

Abstract
Inflammatory hepatocellular adenomas (IHCA) are benign liver tumours defined by the presence of inflammatory infiltrates and by the elevated expression of inflammatory proteins in tumour hepatocytes1,2. Here we show a striking activation of the IL6 signalling pathway in this tumour type, and sequencing candidate genes pinpointed this response to somatic gain-of-function mutations in the IL6ST gene that encodes the signalling co-receptor gp130. Indeed, 60% of IHCA harbour small in-frame deletions that target the binding site of gp130 for IL6, and expression of four different gp130 mutants, in hepatocellular cells, activates STAT3 in absence of ligand. Further, analysis of hepatocellular carcinomas revealed that rare gp130 alterations are always accompanied by β-catenin-activating mutations, suggesting a cooperative effect of these signalling pathways in the malignant conversion of hepatocytes. The recurrent gain-of-function gp130 mutations in these human hepatocellular adenomas fully explains activation of the acute inflammatory phase observed in tumourous hepatocytes, and suggests that similar alterations may occur in other inflammatory epithelial tumours having STAT3 activation.
Keywords: Adenoma, Liver Cell; genetics; pathology; Cell Line, Tumor; Cytokine Receptor gp130; genetics; metabolism; Gene Expression Profiling; Gene Expression Regulation, Neoplastic; Humans; Inflammation; genetics; pathology; Interferons; metabolism; Interleukin-6; metabolism; STAT3 Transcription Factor; metabolism; Sequence Deletion; genetics; Signal Transduction
Keywords: hepatocellular adenoma, heaptocellular carcinoma, gp130, inflammation, oncogene, mutation
Several recent studies have shown STAT3 activation in epithelial tumours, underscoring the importance of IL6 signalling and the inflammatory response in tumourigenesis, which provides an opportunity for therapeutic intervention3. However, the mechanisms that provoke sustained STAT3 activation in tumours are largely unresolved. To define the interaction between the inflammatory response and carcinogenesis in liver tumours, we assessed inflammatory hepatocellular adenomas (IHCA), benign tumours predominately found in women and frequently associated with obesity and alcohol use1,2. Tumour hepatocytes of these adenomas express elevated levels of serum amyloid A (SAA) and C-reactive (CRP) proteins, two members of the acute-phase inflammatory response, whereas SAA and CRP are not expressed in inflammatory cells, Kupffer cells, or other sinusoidal cells in IHCA (Fig. 1a, Supplementary Fig. S1, and Ref. 1). Inflammatory infiltrates were largely localised to arterial vessels, but were also found within the sinusoidal lumens of IHCA. Here, CD45+CD3+ T lymphocytes (CD4:CD8, 2:1) were intermingled with less numerous CD20+CD79A+ B cells, some plasma cells and a few polymorphonuclear cells; no CD30+ lymphocytes, nor CD56+ or CD57+ NK cells were observed. In addition, CD68+ histiocytes were present in infiltrates, as well as prominent Kupffer cells in sinusoidal lumens (Supplementary Fig. S1). Overall, inflammatory infiltrates observed in IHCA were highly polymorphous.
Figure 1. Activation of the interleukin-6 and interferon pathways in inflammatory HCA.

a, Immunohistochemical analysis of CRP expression: high level of expression in tumour hepatocytes (IHCA); adjacent normal non-tumour liver hepatocytes (NTL) and inflammatory cells located in tumour (arrow) are negative. b, qRT-PCR validation of gene array expression data comparing IHCA (n=14, black) to NTL (n=6, white). Graphs plot mean +/− SD. *, **, *** difference between groups at P<0.05; 0.01 and 0.001, respectively (two-tailed Mann-Whitney test). c, Western-blot analysis comparing expression levels of STAT1 (84, 91 kDa), STAT3 (79, 86 kDa), CRP (24 kDa), VEGF (22 kDa), Jak2 and phospho-Jak2 (125 kDa) between IHCA and NTL. d, Schematic representation of the IL6 and interferon pathways showing all genes overexpressed (red) or suppressed (green) in IHCA compared to NTL. Genes validated by qRT-PCR are underlined (IHCA n=14 versus NTL n=6); others were extracted from the microarray analysis (IHCA n=4 versus NTL n=4).
