

Abstract
Cellular adhesion to extracellular matrix is a central phenomenon for the maintenance of tissue integrity and cellular movement. Collectively, these processes are regulated by a fine-tuned balance between the formation and loosening of adhesive contacts, a process involving integrins, and the elevation and diminution of cytoplasmic signalling molecules. We demonstrate that prostaglandin (PG) F2α stimulation rapidly increases the capacity of Ishikawa cells stably expressing the F-prostanoid receptor (FPS) to adhere to vitronectin. Coincident with this elevation in matrix adhesion, we demonstrate a profound PGF2α-induced alteration in cytoskeletal remodelling, characterized by polymerization of the actin cytoskeleton and recruitment of focal adhesion kinase at focal adhesions and enhanced cell migration. Moreover, we show that these PGF2α-induced alterations in adhesion and morphology on vitronectin and migration could be abolished by cultivating FPS cells in the presence of integrin αvβ3 antibody or αvβ3-directed tetrapeptide arg–gly–asp–ser or inhibition of FP receptor signalling with the FP receptor antagonist, chemical disruptors of the phospholipase C-β, protein kinase A, c-Src and epidermal growth factor receptor kinase pathways or inhibition of the monomeric G proteins Rho, Rac and CDC42. These results reveal a mechanism by which prostanoids regulate cell movement, which may be relevant to pathologies of the endometrium.
Keywords: PGF2α, FP receptor, focal adhesions, morphology, migration, chemotaxis
Introduction
Activation of G-protein–coupled receptors (GPCRs) like the F-prostanoid receptor (FPS) regulates heterotrimeric G-protein pathways to control cell growth and behaviour, via the phosphorylation and dephosphorylation of distal-signalling complexes (Daub et al., 1997; Luttrell et al., 1997). These signalling complexes either serve as scaffolds for the plasma membrane recruitment of guanine nucleotide exchange factors (GEFs) for small monomeric G proteins (Rho, Ras, Rac and CDC42) or are recruited to the cytoskeletal contact point with the plasma membrane, where they serve as docking proteins (Lefkowitz, 1993; Luttrell et al., 1997, 1999). One such docking protein at the site where integrin- and proteoglycan-mediated adhesion links to the actin cytoskeleton is focal adhesion tyrosine kinase (FAK) (Wozniak et al., 2004). FAK is a non-receptor tyrosine kinase that forms part of the plasma membrane focal adhesion complex, which assembles on integrin heterodimers following integrin engagement of extracellular matrix (ECM) proteins (Morino et al., 1995; Giancotti and Ruoslahti, 1999). ECM proteins interact with integrins via the arg–gly–asp (RGD) motif at focal adhesion sites to regulate cell signalling and motility (Giancotti and Ruoslahti, 1999; Brakebusch et al., 2002; Wozniak et al., 2004).
Integrin signalling via reorganization of the actin cytoskeleton at focal adhesions has been shown to activate the monomeric G proteins Rho, Rac and CDC42, resulting in membrane extension (Del Pozo et al., 2002; Wozniak et al., 2004) to facilitate cell spreading or migration as well as modulate the signalling of other effector molecules, complexed within the integrin-linked actin-bound scaffold (Clark and Brugge, 1995; Morino et al., 1995). Agents that disrupt cytoskeletal assembly or that block activation of the small monomeric G-protein Rho have been shown to inhibit FAK phosphorylation and focal adhesions (Slack, 1998; Davidson et al., 2004b).
Prostaglandins (PGs) are considered to play a major role in benign and neoplastic endometrial pathologies, where biosynthesis of PGE2 and PGF2α is elevated (Singh et al., 1975; Lundstrom and Green, 1978; Lumsden et al., 1983; Sales and Jabbour, 2003b). We have demonstrated elevated expression of FP receptor in human endometrial adenocarcinomas and ascertained a role for FP receptor in promoting angiogenic and tumourigenic gene expression both in Ishikawa FPS cells and endometrial adenocarcinoma biopsies (Sales et al., 2004b, 2005; Jabbour et al., 2005). Moreover in these studies we have shown that PGF2α–FP receptor signalling establishes a positive feedback loop to sustain the biosynthesis of prostanoids and transcription of cyclooxygenase-2 (COX-2) and fibroblast growth factor via the activation of Gq/11 and hydrolysis of phosphatidylinositol (Jabbour et al., 2005; Sales et al., 2007). In many cell types Gq/11 coupling leads to the activation of FAK in an adhesion-dependent manner via the activation of small monomeric G proteins such as Rho (Rodriguez-Fernandez and Rozengurt, 1996; Pierce et al., 1999; Fujino et al., 2000; Davidson et al., 2004b). Disruption of focal adhesions can abolish activation of FAK (Slack, 1998), leading to an ablation of agonist-mediated cell signalling (Davidson et al., 2004b). In colonic cancers, breast cancers and sarcoma, FAK is overexpressed and plays a role in cancer cell–ECM interactions, adhesion, cell spreading, invasion and metastasis (Weiner et al., 1993).
In the present study we investigated the role of PGF2α in mediating adhesion, morphology and migration of endometrial adenocarcinoma cells via the FP receptor using Ishikawa cells stably expressing FP receptor to the levels observed in endometrial adenocarcinomas.
Results
PGF2α–FP receptor stimulation of FPS cells induces alterations in cell morphology
In the present study we observed that upon stimulation of FPS cells, but not wild-type (WT) cells, with PGF2α there are rapid changes in cellular morphology and lamellipodia formation coincident with reorganization of the actin cytoskeleton (Figure 1a, panel vi) and redistribution of microtubules within the cell (Figure 1b, panel vi). These morphological changes in response to PGF2α were inhibited by co-incubation of FPS cells with the specific FP receptor antagonist AL8810 (Figures 1a and b, panel vii). These alterations in cellular morphology were not observed when WT or FPS cells were treated with 100 nM Iloprost, used as a control eicosanoid (Figures 1a and b, panel viii).
Figure 1.

PGF2α induces ultrastructural changes in FPS cells. Confocal laser-scanning images showing the localization of β-actin and β-tubulin ((a) and (b), respectively) in methanol-fixed wild-type (WT) and FPS Ishikawa cells. Cells were serum starved before stimulation with vehicle (panels i and v for WT and FPS cells, respectively) or 100 nM PGF2α (panels ii and vi for WT and FPS cells, respectively) or 100 nM PGF2α and AL8810 (panels iii and vii for WT and FPS cells, respectively) or 100 nM Iloprost (panels iv and viii for WT and FPS cells, respectively) for 4 h. Data are shown as representative cells from at least four fields of view from three independent experiments.
