrane, although this protein has no predicted transmembrane helices. The localization appeared identical to that of the NDUFAF4 protein. As controls, integral membrane protein ND1 also remained in the pellet, whereas cytochrome c and complex I subunit NDUFS3 did not. MtHSP70 and NDUFS2 display partial membrane association.
Figure 3.

Mitochondrial Localization and High-Molecular-Weight Associations of NDUFAF3
(A) NDUFAF3-GFP, mitotracker, and Hoechst staining of NDUFAF3-GFP-inducible HEK293 Flp-in cells at different magnifications (a and b).
(B) SDS-PAGE immunodetection analysis after subcellular and mitochondrial fractionation of HEK293 cells. Cyto: cytoplasm. Mito: mitochondria. After sonication of purified mitochondria, the pellet (P) contains membrane structures and the supernatant (S) represents the soluble fraction. Carbonate extraction unfolds membrane structures, releasing peripherally associated proteins to the supernatant.
(C) Detection of NDUFS3-GFP (S3-GFP) and NDUFAF3-GFP (AF3-GFP) in native protein complexes via fluorscan of a 5%–15% BN-PAGE gel. Previously identified complex I intermediates are indicated as by Vogel et al.8
(D) Two-dimensional BN- and SDS-PAGE immunodetection of NDUFAF3 and complex I subunits NDUFS2, NDUFS3, and ND1.
(E) TAP identification of binding partners of NDUFAF3. SDS-PAGE immunodetections of proteins after NDUFS3-, NDUFAF3-, and NDUFAF4-TAP purifications indicated on the left. L: lysate. E: eluate. “−“: without doxycycline. “+”: with 1 μg/ml doxycycline for 24 hr.
The NDUFAF3 protein accurately comigrates with previously described complex I assembly intermediates 3, 4, and 5.8 (Figure 3C for NDUFAF3-GFP, Figure 3D for endogenous NDUFAF3). In addition, multiple bands are observed at the approximate size of intermediate 6 (Figure 3C). The same comigration is observed for NDUFAF4 (Figure 4C). To investigate putative interactions between NDUFAF3 and NDUFAF4, we constructed stable HEK293 Trex Flp-in clones containing TAP-tagged NDUFS3-, NDUFAF3-, and NDUFAF4-inducible constructs. Each protein was purified after 24 hr of expression. TAP of NDUFS3, NDUFAF3, and NDUFAF4 resulted in copurification of all three proteins, plus NDUFS2 (Figure 3E). Subunits NDUFA9, ND1, and NDUFA2 were not detected in the eluates, suggesting that NDUFAF3 and NDUFAF4 predominantly interact with complex I core subunits NDUFS2 and NDUFS3. To detect additional proteins, we analyzed the eluates by mass spectrometry (Table S6). In addition to the copurification observed for NDUFS2, NDUFS3, NDUFAF3, and NDUFAF4, this analysis revealed the copurification of NDUFA5 and NDUFS8.
Figure 4.
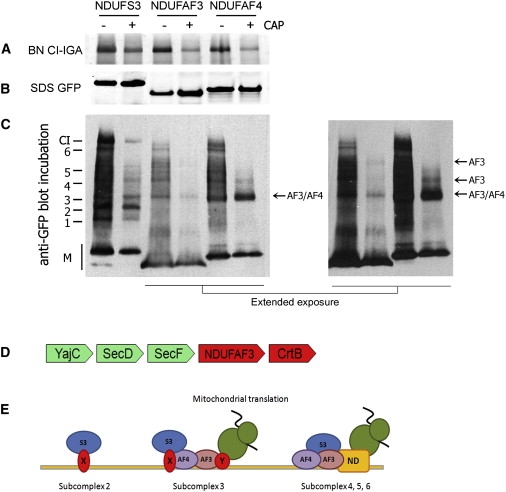
Inhibition of Translation and Proposed Role of NDUFAF3 during Complex I Assembly
HEK293 cells were induced for NDUFS3-, NDUFAF3-, and NDUFAF4-GFP expression with and without prior chloramphenicol treatment (± CAP) for 3 days.
(A) Complex I in-gel activity (CI-IGA) assay.