To resolve the underlying pathogenesis of these inflammatory lesions, a genome-wide transcriptome analysis of four IHCA was compared to four normal liver tissue samples. Among the 285 genes significantly overexpressed in IHCA (Supplementary Table S1), gene ontology analysis identified a strong enrichment for genes associated with inflammation and the immune response, accounting for 40% of the overall terms significantly enriched (Supplementary Table S2). High levels of significance were found for “antigen processing and presentation of peptide antigen” (P=2.10−11) and “regulation of the JAK/STAT cascade” (P=10−5) (Supplementary Table S2). We confirmed this inflammatory signature in an additional 14 IHCA with a clear activation of the acute-phase inflammatory response affecting both type-1 and type-2 acute-phase genes (Fig. 1b and 1c, Supplementary Table S3). Consistent with the known roles of IL6 and JAK-STAT signalling in the acute-phase response4,5, STAT3 mRNA and protein were significantly elevated in IHCA (Fig. 1b and 1c). IHCA also overexpressed several effectors of type-1 and type-2 interferon signalling pathways (e.g., JAK2, STAT1 and STAT2) and their downstream targets (Fig. 1b, 1c). Collectively, these data suggest that IL6 and interferon signalling are the main inflammatory pathways activated in IHCA (Fig. 1d).
Since IL6 was not overexpressed in IHCA and because the inflammatory response was restricted to tumour hepatocytes (Fig. 1a), we reasoned that somatic genetic mutation(s) might account for activation of IL6 receptor signalling in IHCA. We selected IL6ST as a candidate gene since it encoded the cell surface signalling receptor gp130 shared by at least six different cytokines including IL6, IL11, LIF, OSM, CTNF and CT-14,6. We sequenced the entire gp130 coding region in 43 IHCA and 33 non-inflammatory hepatocellular adenomas. Remarkably, 26 mutations in gp130 were identified specifically in 60% (26/43) of IHCA, and these included 16 unique, small in-frame deletions and one 33 bp in-frame duplication in exon 6 (Fig. 2a and Table 1). Notably, all IL6ST mutations were found in IHCA and all were of somatic origin, as they were not observed in adjacent normal liver tissues. In all cases, IL6ST mutations were monoallelic, and IHCA with these mutations expressed both the wild type and mutated alleles at comparable levels, as judged by sequencing RT-PCR products of IL6ST mRNA (Supplementary Fig. S2).
Figure 2. gp130 mutations in inflammatory hepatocellular tumours.
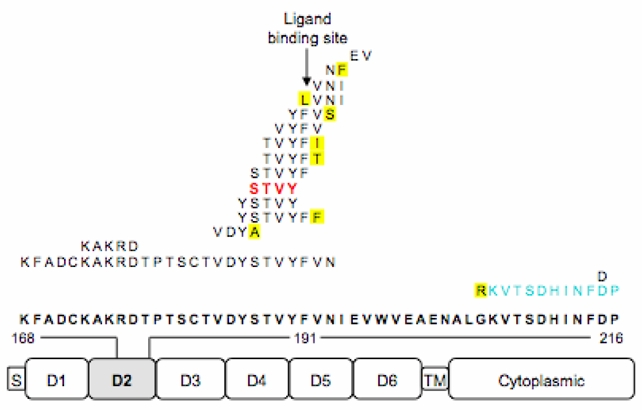
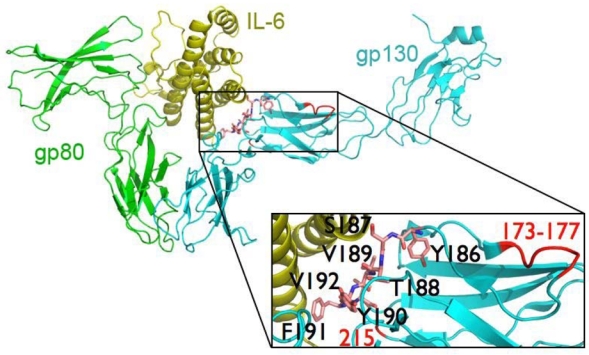
a, The spectrum of mutations in gp130 detected in IHCA (S, signal peptide; TM, transmembrane domain). The location of the 16 deletions, the duplication (in blue), and the resulting amino acid substitutions (in yellow) identified in the D2 domain of gp130 are indicated above the wild type sequence (in bold font). The most frequent mutation is indicated in red. b, Residues 186–192 (shown in pink stick presentation) of the D2 domain of gp130 that are deleted in IHCA are intimately involved in interactions with IL6, as shown in the crystal structure of the IL6R:IL6:gp130 complex (PDB 1P9M)7. IHCA deletions 173–177 and 215 (labelled in red) are also predicted to disrupt the IL6-gp130 interface. The numbering of residues corresponds to the IL6ST cDNA, which has 22 additional N-terminal residues compared to the polypeptide chain due to the nuclear export signal.