PGF2α–FP receptor stimulation enhances the adhesive capacity of FPS cells to ECM
Coincident with the agonist-induced cytoskeletal remodelling in FPS cells, we observed an enhanced capacity of FPS cells, but not WT cells, to adhere to the ECM vitronectin (Figure 2a). No alteration in adherence capacity of WT or FPS cells was observed on fibronectin, laminin, collagen I or IV in response to PGF2α treatment (Figure 2a). The cellular adherence effects of FPS cells to vitronectin in response to PGF2α were abolished by co-incubation of FPS cells with the FP receptor antagonist AL8810 (Figure 2b). In addition to the morphological and adhesion alterations, PGF2α also enhanced the migratory capacity of FPS cells via the FP receptor, since incubation of FPS cells with the specific FP receptor antagonist AL8810 abolished the PGF2α-induced increase in cell migration (Figure 2c).
Figure 2.

Prostaglandin F2α (PGF2α) stimulation induces an increase in the adherent capacity of F-prostanoid receptor (FPS) cells to vitronectin and enhances cell migration. (a) Cell adhesion to extracellular matrix (ECM) as determined by CytoMatrix cell adhesion assay. Wild-type (WT) (open bars) and FPS cells (closed bars) were incubated with vehicle or 100 nM PGF2α for 1h and cellular adherence to the ECM fibronectin, vitronectin, laminin, collagen I or IV was measured. (b) FPS cells were pre-incubated with either vehicle, the FP receptor antagonist AL8810, arg–gly–asp–ser (RGDS) or arg–gly–glu–ser (RGES) tetrapeptides, integrin αvβ3 antibody or immunoglobulin G (IgG) for 1h and then seeded onto vitronectin-coated plates in the presence of vehicle or 100 nM PGF2α for 1h. Cell adhesion induced by PGF2α was calculated as fold increase above vehicle. (c) Cell migration was determined using a Chemotaxis cell migration assay. FPS cells were pre-incubated with either vehicle, the FP receptor antagonist AL8810, RGDS or RGES tetrapeptides, integrin αvβ3 antibody or IgG for 1h and then seeded onto transwell chambers for 24 h. The lower chamber received either vehicle, PGF2α or PGF2α and AL8810, RGDS or RGES tetrapeptides, integrin αvβ3 antibody or IgG as the chemoattractant. In all panels each bar represents the mean±s.e.m. of six independent experiments (b is significantly different from a; P<0.05).
Displacement of the extracellular region of the integrin from the matrix protein with the αvβ3-directed tetrapeptide arg–gly–asp–ser (RGDS) abolished the agonist-induced cellular adherence effects of FPS cells to vitronectin as well as the ability of FPS cells to migrate towards the PGF2α stimulus (Figures 2b and c). In contrast, co-incubation of FPS cells with arg–gly–glu–ser (RGES) tetrapeptide as a negative control did not alter the agonist-induced cell adhesion to vitronectin or increase in FPS cell migration (Figures 2b and c). Similar effects were observed using an integrin αvβ3 antibody or immunoglobulin G (IgG) from the same host species (Figures 2b and c).
PGF2α-induced alterations in cell morphology is dependant on integrin αvβ3 engagement with vitronectin
We investigated whether the PGF2α-induced alterations in cell morphology was dependant on integrin αvβ3 engagement with vitronectin. FPS cells incubated with RGDS tended to round up and become easily dislodged from the vitronectin-coated growth plate. Stimulation of these cells with vehicle (Figure 3Aiv) or 100 nM PGF2α (Figure 3Biv) for 4 h resulted in no apparent alteration in cell morphology as shown in the phase contrast fields. FPS cells treated with the control RGES tetrapeptide showed dramatic alterations in cell morphology after 4 h of PGF2α treatment (Figure 3Div), compared with vehicle-treated cells (Figure 3Civ) as observed in the phase contrast fields.
Figure 3.

Morphological changes are dependant upon integrin αvβ3 engagement with vitronectin. Phase contrast (column iv) and immunofluorescent laser confocal microscopy of FPS cells, showing the localization of β-actin (column i, green channel), focal adhesion tyrosine kinase (FAK) (column ii, red channel) and the co-localization of β-actin with FAK (column iii, yellow channel). FPS cells were seeded onto vitronectin-coated plates, transfected with hemagglutinin (HA)-tagged wild-type FAK cDNA and pre-incubated with 1mM arg–gly–asp–ser (RGDS) or RGES tetrapeptides for 16 h. Cells were stimulated with vehicle (rows A and C) for RGDS- and RGES-treated cells, respectively) or 100 nM prostaglandin F2α (PGF2α) (rows B and D) for RGDS- and RGES-treated cells, respectively) for 4 h. In the PGF2α-treated FPS cells, the generation of β-actin and FAK-rich membrane extensions (lamellipodia) and change in cell morphology are evident in the RGES (D (i) and D (ii) for β-actin and FAK, respectively)-treated cells compared with vehicle-treated cells (C (i) for RGES-treated cells stained for β-actin and C (ii) for RGES-treated cells stained for FAK, respectively). These cytoskeletal alterations in β-actin and FAK staining were not observed in cells treated with PGF2α and RGDS tetrapeptide (figure B (i) and B (ii) for β-actin and FAK, respectively). Data are shown as representative cells from at least four fields of view from three independent experiments.
The alteration in FPS cell morphology in response to agonist treatment in the presence of the control RGES tetrapeptide was associated with redistribution of the actin cytoskeleton at focal adhesions (Figure 3Di) compared to cells treated with vehicle (Figure 3Ci). This alteration in actin distribution at focal adhesions in the PGF2α-treated FPS cells in the presence of RGES was also associated with the coincident expression of FAK at focal adhesions. Here FAK (Figure 3Dii) was observed to co-localize (Figure 3Diii) with actin (Figure 3Di) in the PGF2α-treated FPS cells.
Disruption of the integrin–ECM interaction using the RGDS tetrapeptide caused a destabilization of the integrin α–β dimer and destruction of the focal adhesion complex. Under these conditions, actin and FAK were localized in separate subcellular compartments in presence (Figure 3Biii) or absence (Figure 3Aiii) of PGF2α. Similar effects in cellular morphology were observed using IgG or integrin αvβ3 antibody in place of RGDS or RGES tetrapeptides (data not shown).
PGF2α–FP receptor interaction in FPS cells activates cellular structural-associated proteins
We investigated the signalling pathways activating various cellular structural-associated proteins involved in reorganization of the cytoskeleton. We found that PGF2α–FP receptor stimulation of FPS cells rapidly mobilized the soluble cytoplasmic second messenger system inositol 1,4,5 trisphosphate (IP3) to a greater extent in FPS cells compared with WT cells. This effect was abolished by co-incubation of cells with the FP receptor antagonist AL8810 or chemical inhibitor of phospholipase C-β (PLC-β) U73122 (Figure 4a). Stimulation of FPS cells with PGF2α caused a dramatic time-dependent activation of the non-receptor tyrosine kinase c-Src (Figure 4b), the microtubule-associated protein kinase (also called mitogen-activated protein kinase; MAPK) extracellular signal-regulated kinase (ERK; Figure 4c) and FAK (Figure 4d). These effects of PGF2α stimulation on c-Src, ERK and FAK phosphorylation were significantly elevated in FPS cells compared with WT cells. Within our experimental paradigms, the activation of c-Src and ERK were very rapid, occurring maximally within 5 and 10 min, respectively. However the activation of FAK occurred at a much later time, with maximal phosphorylation occurring at 4 h. This time of activation of FAK by PGF2α coincides with the agonist-induced alteration in cellular morphology and the co-expression of FAK at focal adhesions.