(B) SDS-PAGE fluorscan for GFP-tagged protein.
(C) BN-PAGE immunodetection with antibody to GFP. Previously described NDUFS3-containing intermediates are indicated on the left as by Vogel et al.8 M: monomer. Accumulation is indicated with arrows, referring to subcomplexes of NDUFAF3 (AF3) and of NDUFAF4 (AF4).
(D) The conserved gene cluster in alpha-proteobacteria that links orthologs of NDUFAF3 to the bacterial preprotein translocase complex SecD/SecF/YajC, involved in targeting proteins to the membrane, and to CrtB, a phytoene/squalene synthetase of which the human ortholog C8ORF38 has been implicated in complex I assembly.13 Green: proteins only observed in bacteria. Red: proteins observed in eukaryotes and bacteria.
(E) Tentative model for the role of NDUFAF3 and NDUFAF4 during early complex I assembly. Subcomplex 2 is loosely associated to the mitochondrial inner membrane, as demonstrated by Dieteren et al.,33 by an unknown factor (X). NDUFAF3 and NDUFAF4 are codependent, membrane localized, and present in subcomplex 3, which contains at least subunits NDUFS2 and NDUFS3, and possibly NDUFS8 and NDUFA5. This complex requires mitochondrial translation for progression to higher-molecular-weight subcomplexes. Without mitochondrial translation, NDUFAF3 no longer associates with subcomplex 3 and the complex, with NDUFAF4, accumulates. With mitochondrial translation, both proteins are present until subcomplex 6, close to finalization of holo-complex I. It is yet unknown which factors aid membrane insertion of mitochondrial translation products (indicated with Y). AF3: NDUFAF3. AF4: NDUFAF4. S3: subcomplex 3. X and Y: unknown factors. ND: ND subunits. Green structure: mitochondrial ribosome.
Role of NDUFAF3 in Complex I Assembly
Inhibition of mitochondrial translation disturbs complex I assembly because the mitochondrial DNA-encoded ND subunits of complex I are no longer formed and incorporated. Such inhibition can be achieved by culturing cells in the presence of chloramphenicol for >24 hr, which typically results in accumulation of NDUFS3-containing assembly intermediates 2 and 3 (see Vogel et al.8 and Figure 4C, NDUFS3-GFP + CAP). Given the tight relation among NDUFS3, NDUFAF3, and NDUFAF4, the effect of such inhibition on the incorporation of GFP-tagged NDUFAF3 and NDUFAF4 was investigated. Upon inhibition, complex I in-gel activity was decreased (Figure 4A), whereas the amount of GFP-tagged NDUFAF3 and NDUFAF4 protein remained similar (Figure 4B). Nevertheless, inhibition resulted in disturbed incorporation of NDUFAF3 (Figure 4C) and in accumulation of NDUFAF4 in a complex similar to subcomplex 3 (Figure 4C, arrow).
To obtain additional clues about the function of NDUFAF3, we performed gene-order-conservation analysis. Complex I has been lost six times in the evolution of the currently sequenced eukaryotic genomes.17 Profile-based homology searches with PSI-Blast confirm the observation that NDUFAF3 has also been lost from these lineages13 and is present in the complex I-containing lineages, supporting an interaction with complex I. We examined whether the bacterial orthologs of NDUFAF3 show gene-order conservation in the alpha-proteobacterial ancestors of the mitochondria, a reliable indicator of protein interaction.31 We detected significant gene-order conservation with the Squalene/phytoene synthase (CrtB) ortholog (COG1562, C8ORF38) (Figure 4D) that has been implicated in complex I assembly in man,13 supporting the role for NDUFAF3 in complex I assembly. Furthermore, we detected significant gene-order conservation with the SecF/SecD/YajC gene cluster. The gene cluster of NDUFAF3 with SecD/SecF/YajC is conserved among 20 genera in the current version of STRING. In bacteria, proteins of this cluster are located in the inner membrane, where they are involved in membrane-protein insertion and interact with YidC,32 the bacterial ancestor of the mitochondrial membrane protein OXA1. The link with CrtB is a bit less conserved, occurring in 13 genera, but still significant (likelihood of interaction SecD – NDUFAF3 = 0.8, likelihood of interaction CrtB – NDUFAF3 = 0.5).