Table 1.
gp130 somatic mutations identified in inflammatory hepatocellular tumours
| Number of mutated tumours | Nucleotide change* | Amino acid change* |
|---|---|---|
| 6** | 560_571del | S187_Y190del |
| 4** | 557_571del | Y186_Y190del |
| 3 | 565_576del | V189_V192del |
| 2 | 563_574del | T188_F191del, V192I |
| 1 | 503_580del | K168_N192del |
| 1** | 518_532del | K173_D177del |
| 1** | 551_559del | V184_Y186del, S187A |
| 1 | 557_574del | Y186_F191del, V192F |
| 1 | 559_573del | S187_F191del |
| 1 | 564_575del | T188_F191del, V192T |
| 1 | 569_577del, 578A>C | Y190_V192del, N193S |
| 1 | 573T>G, 574_582del | F191L, V192_I194del |
| 1 | 575_583del | V192_I194del |
| 1 | 577_579del, 580A>T | N193del, I194F |
| 1 | 583_588del | E195_V196del |
| 1 | 614_646dup | K206_P216dup, G205R |
| 1 | 643_645del | D215del |
Codons and mutated nucleotides are numbered according to IL6ST cDNA ORF
gp130 mutants analysed in Hep3B cells
Binding of IL-6 to its cognate receptor gp80 (encoded by IL6R) induces formation of a high affinity ternary hexameric complex consisting of two molecules each of IL6, IL6R and gp1307,8. Gp130 engagement then activates JAK/Tyk tyrosine kinases and the STAT family of transcription factors9–11. The consequences of the IL6ST in-frame deletions observed in IHCA included the removal of 1 to 26 amino acids neighbouring the IL6/IL6R binding site (also known as CHR E-F loop) located in D2 domain of gp130 (Fig. 2b). We modelled the different deletions and the duplication in the known crystal structure of the wild type IL6/IL6R/gp130 ternary complex (PDB 1P9M)7. All of these mutations are predicted to disrupt key residues involved in the gp130-IL6 interface. Specifically, the most frequent alterations target residues 186–191, which direct the gp130-IL6 interaction, whereas the remaining deletions and duplication affect the other two loops that contribute to gp130-IL6 interactions (Fig. 2b). Therefore, the gp130-IL6 interface is targeted in IHCA.
To investigate possible functional consequences of these gp130 mutations, we tested the effects of enforced expression of two frequent deletions (S187_Y190del and Y186_Y190del) and two infrequent mutants (V184_Y186del, S187A and K173_D177del) in Hep3B cells, a hepatocellular carcinoma line that activates the acute inflammatory phase following IL6 treatment12. In the absence of IL6 ligand and serum, overexpression of wild type gp130 alone was not sufficient to activate STAT3 and the downstream acute-phase inflammatory genes (Fig. 3 and Refs. 7,13). In contrast, all gp130 IHCA mutants activated an acute phase inflammatory response and induced typical targets of this response, including CRP, SAA2, SPINK1 and FBG (Fig. 3a, 3c). Further, as observed in IHCA, all of these gp130 mutants induced the expression of SOCS3, which normally serves to harness cytokine signalling (Fig. 3a). Mutant gp130 S187_Y190del was constitutively tyrosine phosphorylated and the activity of STAT3 was clearly increased in gp130 S187_Y190del-expressing Hep3B cells (Fig. 3b). Similarly, immunohistochemical analyses of IHCA demonstrated marked increases in nuclear STAT3 phosphorylated at Tyr-705 (Supplementary Fig. S4e–f). Finally, IL6 augmented the induction of CRP in gp130 S187_Y190del-expressing cells, but mutant receptors were not hypersensitive to low doses of IL6 (Fig. 3d). Therefore, gp130 mutants are constitutively active, and they activate STAT3 and inflammatory response genes in the absence of IL6.
Figure 3. Gain-of-function mutations of gp130.