Figure 4.

Prostaglandin F2α (PGF2α) stimulation of F-prostanoid receptor (FPS) cells activates numerous structurally regulated proteins. (a) Total inositol phosphate hydrolysis was measured in wild-type (WT) (open bars) and FPS cells (closed bars) in response to administration of vehicle or 100 nM PGF2α in the absence or presence of 50 μM AL8810 or 10 μM U73122. In figures (b–d), WT and FPS cells were stimulated with 100 nM PGF2α for the time indicated or left unstimulated (0 min). (b) Cells were immunoprecipitated (IP) with anti-phosphotyrosine antibody (p-Tyr) and subjected to immunoblot analysis (WB) using specific antibodies recognizing the activated form of c-Src non-receptor tyrosine kinase phosphorylated at tyrosine 418. (c) Cell lysates were subjected to immunoblot analysis using antibody against phosphorylated extracellular signal-regulated kinase (ERK). Immunoblots were stripped and reprobed with antibody recognizing total ERK. (d) Cells were transiently transfected with hemagglutinin (HA)-tagged wild-type FAK cDNA. Cell lysates were immunoprecipitated with anti-HA-agarose antibody (HA) and subjected to immunoblot analysis using antibody recognizing phosphorylated FAK. For each, a representative immunoblot is shown, with semi-quantitative analysis determined as described in the ‘Materials and methods’ section. In all panels each bar represents the mean±s.e.m. of at least three independent experiments (b is significantly different from a and c is significantly different from a and b; P<0.05).
PGF2α-mediated activation of c-Src, ERK and FAK occurs in a sequential manner via the FP receptor
We investigated PGF2α–FP receptor signalling to c-Src at Tyr418. We found that phosphorylation of c-Srcy418, after 5 min of treatment with PGF2α, was sensitive to inhibition of cell signalling with the specific FP receptor antagonist AL8810 (Figure 5a, lane 3) or small molecule chemical inhibitors of PLC-β (U73122; Figure 5a, lane 4) or protein kinase A (PKA) (4C3MQ; Figure 5a, lane 5). However phosphorylation of c-Srcy418 by PGF2α was insensitive to inhibition of cell signalling with the chemical inhibitor of the epidermal growth factor receptor (EGFR) tyrosine kinase, AG1478 (Figure 5a, lane 6).
Figure 5.

Prostaglandin F2α (PGF2α) activation of c-Src, extracellular signal-regulated kinase (ERK) and focal adhesion tyrosine kinase (FAK) are dependant on multiple signalling pathways and cell attachment to ECM. (a) FPS cells were stimulated with vehicle (lane 1), 100 nM PGF2α (lane 2), 100 nM PGF2α and AL8810 (lane 3), 100 nM PGF2α and U73122 (lane 4), 100 nM PGF2α and 4C3MQ (lane 5) or 100 nM PGF2α and AG1478 (lane 6) for 5 min, immunoprecipitated with anti-phosphotyrosine antibody (p-Tyr) and immunoblotted for phosphorylated c-Srcy418, (b) FPS cells were transiently transfected with Myc-ERK cDNA and either empty vector cDNA (pcDNA3; lanes 1–7) or dominant-negative (Dn) mutant isoforms of FAK (lane 8), EGFR (lane 9), Rho (lane 10), Ras (lane 11), Rac (lane 12), CDC42 (lane 13), c-Src (lane 14) and MEK (lane 15) and subjected to stimulation with vehicle (lane 1) or 100 nM PGF2α for 10 min. Empty vector transfected cells were stimulated with 100 nM PGF2α (lane 2) 100 nM PGF2α and AL8810 (lane 3), 100 nM PGF2α and U73122 (lane 4), 100 nM PGF2α and 4C3MQ (lane 5), 100 nM PGF2α and PP2 (lane 6) or 100 nM PGF2α and AG1478 (lane 7) for 10 min. Cells were immunoprecipitated with Myc-agarose-conjugated antibody and immunoblotted for phosphorylated ERK (upper panel). Immunoblots were stripped and re-probed with antibody recognizing total ERK (lower panel). (c) FPS cells were transiently transfected with HA-tagged wild-type FAK cDNA and empty vector cDNA (pcDNA3; lanes 1and 2) or dominant-negative (Dn) Rho (lane 3), Rac (lane 4), CDC42 (lane 5), EGFR (lane 6), MEK (lane 7) and c-Src (lane 8) cDNA and stimulated with vehicle or 100 nM PGF2α for 4 h. Cells were immunoprecipitated (IP) with HA-agarose-conjugated antibody and subjected to immunoblot analysis (WB) for phosphorylated FAK. (d) FPS cells were transiently transfected with HA-tagged wild-type FAK cDNA and empty vector cDNA (pcDNA3; lanes 1–8). Cells were stimulated with vehicle (lane 1), 100 nM PGF2α (lane 2), 100 nM PGF2α and AL8810 (lane 3), 100 nM PGF2α and U73122 (lane 4), 100 PGF2α and 4C3MQ (lane 5), 100 nM PGF2α and PP2 (lane 6), 100 nM PGF2α and AG1478 (lane 7) or 100 nM PGF2α and PD98059 (lane 8) for 4 h. Cells were immunoprecipitated (IP) with HA-agarose-conjugated antibody and subjected to immunoblot analysis (WB) for phosphorylated FAK. In all panels each bar represents the mean±s.e.m. of at least four independent experiments (+ denotes addition of agent and − denotes absence of agent; b is significantly different from a; P<0.05).