Discussion
We describe five patients with a severe complex I deficiency presenting with lactic acidemia soon after birth. All children died before the age of six months. The deficiencies were caused by three different mutations in the NDUFAF3 gene. We show that NDUFAF3 is essential for human complex I assembly, in line with its predicted function based on correlated genomic occurrence with complex I proteins.13
With NDUFAF3, the sixth assembly protein implicated in complex I deficiency,10–14 we show cooperation of multiple assembly chaperones in similar assembly steps of complex I. The interaction among NDUFAF3, NDUFAF4, NDUFS2, NDUFS3, and, most likely, subunits NDUFS8 and NDUFA5 shows that NDUFAF3 and NDUFAF4 are bound to similar complex I substructures. In addition, NDUFAF3 and NDUFAF4 colocalize in the mitochondrial inner membrane, comigrate in at least three previously described assembly intermediates, and are codependent, as shown by RNA interference. Taken altogether, these data show that NDUFAF3 and NDUFAF4 cooperate from early to late stages of complex I assembly in association with at least the highly conserved subunits NDUFS2, NDUFS3, and NDUFS8, and accessory subunit NDUFA5 until step 6 in our assembly scheme.8
The NDUFAF3-NDUFAF4 interaction provides insights into the mechanism of early complex I assembly. The requirement of mitochondrial translation for NDUFAF3 to occur in high-molecular-weight associations supports a function related to mitochondrial translation or steps thereafter, such as early assembly of the mitochondrial DNA-encoded ND subunits of complex I. In contrast, the same inhibition causes NDUFAF4 to accumulate in a small nuclear-DNA-encoded intermediate similar to that observed for NDUFS3.8 One explanation for the combined results is that NDUFAF4, bound to NDUFAF3, recruits a nuclear-DNA-encoded early assembly intermediate that contains at least NDUFS2 and NDUFS3 at the mitochondrial inner membrane (Figure 4E). Without mitochondrial translation, NDUFAF3 can no longer efficiently associate to subcomplex 3, and an intermediate that consists of NDUFAF4 and at least NDUFS2 and NDUFS3 accumulates.
The predicted interaction of the ancestors of NDUFAF3 with the Sec membrane-translocation machinery in alpha-proteobacteria is exciting, because a Sec-like translocase has not yet been demonstrated for the mammalian OXPHOS system. It supports a role for NDUFAF3 in coupling mitochondrial translation to membrane insertion during assembly. In addition, the gene-order-conservation analysis links NDUFAF3 to C8ORF38, another assembly chaperone implicated in complex I deficiency.13
In conclusion, we show that mutations in NDUFAF3 cause complex I deficiency. Furthermore, characterization of NDUFAF3 demonstrates its role as a NDUFAF4-interacting complex I assembly protein and highlights a translation-dependent early assembly mechanism. Discovery of the involvement of multiple, disease-implicated chaperones in similar assembly stages constitutes an important step forward in the understanding of the molecular basis of complex I assembly defects.
Supplemental Data
Supplemental Data include three figures and six tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Modeling details, http://www.cmbi.ru.nl/∼hvensela/C3ORF60/
MSQuant, http://www.msquant.sourceforge.net
NCBInr database, http://www.ncbi.nlm.nih.gov
Online Medelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
STRING, http://string.embl.de
WHAT IF web server, http://swift.cmbi.ru.nl/
Acknowledgments
We thank Orly Elpeleg for critical evaluation of this manuscript. We are grateful to H. Lorberboum-Galski, B. Robinson, A. Lombes, and Frank van Kuppeveld for providing, respectively, the NDUFAF4 (C6ORF66), NDUFS2, ND1, and EGFP antibodies; to H. Swarts for generating GFP-tagged baculovirus, and to C. Dieteren for confocal imaging. A part of this work was supported by a grant from the Israeli Ministry of Health (A.S.), by the Research Council for Earth and Life Sciences (ALW) with the financial aid from the Netherlands Organization for Scientific Research (NWO) (R.V.), and by the European Community's Sixth Framework Programme for Research, Priority 1 “Life sciences, genomics and biotechnology for health,” contract number LSHM-CTY-2004-005260 (MITOCIRCLE) (S.H. and L.vd.H.). H.V. acknowledges support from the Netherlands Bioinformatics Centre (NBIC).