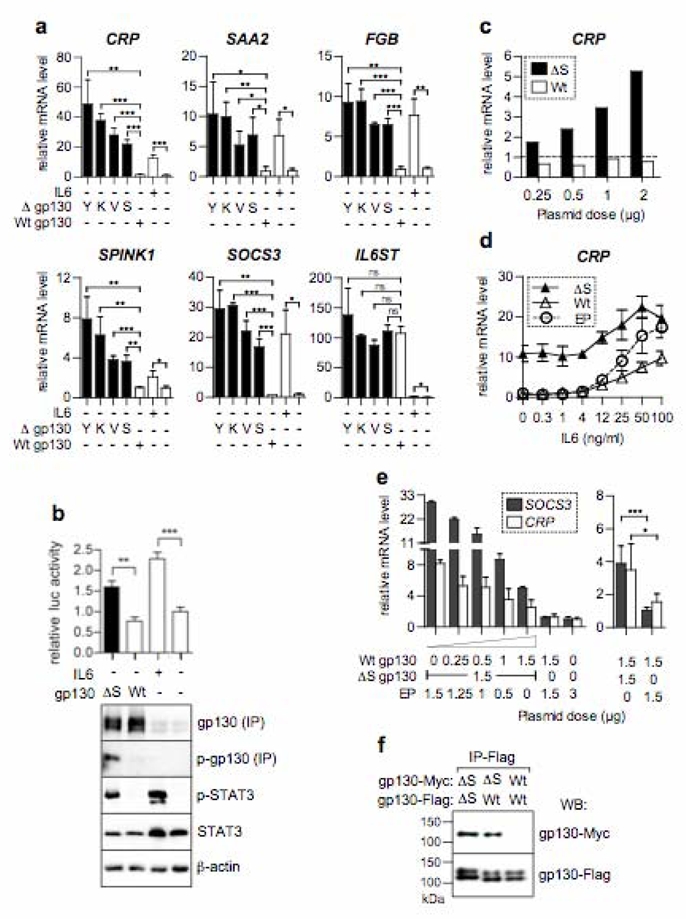
a, Gp130 mutants (Δ) including S187_Y190del (S), Y186_Y190del (Y), V184_Y186del, S187A (V) and K173_D177del (K) or controls including gp130 wild type (Wt) and empty plasmid (EP) were transfected in Hep3B. Graphs plot qRT-PCR results relative to EP transfected unstimulated cells, mean +/− SD (transfections in triplicate). b, Graph plots luciferase activity (mean) measured in triplicate transfections (+/−SD) with pSTAT3-luc alone or together with Wt or the ΔS gp130. Expression of total and phosphorylated (p-STAT3 and p-gp130) proteins were analysed using western blotting, after immunoprecipitation (IP) for gp130. c, CRP mRNA expression after transfection with increasing amounts of plasmids expressing Wt or ΔS gp130 (baseline, mock-transfected cells). d, Hep3B transfected with EP, Wt or ΔS gp130 and exposed to increasing concentrations of IL6. Graph plots mean CRP expression value relative to cells transfected with EP +/− SD (transfections in triplicate). e, Effect of increasing amounts of Wt gp130 on the ΔS gp130 mutant activity, without IL6. Graph plots mean level of expression of SOCS3 and CRP relative to cells transfected with EP +/− SD in duplicate (left) or in 5 independent transfections (right). f, Flag and Myc-tagged constructs expressing either the wild-type or the mutant ΔS gp130 were co-transfected (1:1) into Hep3B. gp130 dimers formation was detected after immunoprecipitation using the anti-flag antibody, followed by western blot analysis with the anti-myc and anti-flag antibodies. Shown is one representative of three independent experiments. *, **, *** indicate differences between groups at P<0.05; 0.01 and 0.001, respectively (two-tailed t-test).
A critical step in the activation of intracellular signalling after IL6 binding on gp130 is the formation a hexameric structure that juxtaposes the membrane proximal domains of two gp130 molecules at the cell surface7,14,15. Using co-immunoprecipitation, we showed that the gp130 S187_Y190del IHCA mutant was able to homodimerize or heterodimerize with wild-type gp130 independently of IL6, whereas wild-type gp130 cannot homodimerize (Fig. 3f). Homodimerization of gp130 in absence of ligand has also been previously described for two other gp130 mutants, Y190FV to AAA and Y190A16. Moreover, overexpression of wild-type gp130 impaired the activity of the mutant gp130 S187_Y190del in a dose-dependent manner (Fig 3e); therefore, mutant gp130 activity appears to be driven by its homodimerization that can be competed by the wild-type protein. Interestingly, using a reverse genetic approach in mice, Ernst and collaborators have recently shown that IL11 promotes chronic gastric inflammation and associated tumourigenesis mediated by gp130 and STAT3 activation17. In IHCA, we also found significant increases (6-fold) in the levels of IL11 mRNA (P<0.01) that may serve to amplify gp130/IL6 signalling, despite a modest decrease in the expression of its specific receptor (IL11RA) (Supplementary Fig. S5a). However, in mutant gp130-expressing Hep3B cells IL11 was not induced (Supplementary Fig. S5b), suggesting that elevation of IL11 in IHCA in vivo may occur through secondary effects.
Relationships between activation of IL6 signalling and hepatic tumourigenesis have also been suggested from studies in mice, where transgenics that overexpress both IL6 and IL6R in hepatocytes develop large liver adenomas18. Further, in knock-in mice expressing gp130Y757 mutant, gastric inflammation and subsequent carcinoma development result from STAT3 activation19–21. The IL6 pathway is also known to activate SHP-2/RAS/ERK signalling22. While in humans STAT3 activation is a clear hallmark of gp130-mutated IHCA, phosphorylated ERK1/2 was only detected in 50% of these tumours (Supplementary Fig. S6a), and phospho-ERK1/2 was not observed in Hep3B overexpressing mutant gp130 (Supplementary Fig. S6b). Thus, in contrast to STAT3 activation, activation of the SHP-2/RAS/ERK pathway does not appear to contribute to all IHCA.