We subsequently investigated the signalling pathways mediating the PGF2α–FP receptor activation of ERK. FPS cells were transiently transfected with an Myc-tagged ERK cDNA construct and either empty vector cDNA (pcDNA3) or cDNA encoding a dominant-negative (Dn) mutant isoform of FAK, EGFR, c-Src, ERK kinase (mitogen-activated protein kinase kinase (MEK)) or small monomeric G-protein Rho, Ras, Rac or CDC42. Cells were either treated with vehicle or 100 nM PGF2α following transfection or pre-treated with the same panel of chemical inhibitors used in Figure 5a, with the inclusion of the c-Src kinase inhibitor PP2, for 1h prior to agonist stimulation for 10 min. As shown in Figure 5b, PGF2α stimulation of FPS cells and immunoprecipitation of Myc-ERK showed a profound elevation of ERK activity (Figure 5b, lane 2) compared with vehicle-treated cells (Figure 5b, lane 1). This PGF2α-mediated phosphorylation of Myc-ERK was abolished when cells were co-incubated with AL8810 (Figure 5b, lane 3), U73122 (Figure 5b, lane 4), 4C3MQ (Figure 5b, lane 5), PP2 (Figure 5b, lane 6) and AG1478 (Figure 5b, lane 7) or by co-expression of the Dn-EGFR (Figure 5b, lane 9), Rho (Figure 5b, lane 10), Ras (Figure 5b, lane 11), Rac (Figure 5b, lane 12), CDC42 (Figure 5b, lane 13), c-Src (Figure 5b, lane 14) and MEK (Figure 5b, lane 15) cDNA isoforms with Myc-ERK. However when the tyrosine mutant FAK isoform, which displays an inability to bind c-Src (Schlaepfer and Hunter, 1997), was co-expressed with the Myc-ERK, there was no attenuation in ERK phosphorylation (Figure 5b, lane 8), demonstrating that tyrosine phosphorylation of FAK is not an inherent requirement for ERK activation in FPS cells by PGF2α. Previously we showed that PGF2α activation of ERK in FPS cells, occurred independently of protein kinase C (Sales et al., 2005). Interestingly in the present study we show a requirement for PKA activity in transducing the signal from the FP receptor to ERK.
We next investigated the signalling pathways mediating the phosphorylation of FAK by PGF2α in FPS cells by co-transfecting a hemagglutinin (HA)-tagged WT FAK cDNA construct with either empty vector cDNA or cDNA encoding the Dn isoforms of the small monomeric G-protein Rho, Rac, CDC42 or Dn isoforms of EGFR, MEK or c-Src. Following transfection, cells were stimulated with vehicle or 100 nM PGF2α for 4 h or pre-incubated with the FP receptor antagonist AL8810 or small molecule chemical inhibitors of PLC-β (U73122), PKA (4C3MQ), c-Src (PP2), EGFR (AG1478) or MEK (PD98059) for 1 h and then subjected to agonist stimulation for 4 h.
Agonist stimulation of FPS cells significantly phosphorylated FAK (Figure 5c, lane 2) compared with vehicle-treated cells (Figure 5c, lane 1). Co-expression of Dn-Rho (Figure 5c, lane 3), Rac (Figure 5c, lane 4), CDC42 (Figure 5c, lane 5), EGFR (Figure 5c, lane 6), MEK (Figure 5c, lane 7) and c-Src (Figure 5c, lane 8) attenuated the PGF2α-induced FAK phosphorylation. Similarly, FAK phosphorylation in FPS cells in response to PGF2α (Figure 5d, lane 2) could be abolished by co-incubation of cells with AL8810 (Figure 5d, lane 3), U73122 (Figure 5d, lane 4), 4C3MQ (Figure 5d, lane 5), PP2 (Figure 5d, lane 6), AG1478 (Figure 5d, lane 7) and PD98059 (Figure 5d, lane 8). The temporal change in activation of FAK (after 4 h) compared with c-Src (5 min) and ERK (10 min) in FPS cells and the attenuation of FAK phosphorylation by Dn-MEK and PD98059 suggest that FAK activation occurs downstream of c-Src and ERK activation to coincide with the PGF2α-induced phosphorylation of FAK and actin at focal adhesions and alteration in cell morphology.
To determine whether PGF2α signalling to ERK and FAK in FPS cells was dependant on engagement of integrin receptors to ECM, we incubated FPS cells with 1mM RGDS or RGES tetrapeptides for 16 h and then subjected cells to agonist stimulation. As shown in Figures 6a and b, respectively, active displacement of the extracellular region of the integrin from the ECM and disruption of the focal adhesion protein complex with RGDS abolished the PGF2α-induced phosphorylation of ERK (Figure 6a) and FAK (Figure 6b). Similar results were achievable using the integrin αvβ3 antibody (data not shown). However co-treatment of FPS cells with the chemically similar RGES tetrapeptide, which fails to bind integrin receptors (or IgG, data not shown), had no effect on the PGF2α-induced phosphorylation of ERK and FAK. Thus the selective disruption of the focal adhesion complexes attenuated the ability of PGF2α to coordinate the activation of the cytoskeleton-linked proteins microtubule-associated protein kinase (ERK) and FAK and direct the change in cell morphology.
Figure 6.

Morphological changes in FPS cells is dependant on FP receptor, EGFR and extracellular signal-regulated kinase (ERK) signalling. (a) FPS cells were pre-incubated with 1mM arg–gly–asp–ser (RGDS) or arg–gly–glu–ser (RGES) tetrapeptides for 16 h and then stimulated with vehicle or 100 nM prostaglandin F2α (PGF2α) for 10 min. ERK phosphorylation was detected by immunoblot analysis (upper panel) and normalized for protein loading against total ERK protein (lower panel) on the same blot. (b) FPS cells were transiently transfected with hemagglutinin (HA)-tagged wild-type FAK cDNA. FPS cells were preincubated for 16 h with 1 mM RGDS or RGES tetrapeptides and stimulated for 4 h with vehicle or 100 nM PGF2α. Cells were immunoprecipitated (IP) with HA-agarose-conjugated antibody and subjected to immunoblot analysis (WB) for phosphorylated FAK. In all panels each bar represents the mean±s.e.m. of at least four independent experiments (b is significantly different from a; P<0.05). (c) Phase contrast microscopy (panels iv, viii, xii, xvi and xx) and confocal laser-scanning images showing the localization of β-actin (panels i, v, ix, xiii and xvii), HA-tagged FAK (panels ii, vi, x, xiv and xvii) and co-localization of FAK with actin (panels iii, vii, xi, xv and xix) in FPS cells. Cells were transiently transfected with HA-tagged wild-type FAK cDNA construct, serum starved and treated with vehicle (panels i–iv), 100 nM PGF2α (panels v–viii), 100 nM PGF2α and AL8810 (panels ix–xii), 100 nM PGF2α and AG1478 (panels xiii–xvi) or 100 nM PGF2α and PD98059 (panels xvii–xx) for 4 h. In the PGF2α-treated FPS cells, the generation of β-actin (panel v) and FAK-rich membrane extensions (panel vi; lamellipodia) are consistent with change in cell morphology. Here FAK and actin co-localize at focal adhesions (panel vii) compared with vehicle-treated cells where FAK (panel ii) and actin (panel i) are localized in separate compartments (panel iii). The cytoskeletal alterations and co-localization of FAK with actin in PGF2α-treated cells were abolished by co-treatment of cells with AL8810 (panel xi), AG1478 (panel xv) and PD98059 (panel xix). Data are shown as representative cells from at least four fields of view from three independent experiments.