References
- 1.Smeitink J.A., Zeviani M., Turnbull D.M., Jacobs H.T. Mitochondrial medicine: A metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 2006;3:9–13. doi: 10.1016/j.cmet.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Smeitink J., van Den Heuvel L., DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001;2:342–352. doi: 10.1038/35072063. [DOI] [PubMed] [Google Scholar]
- 3.Thorburn D.R. Mitochondrial disorders: Prevalence, myths and advances. J. Inherit. Metab. Dis. 2004;27:349–362. doi: 10.1023/B:BOLI.0000031098.41409.55. [DOI] [PubMed] [Google Scholar]
- 4.Vogel R.O., Smeitink J.A., Nijtmans L.G. Human mitochondrial complex I assembly: A dynamic and versatile process. Biochim. Biophys. Acta. 2007;1767:1215–1227. doi: 10.1016/j.bbabio.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Antonicka H., Ogilvie I., Taivassalo T., Anitori R.P., Haller R.G., Vissing J., Kennaway N.G., Shoubridge E.A. Identification and characterization of a common set of complex I assembly intermediates in mitochondria from patients with complex I deficiency. J. Biol. Chem. 2003;278:43081–43088. doi: 10.1074/jbc.M304998200. [DOI] [PubMed] [Google Scholar]
- 6.Ugalde C., Vogel R., Huijbens R., van den Heuvel L.P., Smeitink J., Nijtmans L. Human mitochondrial complex I assembles through the combination of evolutionary conserved modules: A framework to interpret complex I deficiencies. Hum. Mol. Genet. 2004;13:2461–2472. doi: 10.1093/hmg/ddh262. [DOI] [PubMed] [Google Scholar]
- 7.Lazarou M., McKenzie M., Ohtake A., Thorburn D.R., Ryan M.T. Analysis of the assembly profiles for mitochondrial and nuclear encoded subunits into complex I. Mol. Cell. Biol. 2007;27:4228–4237. doi: 10.1128/MCB.00074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogel R.O., Dieteren C.E., van den Heuvel L.P., Willems P.H., Smeitink J.A., Koopman W.J., Nijtmans L.G. Identification of mitochondrial complex i assembly intermediates by tracing tagged NDUFS3 demonstrates the entry point of mitochondrial subunits. J. Biol. Chem. 2007;282:7582–7590. doi: 10.1074/jbc.M609410200. [DOI] [PubMed] [Google Scholar]
- 9.Lazarou M., Thorburn D.R., Ryan M.T., McKenzie M. Assembly of mitochondrial complex I and defects in disease. Biochim. Biophys. Acta. 2008;1793:78–88. doi: 10.1016/j.bbamcr.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 10.Dunning C.J., McKenzie M., Sugiana C., Lazarou M., Silke J., Connelly A., Fletcher J.M., Kirby D.M., Thorburn D.R., Ryan M.T. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J. 2007;26:3227–3237. doi: 10.1038/sj.emboj.7601748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogilvie I., Kennaway N.G., Shoubridge E.A. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Invest. 2005;115:2784–2792. doi: 10.1172/JCI26020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saada A., Edvardson S., Rapoport M., Shaag A., Amry K., Miller C., Lorberboum-Galski H., Elpeleg O. C6ORF66 is an assembly factor of mitochondrial complex I. Am. J. Hum. Genet. 2008;82:32–38. doi: 10.1016/j.ajhg.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugiana C., Pagliarini D.J., McKenzie M., Kirby D.M., Salemi R., Abu-Amero K.K., Dahl H.H., Hutchison W.M., Vascotto K.A., Smith S.M. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am. J. Hum. Genet. 2008;83:468–478. doi: 10.1016/j.ajhg.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogel R.O., Janssen R.J., van den Brand M.A., Dieteren C.E., Verkaart S., Koopman W.J., Willems P.H., Pluk W., van den Heuvel L.P., Smeitink J.A. Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev. 2007;21:615–624. doi: 10.1101/gad.408407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vahsen N., Cande C., Briere J.J., Benit P., Joza N., Larochette N., Mastroberardino P.G., Pequignot M.O., Casares N., Lazar V. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bych K., Kerscher S., Netz D.J., Pierik A.J., Zwicker K., Huynen M.A., Lill R., Brandt U., Balk J. The iron-sulphur protein Ind1 is required for effective complex I assembly. EMBO J. 2008;27:1736–1746. doi: 10.1038/emboj.2008.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saada A., Shaag A., Elpeleg O. mtDNA depletion myopathy: Elucidation of the tissue specificity in the mitochondrial thymidine kinase (TK2) deficiency. Mol. Genet. Metab. 2003;79:1–5. doi: 10.1016/s1096-7192(03)00063-5. [DOI] [PubMed] [Google Scholar]
- 19.Janssen A.J., Trijbels F.J., Sengers R.C., Smeitink J.A., van den Heuvel L.P., Wintjes L.T., Stoltenborg-Hogenkamp B.J., Rodenburg R.J. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin. Chem. 2007;53:729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- 20.Saada A., Bar-Meir M., Belaiche C., Miller C., Elpeleg O. Evaluation of enzymatic assays and compounds affecting ATP production in mitochondrial respiratory chain complex I deficiency. Anal. Biochem. 2004;335:66–72. doi: 10.1016/j.ab.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 21.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoefs S.J., Dieteren C.E., Distelmaier F., Janssen R.J., Epplen A., Swarts H.G., Forkink M., Rodenburg R.J., Nijtmans L.G., Willems P.H. NDUFA2 complex I mutation leads to Leigh disease. Am. J. Hum. Genet. 2008;82:1306–1315. doi: 10.1016/j.ajhg.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krieger E., Darden T., Nabuurs S.B., Finkelstein A., Vriend G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins. 2004;57:678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- 24.Vogel R.O., Janssen R.J., Ugalde C., Grovenstein M., Huijbens R.J., Visch H.J., van den Heuvel L.P., Willems P.H., Zeviani M., Smeitink J.A. Human mitochondrial complex I assembly is mediated by NDUFAF1. FEBS J. 2005;272:5317–5326. doi: 10.1111/j.1742-4658.2005.04928.x. [DOI] [PubMed] [Google Scholar]
- 25.Nijtmans L.G., Henderson N.S., Holt I.J. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods. 2002;26:327–334. doi: 10.1016/S1046-2023(02)00038-5. [DOI] [PubMed] [Google Scholar]
- 26.Rappsilber J., Ishihama Y., Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 27.Jensen L.J., Kuhn M., Stark M., Chaffron S., Creevey C., Muller J., Doerks T., Julien P., Roth A., Simonovic M. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones S., Thornton J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakrabarti P., Janin J. Dissecting protein-protein recognition sites. Proteins. 2002;47:334–343. doi: 10.1002/prot.10085. [DOI] [PubMed] [Google Scholar]
- 30.Triepels R.H., van den Heuvel L.P., Loeffen J.L., Buskens C.A., Smeets R.J., Rubio Gozalbo M.E., Budde S.M., Mariman E.C., Wijburg F.A., Barth P.G. Leigh syndrome associated with a mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I. Ann. Neurol. 1999;45:787–790. doi: 10.1002/1531-8249(199906)45:6<787::aid-ana13>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 31.Huynen M., Snel B., Lathe W., Bork P. Predicting protein function by genomic context: Quantitative evaluation and qualitative inferences. Genome Res. 2000;10:1204–1210. doi: 10.1101/gr.10.8.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nouwen N., Driessen A.J. SecDFyajC forms a heterotetrameric complex with YidC. Mol. Microbiol. 2002;44:1397–1405. doi: 10.1046/j.1365-2958.2002.02972.x. [DOI] [PubMed] [Google Scholar]
- 33.Dieteren C.E., Willems P.H., Vogel R.O., Swarts H.G., Fransen J., Roepman R., Crienen G., Smeitink J.A., Nijtmans L.G., Koopman W.J. Subunits of mitochondrial complex I exist as part of matrix- and membrane-associated subcomplexes in living cells. J. Biol. Chem. 2008;283:34753–34761. doi: 10.1074/jbc.M807323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.