Surprisingly, type 1 and type 2 interferon targets are overexpressed in gp130-mutated IHCA. Their aberrant activation here might be due to gp130-mediated STAT1/STAT2 activation and inefficient negative feedback by SOCS3, which is thought to prevent activation of type 2 interferon genes under physiological conditions23. Interestingly, the targets in gp130-mutated IHCA include HIF1A that encodes hypoxia-inducible factor-1a which may contribute to VEGF overexpression and the “telangiectatic” phenotype of IHCA (Fig. 1b, 1c)1,2,24. Further, the inflammatory infiltrate that is a hallmark of IHCA may reflect the robust induction (~110-fold) of CCL20 observed in all IHCA (Fig. 1b). Specifically, CCL20 is a chemokine that interacts with CCR6 and that directs chemoattraction of a wide spectrum of immune cells including dendritic cells and B and T lymphocytes25, and such a scenario could direct or augment the inflammatory response seen in IHCA.
To investigate the possible interaction and cooperation of gp130 activation with other pathways altered in hepatocellular adenoma, we searched in the same series of 76 adenomas for CTNNB1 (encoding β-catenin) and HNF1A mutations (encoding Hepatocyte Nuclear Factor 1α), two genetic alterations frequently associated with hepatocellular adenoma (HCA)2,26,27. HNF1α inactivation (22 cases) and gp130 activation (26 cases) were mutually exclusive in HCA (P<10−4, Fisher exact test, Supplementary Table S4). By contrast, β-catenin activating mutations were found in 12 adenomas, and half of these had associated gp130 mutations. For two IHCA associated with a malignant transformation to carcinoma, both activating gp130 and β-catenin mutations were found (cases 469 and 786, Supplementary Table S4). In one of these cases, we were able to genotype the hepatocellular carcinoma (HCC) and this harboured the same mutations in CTNNB1 and IL6ST as the corresponding IHCA. In contrast, no cases of malignant transformation were observed in gp130-mutated IHCA without a β-catenin mutation (P=0.05, two-tailed Fisher exact test). When we sequenced IL6ST exon 6 in 111 HCC lacking a known history of IHCA28, only two cases were mutated. Interestingly, these tumours were compatible with a malignant transformation of a non-diagnosed IHCA since both mutated HCC had inflammatory infiltrates; they harboured β-catenin-activating mutations and developed in non-cirrhotic liver (Supplementary Table S4). Collectively, these findings suggest cooperation between the IL6 and β-catenin pathways in the malignant transformation of IHCA.
IL6ST mutations were identified in 60% of IHCA. The remaining IHCA were, however, similar in their expression profiles as IL6ST-mutated IHCA (Supplementary Fig. S3) and, accordingly, activated nuclear phospho-STAT3 was also found in all non-mutated IHCA tested (Supplementary Fig. S4f). Sequencing of the functional domains of three gp130 co-receptors (IL6R, LIFR, and OSMR), the functional domains of three transducing proteins (JAK2, JAK1, and TYK2), and of the coding sequence of SOCS3, failed to identify mutations in gp130-non-mutated IHCA. Moreover, these tumours lacked mutations in the exons or intron/exon boundaries of IL6ST, and IL6ST mRNA levels were comparable to those expressed in normal hepatocytes (Supplementary Fig. S4a). Finally, no gain of chromosome 5q copy number at the IL6ST locus was observed using CGH-SNP experiments in gp130 mutated and non-mutated IHCA (data not shown). However, by western blotting and immunohistochemistry, we found that most IHCA cases overexpressed gp130 protein (Supplementary Fig. S4b–d), suggesting that the translation or turnover of gp130 is affected in both gp130-non-mutated and mutated tumours. Therefore, gp130 overexpression and STAT3 activation are related to gp130 activating mutation in 60% of the IHCA, whereas in the non-mutated cases, the mechanism of STAT3 activation remains to be defined. Here, for example, activation may reflect mutations in a regulatory protein(s) that controls gp130 stabilisation, or be due to alterations in factors that control its translation.
The present findings reveal that STAT3 activation and the inflammatory phenotype in benign liver tumours can occur through gain-of-function somatic mutations in gp130. To our knowledge, this is the first identification of somatic mutation of gp130 in tumours and they define gp130 as an oncogene with involvement in benign human tumours. Our findings also underscore the important role of inflammatory response in hepatocellular tumourigenesis and are in accord with a recent study demonstrating that IL6 promotes liver tumour onset in mice29. Importantly, our studies also suggest that cooperating mutations of the WNT/β-catenin and IL6/STAT pathways in human liver tumours contribute to the occurrence of inflammatory hepatocellular carcinomas that develop in the absence of cirrhosis. More broadly, these findings indicate that this novel mechanism for activation of the IL6 signalling pathway should be interrogated in other human tumours, particularly those having an inflammatory phenotype and STAT3 activation, a hallmark of many epithelial tumours.