PGF2α-FP receptor-induced change in cell morphology is dependant upon the EGFR-mediated activation of ERK
We found that the PGF2α-mediated changes in the actin cytoskeleton (Figure 6c, panel v) and distribution of FAK (Figure 6c, panel vi), which co-localized at focal adhesions in FPS cells (Figure 6c, panel vii), were abolished when cells are co-incubated with the FP receptor antagonist (Figure 6c, panel xi), EGFR kinase inhibitor AG1478 (Figure 6c, panel xv) or the MEK inhibitor PD98059 (Figure 6c, panel xix). In addition, the cellular distribution of actin and FAK and cell morphology in FPS cells treated with PGF2α and AL8810 (Figure 6c, panel xi), AG1478 (Figure 6c, panel xv) or PD98059 (Figure 6c, panel xix) were similar to cells treated with vehicle alone (Figure 6c, panel iii). Similarly, cells incubated with chemical inhibitor on their own for the same length of time showed no alteration in cell morphology (data not shown).
Finally we have shown that treatment of FPS cells with the EGFR kinase inhibitor AG1478 or the MEK inhibitor PD98059, which abolishes the cytoskeletal reorganization, also abolishes the ability of cells to migrate towards the PGF2α stimulus (Figure 7).
Figure 7.
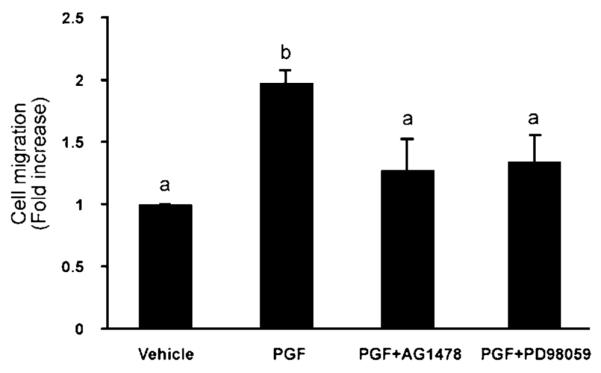
FPS cell migration is dependant on EGFR and ERK signalling. Cell migration was determined using a Chemotaxis cell migration assay. FPS cells were pre-incubated with either vehicle, AG1478 or PD98059 for 1 h and then seeded onto transwell chambers for 24 h. The lower chamber received vehicle, prostaglandin F2α (PGF2α) or PGF2α and AG1478 or PD98059 as the chemoattractant. Each bar represents the mean±s.e.m. of at least four independent experiments (b is significantly different from a; P<0.05).
Discussion
PGs have been associated with endometrial pathologies such as endometriosis and cancer, however the cellular mechanisms whereby they can modulate cell adhesion and movement involved in such disorders is unclear (Singh et al., 1975; Lumsden et al., 1983; Sales and Jabbour, 2003a; Jabbour and Sales, 2004).
Recently, we have demonstrated a role for the FP receptor in endometrial adenocarcinomas (Sales et al., 2004b, 2005, 2007). Here we show that activation of the FP receptor by PGF2α leads to increased adherence of Ishikawa FPS cells to the ECM vitronectin coincident with the redistribution of microtubules within the cell and the formation of actin stress fibres and lamellipodia, resulting in a dramatic alteration in cell morphology and ligand-induced FPS cell migration.
Vitronectin, an adhesive glycoprotein found in the circulation and tissues, regulates cell adhesion, growth, differentiation and migration in vitro and in vivo. There is now much evidence to support a role for vitronectin and its integrin receptors in pathology (Preissner, 1991; Giancotti and Ruoslahti, 1999; Brakebusch et al., 2002). In transformed cells, vitronectin interacts with integrin αvβ3 at focal adhesions via the minimum consensus motif RGD (Preissner, 1991; Giancotti and Ruoslahti, 1999; Brakebusch et al., 2002). In ovarian cancer tissues vitronectin and integrin αvβ3 are abundantly expressed (Cruet et al., 1999). Moreover, in primary ovarian carcinomas integrin αvβ3 is found at a significantly higher rate compared with ovarian tumours of low malignant potential (Liapis et al., 1997), indicating a role for engagement of integrin αvβ3/vitronectin interaction in cancer progression. Similarly, in the endometrium, dysregulation of integrin αvβ3 expression has been associated with endometriosis, a disorder manifest by retrograde menstruation and adhesion of endometrial tissue and outgrowth in the peritoneal cavity and viscera (Lessey et al., 1994; Sales and Jabbour, 2003a; Jabbour and Sales, 2004).
Clusters of integrins assembled at focal adhesion complexes are complexed with scaffold proteins and associate with the cytoskeleton at the plasma membrane contact point with the ECM, where they transduce the signal from GPCRs (Damsky and Werb, 1992; Giancotti and Ruoslahti, 1999). One such protein at the site of integrin–cytoskeleton contact is FAK (Wozniak et al., 2004). In the present study we show that the PGF2α-induced adhesion to vitronectin and re-organization of the actin cytoskeleton also promotes the recruitment of FAK to focal adhesion sites in an EGFR- and ERK1/2-dependent manner.
We used specific small molecule chemical inhibitors of cell signalling and co-transfection studies with Dn mutant proteins targeted to specific signalling pathways to map out the signalling events activated following PGF2α– FP receptor engagement leading to the activation of c-Src, ERK1/2 and FAK. We have shown that PGF2α–FP receptor stimulation of FPS cells rapidly activates c-Src, ERK1/2 and FAK in a PLC-β-IP3, PKA, EGFR-dependent manner. In addition ERK and FAK phosphorylation in FPS cells in response to PGF2α were also sensitive to inhibition of cell signalling with Dn mutant isoforms targeted against the small monomeric G-proteins Rho, Rac and CDC42. The small monomeric G proteins are typically activated through a growth factor receptor-induced association at the plasma membrane with GEFs (Egan et al., 1993) to regulate the polymerization of actin to produce stress fibres and lamellipodia. In addition to stress fibres, Rho controls the assembly of focal adhesion complexes. Agents that block activation of Rho have been shown to inhibit FAK phosphorylation and focal adhesions in other model systems (Slack, 1998; Davidson et al., 2004b). Indeed Pierce et al. (1999) have shown that agonist stimulation of FP receptor in HEK293 cells leads to cellular shape change, induction of actin stress fibre formation and activation of FAK in a Rho-dependent manner. Rac and CDC42 are also present at the periphery of migrating cells and direct the assembly of multimolecular focal complexes at the plasma membrane (Nobes and Hall, 1995; Wozniak et al., 2004).
Disruption of focal adhesions and cytoskeletal assembly has been shown to inhibit FAK phosphorylation and signalling (Slack, 1998; Davidson et al., 2004b). Similarly studies using FAK mutants, which fail to localize to focal adhesions, exhibit impaired autophosphorylation and an inability to bind FAK substrates and induce cell signalling (Wozniak et al., 2004). In the present study, we have shown that disruption of integrin αvβ3–ECM interaction with an excess of an RGDS tetrapeptide abolishes the capacity of FPS cells to adhere to ECM in response to agonist treatment, as well as the agonist-induced alteration in morphology, induction of actin stress fibres and the presence of FAK co-localized with actin at focal adhesions. Coincident with this abolition of integrin engagement, morphological change and cell migration we found that disruption of integrin–ECM interaction with the RGDS tetrapeptide also abolished cell signalling to ERK and FAK. These data suggest that integrin αvβ3–ECM adherence and cytoskeletal reorganization is necessary for FPS cell signalling and migration in response to agonist treatment and suggest that chemical disruptors of integrin–ECM engagement may be potential therapy for inhibiting the adverse effects of PGF2α–FP receptor signalling in vivo. Indeed, blocking antibodies targeted against integrin αvβ3 or cyclic RGD peptide antagonists have been demonstrated to induce tumour regression by influencing cell adhesion/migration and blocking proliferation and reducing tumour angiogenesis (Hynes, 2002). However the feasibility of such strategies remains a matter of debate.
Taken together, our findings herein provide a novel mechanism for the control of endometrial epithelial cell behaviour and movement by the FP receptor via integrin αvβ3-ECM-mediated cell adhesion and migration. We believe that the findings we have presented herein may have relevance for understanding the molecular mechanisms regulating endometrial pathologies, such as endometriosis and cancer expressing aberrant levels of prostanoid receptors, and which are associated with cell movement metastasis and adhesion to distant sites of the body.
Materials and methods
Reagents
The anti-Myc-agarose, anti-phosphotyrosine (PY20) agarose, anti-phospho-c-SrcY418, anti-phospho-FAK, anti-FAK, anti-HA-agarose pre-conjugate, IgG, anti-integrin αvβ3, anti-β-tubulin and anti-β-actin antibodies were purchased from Santa Cruz Biotechnology (Autogen-Bioclear, Wiltshire, UK). RGDS and RGES, alkaline phosphatase secondary antibodies, indomethacin, phosphate-buffered saline, bovine serum albumin, AL8810 (used at a final concentration of 50 μM) and PGF2α (used at a final concentration of 100 nM) were purchased from Sigma Chemical Company (Dorset, UK). The Iloprost was purchased from Cayman chemical company (Axxora, Nottingham, UK). The Src kinase-specific inhibitor PP2 (used at a final concentration of 10 μM), EGFR kinase-specific inhibitor tryphostin AG1478 (used at a final concentration of 100 nM), PLC-β inhibitor U73122 (used at a final concentration of 10 μM), MEK1/2 inhibitor PD98059 (used at a final concentration of 50 μM), PKA inhibitor 4-cyano-3-methylisoquinoline (4C3MQ, used at a final concentration of 1 μM) were purchased from Calbiochem (Nottingham, UK). The Dn–MEK (Seger et al., 1994; Jaaro et al., 1997), Dn–EGFR (Benard et al., 2001), Dn–Rho, Dn–Rac (Harris et al., 2002), Dn–Ras (Benard et al., 2001), Dn–CDC42 (Levi et al., 1998) and Dn–FAK (Benard et al., 2001) cDNA constructs were a kind gift from Prof Zvi Naor (Department of Biochemistry, University of Tel Aviv, Israel). The Myc-tagged ERK-2 construct, HA-tagged WT FAK cDNA and Dn-c-Src cDNA constructs (Davidson et al., 2004a) were obtained from Prof Robert Millar (MRC Human Reproductive Sciences Unit, Edinburgh, UK). The FP receptor antagonist AL-8810 is a specific antagonist of the FP receptor (Griffin et al., 1999). The mean potency of the FP receptor antagonist AL-8810 is EC50=261±44 nM and Emax=19% compared with the FP receptor agonist cloprostenol: EC50=0.84 nM and Emax=100. In this study the authors show that the FP receptor antagonist dose dependently inhibits 100 nM FP receptor agonist with 100% inhibition at 100 μM (Griffin et al., 1999). We have shown that the FP receptor antagonist can inhibit 100 nM PGF2α at a concentration of 10–50 μM (Sales et al., 2005). Moreover at this concentration the FP receptor antagonist does not inhibit responses of TP, DP, EP2 or EP4 receptors (Griffin et al., 1999).
Cell culture and transfection
Ishikawa WT (European Collection of Cell Culture, Wiltshire, UK) and Ishikawa cells stably expressing the FPS to the levels observed in endometrial adenocarcinomas (FPS cells) were maintained as described (Sales et al., 2005). Transient transfections of FPS cells were performed using Superfect (Qiagen, Crawley, UK) as per the manufacturer’s protocol. All experiments were conducted in the presence of 8.3 μM indomethacin (a dual COX enzyme inhibitor used to inhibit endogenous prostanoid biosynthesis). To actively displace integrins from their cell matrix attachments, FPS cells were incubated in serum-free media with 1mM RGDS or RGES tetrapeptides, 50 μg ml-1 final concentration of anti-integrin αvβ3 antibody or equivalent IgG from the same host species for 16 h (or 1 h for cell adhesion assays). Following agonist stimulation, any displaced and detached cells were collected with any adherent cells by centrifugation at 1000g for 5 min.
Protein extraction and western blot analysis
Protein extraction and immunoprecipitation, sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blot analysis were performed as described previously (Sales et al., 2004a, 2005). using specific primary antibodies as described in the figure legend and secondary antibodies conjugated to Alexafluor 680 (Molecular Probes Inc., Eugene, OR, USA) or IRdye 800 (Rockland Immunochemicals, Gilbertsville, PA, USA). Immunoreactive proteins were detected and quantified using the Odyssey infrared imaging system (LI-COR Biosciences, Cambridge, UK). Relative density of immunoblots was calculated by dividing the value obtained from the phosphorylated immunoblots by the value obtained from total protein immunoblots or light-chain IgG and expressed as fold above vehicle controls.
Immunohistochemistry and confocal laser microscopy
Confocal laser microscopy was performed on a Zeiss (Jena, Germany) laser-scanning microscope LM510. Cells were plated upon vitronectin-coated slides, transfected with an HA-tagged WT FAK cDNA construct and serum starved for 16 h in the absence or presence of RGDS/RGES tetrapeptides, αvβ3 antibody or IgG. Thereafter cells were either subjected to agonist stimulation for 4 h or pre-treated with chemical inhibitor or FP receptor antagonist for 1h prior to agonist stimulation as described in the figure legend. After stimulation, cells were fixed with 100% methanol, blocked using 5% normal rabbit serum before incubation with mouse anti-HA antibody at a dilution of 1:100 for 18 h at 4 °C. Thereafter sections were incubated sequentially with biotinylated rabbit anti-mouse (DAKO; Dako Corp., High Wycombe, UK) and fluorochrome streptavidin 546 Alexafluor (Molecular Probes Inc.) diluted 1 in 200. Sections were re-blocked with 5% normal rabbit serum and incubated sequentially with anti-actin/tubulin antibody at a dilution of 1:50 at 4 °C for 18 h and fluorochrome rabbit anti-goat fluorescein isothyocyanate (Molecular Probes Inc.) diluted 1 in 200 at 25 °C for 2 h. Nuclear staining was detected by incubating cells with a 1 in 2000 dilution of ToPro2 (Molecular probes Inc.) for 2 min. Control sections were incubated with IgG from the same host species.