Methods summary
All tumours and corresponding non-tumour liver tissues were frozen after surgical resection. These tumours were clinically and genetically characterized; they were previously included in genetic and phenotypic studies (Supplementary Table S4 and refs. 1, 2 and 28). The study was approved by the local Ethics Committee (Paris Saint-Louis), and informed consent was obtained in accordance with French legislation. Transcriptional profiling were performed using Affymetrix oligonucleotide GeneChips HG-U133A and the microarray data have been deposited in Gene Expression Omnibus (GEO) under accession number GSE11819. Quantitative RT-PCR was performed as described30 using pre-designed primers and probe sets from Applied Biosystems (provided in Supplementary Table S5). In all cases the somatic origin of the mutation found in tumour was verified by sequencing the corresponding adjacent, normal liver sample. In HEP3B cell experiments, all quantitative RT-PCR, luciferase assay and western blotting were performed two days after transfection and after 6 hours of serum starvation.
Methods
A series of 76 HCA was analysed. All samples were collected in Bordeaux hospital. They were extensively characterised and included in previous studies1,2. All the 111 HCC were included and described previously28. For each tumour, a corresponding frozen non-tumour liver sample was available for analyses.
Transcriptome analyses
Transcriptional profiling of four IHCA and four normal liver tissues were performed using Affymetrix oligonucleotide GeneChips HG-U133A (GEO accession number GSE11819). For each case, 5 μg of total RNA were labeled and hybridization was performed using 20 μg of cRNA per hybridization (GeneChip Fluidics Station 400), according to the manufacturer’s instructions (Affymetrix, Santa Clara, CA). Array images were digitized by using Microarray Suite 5.0 (MAS5) software, embedded in the Affymetrix GeneChip Operating Software (Affymetrix, Santa Clara, USA). Gene intensity derivation was carried out from the raw numerical data (CEL files) by using the R package affy31, available as part of the Bioconductor project32, and the DNA-Chip analyzer (dChip) program33. Probe sets corresponding to control genes or having a “_x_” annotation were masked yielding a total of 19,787 probe sets available for further analyses. Background subtraction, normalization and expression summaries for every probe set were calculated with the log-scale robust multi-array analysis (RMA) algorithm using a background adjusted PM intensities model34, and with the model-based expression indexes (MBEI) calculation following a PM/MM difference model33. The log-transformed normalized data was considered for further analysis; and a gene annotation was adjoined using the Unigene cluster identifier as a common primary key (Unigene Build184-June05).
Differential analyses were performed using multiple testing procedures to evaluate statistical significance for differentially expressed genes. The biological groups were compared as independent conditions. A pooled curve-fit error method was used for random error estimation; a range of 3.0 median absolute deviations (MADs) to 4.0 MAD established outlier-detection thresholds automatically. Data were subjected to iterative normalization by centering to the median across biological groups. Statistical analyses were computed using both z- and t-statistics including a p-value ranking. Considering respective tests, a false discovery rate (FDR) adjustment of the p-values was used for multiple testing corrections, and permutation tests (n>1000) were computed to control the proportion of false discoveries (i.e. false positives, > 90% confidence). Only genes whose expression significantly differed between inflammatory HCA versus non-tumour liver tissues (considering absolute fold change > 1.5 and p-values ≤ 0.01) were selected.
Gene ontology (GO) categories that were significantly over-represented in genes significantly up-regulated or suppressed in inflammatory HCA were determined by the hypergeometric test using the web-based tool GOTree Machine (GOTM) (http://bioinfo.vanderbilt.edu/gotm/) by comparison with the distribution of the overall genes included in the HG-U133A Affymetrix array.
Quantitative RT-PCR
Quantitative RT-PCR was performed in duplicate as described30. Ribosomal 18S RNA (R18S) was used to normalize expression data and 2−ΔΔCT method was applied. Final results were expressed as the n-fold differences in target gene expression in tested samples when compared with the mean expression value of non-tumour tissues (for tumour analysis) or with control cell line.
DNA sequencing
DNA sequencing was performed as described26 using primers provided in Supplementary Table S6. All mutations were validated by sequencing a second independent PCR product on both strands.
Cell culture
Hep3B cells (ATCC) were grown in DMEM supplemented with 10% fetal calf serum. For transfections, cells were plated 16h prior to transfection to produce monolayers that were 60% confluent and these were transfected by using Lipofectamine™ LTX according to the manufacturer’s instructions (Invitrogen). Transfection efficiency was monitored by measuring the level of either wild-type and mutated gp130 mRNA using quantitative RT-PCR and western blot analysis. The same gp130 expression level was observed in wild-type and mutant transfected cells. 48h after transfection, cells were maintained in serum-free medium, then cells were either left untreated or were stimulated with rhIL-6 (100 ng/ml) in a serum-free medium for 3 hr, just before cell harvest and protein and RNA extraction. For luciferase assay, Hep3B cells were co-transfected with Stat3 luciferase reporter vector (pSTAT3-luc 0.5μg, Panomics) and the expression plasmid for wild type or S187_Y190del mutant (ΔS) gp130 (1μg). Two days after transfection, the cells were lysed and the luciferase activity was measured according to the manufacturer’s recommendations (Promega). The activities were normalized to protein in each cell lysate.