Phosphatidylinositol hydrolysis
Accumulation of total inositol phosphates was measured in Ishikawa WT and FPS cells treated either with vehicle, 100 nM PGF2α, 100 nM PGF2α and AL8810 or 100 nM PGF2α and U73122 according to published protocols (Sales et al., 2004b).
Cell adhesion assays
Agonist-induced cell-matrix adhesion was assessed using a CytoMatrix screening kit ECM 205 and CytoMatrix Human vitronectin cell adhesion strips ECM 102 (Chemicon, Temecula, CA, USA) according to the manufacturer’s protocol. WT or FPS cells were incubated in serum-free media in the presence/absence of 1mM RGDS, 1mM RGES, 50 μM AL8810, 50 μg IgG or 50 μg anti-integrin αvβ3 antibody for 1h prior to the addition of vehicle or 100 nM PGF2α. Thereafter cells were seeded onto the substrate-coated adhesion strips and incubated at 37 °C for 45 min. Adherent cells were fixed, stained and quantified according to the manufacturer’s protocol. Fold increase was determined by dividing the absorbance of cells treated with PGF2α by the absorbance of cells treated with vehicle.
Cell migration assays
Agonist-induced cell migration was assessed using the QCM Chemotaxis cell migration assay (Chemicon). FPS cells were incubated in 1ml of serum-free media in the presence/absence of 200 nM AG1478, 50 μM PD98059, 1mM RGDS, 1mM RGES, 50 μM AL8810, 50 μg IgG or 50 μg anti-integrin αvβ3 antibody for 1h. Thereafter 100 μl of each treatment containing 50 000 cells were placed into the migration (upper) chamber in triplicate. The lower chamber received either vehicle, PGF2α or PGF2α and 200 nM AG1478, 50 μM PD98059, 1mM RGDS, 1mM RGES, 50 μM AL8810, 50 μg IgG or 50 μg anti-integrin αvβ3 antibody for 24 h at 37 °C. Migratory cell number was determined according to the manufacturer’s instruction. Fold increase was determined by dividing the fluorescence of cells treated with PGF2α or PGF2α and inhibitor by the fluorescence of cells treated with vehicle or vehicle and inhibitor.
Statistics
Where appropriate, data were subjected to statistical analysis with analysis of variance and Fisher’s protected least significant difference tests (Statview 5.0; Abacus Concepts Inc., USA).
References
- Benard O, Naor Z, Seger R. Role of dynamin, Src, and Ras in the protein kinase C-mediated activation of ERK by gonadotropin-releasing hormone. J Biol Chem. 2001;276:4554–4563. doi: 10.1074/jbc.M006995200. [DOI] [PubMed] [Google Scholar]
- Brakebusch C, Bouvard D, Stanchi F, Sakai T, Fassler R. Integrins in invasive growth. J Clin Invest. 2002;109:999–1006. doi: 10.1172/JCI15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Cruet S, Salamanca C, Mitchell GW, Auersperg N. Alphavbeta3 and vitronectin expression by normal ovarian surface epithelial cells: role in cell adhesion and cell proliferation. Gynecol Oncol. 1999;75:254–260. doi: 10.1006/gyno.1999.5572. [DOI] [PubMed] [Google Scholar]
- Damsky CH, Werb Z. Signal transduction by integrin receptors for extracellular matrix: cooperative processing of extracellular information. Curr Opin Cell Biol. 1992;4:772–781. doi: 10.1016/0955-0674(92)90100-q. [DOI] [PubMed] [Google Scholar]
- Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997;16:7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L, Pawson AJ, De Maturana RL, Freestone SH, Barran P, Millar RP, et al. Gonadotropin-releasing hormone-induced activation of diacylglycerol kinase-zeta and its association with active c-src. J Biol Chem. 2004a;279:11906–11916. doi: 10.1074/jbc.M310784200. [DOI] [PubMed] [Google Scholar]
- Davidson L, Pawson AJ, Millar RP, Maudsley S. Cytoskeletal reorganization dependence of signaling by the gonadotropin-releasing hormone receptor. J Biol Chem. 2004b;279:1980–1993. doi: 10.1074/jbc.M309827200. [DOI] [PubMed] [Google Scholar]
- Del Pozo MA, Kiosses WB, Alderson NB, Meller N, Hahn KM, Schwartz MA. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat Cell Biol. 2002;4:232–239. doi: 10.1038/ncb759. [DOI] [PubMed] [Google Scholar]
- Egan SE, Giddings BW, Brooks MW, Buday L, Sizeland AM, Weinberg RA. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature. 1993;363:45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- Fujino H, Pierce KL, Srinivasan D, Protzman CE, Krauss AH, Woodward DF, et al. Delayed reversal of shape change in cells expressing FP(B) prostanoid receptors. Possible role of receptor resensitization. J Biol Chem. 2000;275:29907–29914. doi: 10.1074/jbc.M003467200. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Griffin BW, Klimko P, Crider JY, Sharif NA. AL-8810: a novel prostaglandin F2 alpha analog with selective antagonist effects at the prostaglandin F2 alpha (FP) receptor. J Pharmacol Exp Ther. 1999;290:1278–1284. [PubMed] [Google Scholar]
- Harris D, Bonfil D, Chuderland D, Kraus S, Seger R, Naor Z. Activation of MAPK cascades by GnRH: ERK and Jun N-terminal kinase are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHbeta-subunit promoter. Endocrinology. 2002;143:1018–1025. doi: 10.1210/endo.143.3.8675. [DOI] [PubMed] [Google Scholar]
- Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8:918–921. doi: 10.1038/nm0902-918. [DOI] [PubMed] [Google Scholar]
- Jaaro H, Rubinfeld H, Hanoch T, Seger R. Nuclear translocation of mitogen-activated protein kinase kinase (MEK1) in response to mitogenic stimulation. Proc Natl Acad Sci USA. 1997;94:3742–3747. doi: 10.1073/pnas.94.8.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbour HN, Sales KJ. Prostaglandin receptor signalling and function in human endometrial pathology. Trends Endocrinol Metab. 2004;15:398–404. doi: 10.1016/j.tem.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Jabbour HN, Sales KJ, Boddy SC, Anderson RA, Williams AR. A positive feedback loop that regulates cyclooxygenase-2 expression and prostaglandin F2alpha synthesis via the F-series-prostanoid receptor and extracellular signal-regulated kinase 1/2 signaling pathway. Endocrinology. 2005;146:4657–4664. doi: 10.1210/en.2005-0804. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. G protein-coupled receptor kinases. Cell. 1993;74:409–412. doi: 10.1016/0092-8674(93)80042-d. [DOI] [PubMed] [Google Scholar]
- Lessey BA, Castelbaum AJ, Sawin SW, Buck CA, Schinnar R, Bilker W, et al. Aberrant integrin expression in the endometrium of women with endometriosis. J Clin Endocrinol Metab. 1994;79:643–649. doi: 10.1210/jcem.79.2.7519194. [DOI] [PubMed] [Google Scholar]
- Levi NL, Hanoch T, Benard O, Rozenblat M, Harris D, Reiss N, et al. Stimulation of Jun N-terminal kinase (JNK) by gonadotropin-releasing hormone in pituitary alpha T3-1cell line is mediated by protein kinase C, c-Src, and CDC42. Mol Endocrinol. 1998;12:815–824. doi: 10.1210/mend.12.6.0120. [DOI] [PubMed] [Google Scholar]
- Liapis H, Adler LM, Wick MR, Rader JS. Expression of alpha(v)beta3 integrin is less frequent in ovarian epithelial tumors of low malignant potential in contrast to ovarian carcinomas. Hum Pathol. 1997;28:443–449. doi: 10.1016/s0046-8177(97)90033-2. [DOI] [PubMed] [Google Scholar]
- Lumsden MA, Kelly RW, Baird DT. Primary dysmenorrhoea: the importance of both prostaglandins E2 and F2 alpha. Br J Obstet Gynaecol. 1983;90:1135–1140. doi: 10.1111/j.1471-0528.1983.tb06460.x. [DOI] [PubMed] [Google Scholar]
- Lundstrom V, Green K. Endogenous levels of prostaglandin F2alpha and its main metabolites in plasma and endometrium of normal and dysmenorrheic women. Am J Obstet Gynecol. 1978;130:640–646. doi: 10.1016/0002-9378(78)90320-4. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Daaka Y, Lefkowitz RJ. Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr Opin Cell Biol. 1999;11:177–183. doi: 10.1016/s0955-0674(99)80023-4. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, van Biesen T, Hawes BE, Koch WJ, Krueger KM, Touhara K, et al. G-protein-coupled receptors and their regulation: activation of the MAP kinase signaling pathway by G-protein-coupled receptors. Adv Second Messenger Phosphoprotein Res. 1997;31:263–277. [PubMed] [Google Scholar]
- Morino N, Mimura T, Hamasaki K, Tobe K, Ueki K, Kikuchi K, et al. Matrix/integrin interaction activates the mitogen-activated protein kinase, p44erk-1and p42erk-2. J Biol Chem. 1995;270:269–273. doi: 10.1074/jbc.270.1.269. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Fujino H, Srinivasan D, Regan JW. Activation of FP prostanoid receptor isoforms leads to Rho-mediated changes in cell morphology and in the cell cytoskeleton. J Biol Chem. 1999;274:35944–35949. doi: 10.1074/jbc.274.50.35944. [DOI] [PubMed] [Google Scholar]
- Preissner KT. Structure and biological role of vitronectin. Annu Rev Cell Biol. 1991;7:275–310. doi: 10.1146/annurev.cb.07.110191.001423. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Fernandez JL, Rozengurt E. Bombesin, bradykinin, vasopressin, and phorbol esters rapidly and transiently activate Src family tyrosine kinases in Swiss 3T3 cells. Dissociation from tyrosine phosphorylation of p125 focal adhesion kinase. J Biol Chem. 1996;271:27895–27901. doi: 10.1074/jbc.271.44.27895. [DOI] [PubMed] [Google Scholar]
- Sales KJ, Boddy SC, Williams AR, Anderson RA, Jabbour HN. F-prostanoid receptor regulation of fibroblast growth factor 2 signalling in endometrial adenocarcinoma cells. Endocrinology. 2007;148:3635–3644. doi: 10.1210/en.2006-1517. [DOI] [PubMed] [Google Scholar]
- Sales KJ, Jabbour HN. Cyclooxygenase enzymes and prostaglandins in pathology of the endometrium. Reproduction. 2003a;126:559–567. doi: 10.1530/rep.0.1260559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales KJ, Jabbour HN. Cyclooxygenase enzymes and prostaglandins in reproductive tract physiology and pathology. Prostaglandins Other Lipid Mediat. 2003b;71:97–117. doi: 10.1016/s1098-8823(03)00050-9. [DOI] [PubMed] [Google Scholar]
- Sales KJ, List T, Boddy SC, Williams AR, Anderson RA, Naor Z, et al. A novel angiogenic role for prostaglandin F2alpha-FP receptor interaction in human endometrial adenocarcinomas. Cancer Res. 2005;65:7707–7716. doi: 10.1158/0008-5472.CAN-05-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales KJ, Maudsley S, Jabbour HN. Elevated prostaglandin EP2 receptor in endometrial adenocarcinoma cells promotes vascular endothelial growth factor expression via cyclic 3′,5′-adenosine monophosphate-mediated transactivation of the epidermal growth factor receptor and extracellular signal-regulated kinase 1/2 signaling pathways. Mol Endocrinol. 2004a;18:1533–1545. doi: 10.1210/me.2004-0022. [DOI] [PubMed] [Google Scholar]
- Sales KJ, Milne SA, Williams AR, Anderson RA, Jabbour HN. Expression, localization, and signaling of prostaglandin F2 alpha receptor in human endometrial adenocarcinoma: regulation of proliferation by activation of the epidermal growth factor receptor and mitogen-activated protein kinase signaling pathways. J Clin Endocrinol Metab. 2004b;89:986–993. doi: 10.1210/jc.2003-031434. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c-Src. J Biol Chem. 1997;272:13189–13195. doi: 10.1074/jbc.272.20.13189. [DOI] [PubMed] [Google Scholar]
- Seger R, Seger D, Reszka AA, Munar ES, Eldar-Finkelman H, Dobrowolska G, et al. Overexpression of mitogen-activated protein kinase kinase (MAPKK) and its mutants in NIH 3T3 cells. Evidence that MAPKK involvement in cellular proliferation is regulated by phosphorylation of serine residues in its kinase subdomains VII and VIII. J Biol Chem. 1994;269:25699–25709. [PubMed] [Google Scholar]
- Singh EJ, Baccarini I, Zuspan FP. Levels of prostaglandins F-2alpha and E-2 in human endometrium during the menstrual cycle. Am J Obstet Gynecol. 1975;121:1003–1006. doi: 10.1016/0002-9378(75)90927-8. [DOI] [PubMed] [Google Scholar]
- Slack BE. Tyrosine phosphorylation of paxillin and focal adhesion kinase by activation of muscarinic m3 receptors is dependent on integrin engagement by the extracellular matrix. Proc Natl Acad Sci USA. 1998;95:7281–7286. doi: 10.1073/pnas.95.13.7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner TM, Liu ET, Craven RJ, Cance WG. Expression of focal adhesion kinase gene and invasive cancer. Lancet. 1993;342:1024–1025. doi: 10.1016/0140-6736(93)92881-s. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–119. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]