Generation of gp130 mutants
A full-length gp130 open reading frame cloned in pORF9 vector was purchased from Invivogen (pORF9-hIL6ST). Mutagenesis reactions were performed using the QuickChange site-directed mutgenesis kit (Stratagen). All constructs were verified by sequencing. Using the same method, we also introduced two different epitope tags (myc and flag) at the C-terminal end in the wild-type and S187_Y190del constructs.
Western blot analysis and immunoprecipitation
Western blot analyses were performed as described30 using the antibodies specific for STAT3, phospho-STAT3 Tyr705, STAT1, phospho-JAK2 Tyr1007/1008 (Cell Signaling Technology, diluted 1:500), JAK2 (Santa Cruz Biotechnology, 1:500), CRP (Sigma, 1:500), gp130 (C-20 Santa Cruz Biotechnology, 1:200), and VEGF (Novus Biologicals, 1:100). The phosphorylated form of gp130 (p-gp130) was analysed by SDS-PAGE after immunoprecipitation from cell lysates containing 750 μg of proteins using 5μg of the C-20 anti-gp130 antibody. The membrane was then probed with antibodies directed against phosphotyrosine (clone 4G10, Upstate Biotechnology) and gp130 (C20). For dimerization assays, cell lysates were incubated with Protein G agarose (Pierce) and anti-flag antibody (Cell signaling technology, 1:50), at 4°C overnight. The immune complexes were sedimented, washed, separated by SDS-PAGE and analysed by Western blot using anti-flag (1:1000) and anti-myc (1:1000) antibodies. In western blot, red ponceau staining or actin (Sigma, 1:3000) expression was analysed to appreciate protein loading.
Immunohistochemistry
Immunohistochemistry was performed using a Dako autostainer, on paraffin sections of 10% fixed tumour tissue using 2 monoclonal antibodies against inflammatory proteins: anti-C-reactive protein (CRP), (Abcam, 1:1500), anti-serum amyloid A (SAA), (Dako, 1:50), anti-phospho-STAT3 Tyr705 (Cell signaling Technology, 1:50), anti-gp130 (C-20 Santa Cruz Biotechnology, 1:200). For inflammatory cells immunotyping, the following antibodies (Dako) were used: CD45 (clone EB11+PD7/26, 1:300), CD3 (polyclonal, 1:100), CD4 (clone OPD4, 1:100), CD8 (clone C8/144B, 1:20), CD20 (clone L26, 1:100), CD68 (clone PG-M1, 1:50). For each immunohistochemical procedure, antigen retrieval was performed in citrate buffer, detection was amplified by the Dako Envision system.
Supplementary Material
Acknowledgments
We are indebted to Philippe Bois (The Scripps Research Institute, Jupiter) and Olivier Bernard (Inserm E0210, Paris) for scientific discussion and critical reading of this manuscript. We warmly thank Cristel Thomas and Gaelle Cubel for their participation to this work that is dedicated to Jean-Philippe Salier (Inserm U519, Rouen). We also thank Jean Saric, Christophe Laurent, Antonio Sa Cunha, Brigitte Le Bail, Anne Rullier for contributing to the tissue collection (CHU Bordeaux). This work was supported by Inserm (Réseaux de recherche clinique et réseaux de recherche en santé des populations), the Ligue Nationale Contre le Cancer (“Cartes d’identité des tumeurs” program), ARC (grant 5158), and the Fondation de France. S.R. and M.A. are supported by a fellowship from la Ligue Nationale Contre le Cancer and the Inca, respectively. JZR is supported by an interface contract between Inserm and Bordeaux hospital. TI is supported by the National Institutes of Health grants GM071596, AI055894, and AI067949.
References
- 1.Bioulac-Sage P, et al. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology. 2007;46:740–8. doi: 10.1002/hep.21743. [DOI] [PubMed] [Google Scholar]
- 2.Zucman-Rossi J, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43:515–24. doi: 10.1002/hep.21068. [DOI] [PubMed] [Google Scholar]
- 3.Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumourigenesis? Cancer Cell. 2008;13:7–9. doi: 10.1016/j.ccr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 4.Akira S, et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 5.Wegenka UM, Buschmann J, Lutticken C, Heinrich PC, Horn F. Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly activated by interleukin-6 at the posttranslational level. Mol Cell Biol. 1993;13:276–88. doi: 10.1128/mcb.13.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hibi M, et al. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990;63:1149–57. doi: 10.1016/0092-8674(90)90411-7. [DOI] [PubMed] [Google Scholar]
- 7.Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300:2101–4. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- 8.Ward LD, et al. High affinity interleukin-6 receptor is a hexameric complex consisting of two molecules each of interleukin-6, interleukin-6 receptor, and gp-130. J Biol Chem. 1994;269:23286–9. [PubMed] [Google Scholar]
- 9.Lutticken C, et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. 1994;263:89–92. doi: 10.1126/science.8272872. [DOI] [PubMed] [Google Scholar]
- 10.Murakami M, et al. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science. 1993;260:1808–10. doi: 10.1126/science.8511589. [DOI] [PubMed] [Google Scholar]
- 11.Stahl N, et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science. 1994;263:92–5. doi: 10.1126/science.8272873. [DOI] [PubMed] [Google Scholar]
- 12.Coulouarn C, et al. Genome-wide response of the human Hep3B hepatoma cell to proinflammatory cytokines, from transcription to translation. Hepatology. 2005;42:946–55. doi: 10.1002/hep.20848. [DOI] [PubMed] [Google Scholar]
- 13.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–54. [PubMed] [Google Scholar]
- 14.Chow D, He X, Snow AL, Rose-John S, Garcia KC. Structure of an extracellular gp130 cytokine receptor signaling complex. Science. 2001;291:2150–5. doi: 10.1126/science.1058308. [DOI] [PubMed] [Google Scholar]
- 15.Skiniotis G, Boulanger MJ, Garcia KC, Walz T. Signaling conformations of the tall cytokine receptor gp130 when in complex with IL-6 and IL-6 receptor. Nat Struct Mol Biol. 2005;12:545–51. doi: 10.1038/nsmb941. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Nicholas J. Identification of amino acid residues of gp130 signal transducer and gp80 alpha receptor subunit that are involved in ligand binding and signaling by human herpesvirus 8-encoded interleukin-6. J Virol. 2002;76:5627–36. doi: 10.1128/JVI.76.11.5627-5636.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ernst M, et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumourigenesis in gp130 receptor mutant mice. J Clin Invest. 2008;118:1727–38. doi: 10.1172/JCI34944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maione D, et al. Coexpression of IL-6 and soluble IL-6R causes nodular regenerative hyperplasia and adenomas of the liver. Embo J. 1998;17:5588–97. doi: 10.1093/emboj/17.19.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Judd LM, et al. Gastric cancer development in mice lacking the SHP2 binding site on the IL-6 family co-receptor gp130. Gastroenterology. 2004;126:196–207. doi: 10.1053/j.gastro.2003.10.066. [DOI] [PubMed] [Google Scholar]
- 20.Judd LM, et al. STAT3 activation regulates growth, inflammation, and vascularization in a mouse model of gastric tumourigenesis. Gastroenterology. 2006;131:1073–85. doi: 10.1053/j.gastro.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 21.Tebbutt NC, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med. 2002;8:1089–97. doi: 10.1038/nm763. [DOI] [PubMed] [Google Scholar]
- 22.Boulton TG, Stahl N, Yancopoulos GD. Ciliary neurotrophic factor/leukemia inhibitory factor/interleukin 6/oncostatin M family of cytokines induces tyrosine phosphorylation of a common set of proteins overlapping those induced by other cytokines and growth factors. J Biol Chem. 1994;269:11648–55. [PubMed] [Google Scholar]
- 23.Croker BA, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–5. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 24.Bioulac-Sage P, et al. Clinical, morphologic, and molecular features defining so-called telangiectatic focal nodular hyperplasias of the liver. Gastroenterology. 2005;128:1211–8. doi: 10.1053/j.gastro.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Schutyser E, Struyf S, Van Damme J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003;14:409–26. doi: 10.1016/s1359-6101(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 26.Bluteau O, et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet. 2002;32:312–5. doi: 10.1038/ng1001. [DOI] [PubMed] [Google Scholar]
- 27.Chen YW, Jeng YM, Yeh SH, Chen PJ. P53 gene and Wnt signaling in benign neoplasms: beta-catenin mutations in hepatic adenoma but not in focal nodular hyperplasia. Hepatology. 2002;36:927–35. doi: 10.1053/jhep.2002.36126. [DOI] [PubMed] [Google Scholar]
- 28.Boyault S, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 29.Naugler WE, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–4. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 30.Rebouissou S, et al. HNF1alpha inactivation promotes lipogenesis in human hepatocellular adenoma independently of SREBP-1 and carbohydrate-response element-binding protein (ChREBP) activation. J Biol Chem. 2007;282:14437–46. doi: 10.1074/jbc.M610725200. [DOI] [PubMed] [Google Scholar]
- 31.Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–15. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 32.Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:Research0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irizarry RA, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.