

Abstract
The ability to charge huge biomolecules without breaking them apart has made matrix-assisted laser desorption/ionization (MALDI) mass spectrometry an indispensable tool for biomolecular analysis. Conventional, empirically selected matrices produce abundant matrix ion clusters in the low-mass region (<500 Da), hampering the application of MALDI-MS to metabolomics. An ionization mode of MAILD, a rational protocol for matrix selection based on Brønsted–Lowry acid–base theory and its application to metabolomics, biological screening/profiling/imaging, and clinical diagnostics is illustrated. Numerous metabolites, covering important metabolic pathways (Krebs' cycle, fatty acid and glucosinolate biosynthesis), were detected in extracts, biofluids, and/or in biological tissues (Arabidopsis thaliana, Drosophila melanogaster, Acyrthosiphon pisum, and human blood). This approach moves matrix selection from “black art” to rational design and sets a paradigm for small-molecule analysis via MALDI-MS.
Keywords: clinical diagnostic, matrix design, positive/negative ions, ion-formation model, MALDI
Owing to their “soft” nature of ionization, MALDI-MS (1, 2) and electrospray ionization- mass spectrometry (3) (ESI-MS) have been at the forefront of bioanalytical research with far-reaching applications in proteomics (4), genomics (5), biological imaging (6), and metabolomics (7). In MALDI-MS, biomolecules mixed with matrices (small, UV-absorbing compounds) and exposed to laser pulses form gas-phase ions that are typically measured in time-of-flight (TOF) mass analyzers. Although the high-throughput nature of MALDI-MS makes it an ideal tool for large-scale metabolomic studies, its application in the field has been rather limited. This is because all conventional matrices (8–10) produce a forest of interfering low-mass ions (<500 Da) obscuring the detection of metabolites in the range. Despite several approaches (11–13), challenging MALDI-MS-based metabolomic tasks such as direct biofluid analysis, on-tissue metabolite screening, and generation of snapshots of the metabolic machinery of biological systems remains an unmet challenge.
These limitations call for matrices devoid of interfering ions (“ionless matrices”), yet still assisting an efficient ionization/desorption of the analytes. An ideal solution would be to have a rational selection protocol for such matrices, whereby depending on the properties of the analytes of interest, appropriate matrices could be designed. Such a development would not only cross a long-lasting hurdle of empirical selection of MALDI matrices but would also provide a powerful, fast, and easy-to-use tool to the biological community to selectively probe into the metabolomes of living organisms.
Here, we report on a first-ever rational selection protocol for matrix-assisted mass spectrometry matrices based on the classical Brønsted–Lowry acid–base theory (14) and density functional theory (DFT) quantum chemical calculations. The matrices developed herein are ionless, in other words, the matrices produce no interfering matrix- related ions, thus overcoming the problem of most conventional matrices and allowing the detection of small molecules (0–1,000 Da). Furthermore, the enormous applicability of our matrices in performing targeted metabolomic studies is demonstrated.
Results and Discussion
Brønsted–Lowry Concept Was Successfully Applied for the Selection of Ionless Matrices with the Potential to Study a Wide Variety of Metabolites.
Conventional matrices such as 4-hydoxy-α-cyanocinnamic acid (α-CHCA) and 2,5-dihydroxybenzoic acid (DHB) produce copious interfering ions in the m/z < 500 Th range, making detection of metabolites in that range rather difficult, if not impossible (Fig. 1A, B, D, and E). We reasoned that mixing an acidic or basic analyte with a strong base or acid, respectively, used as a matrix and crystallization of the salt formed on a target, followed by desorption of the formed salt by a laser pulse having frequency similar to the absorption maximum of base/acid matrix, should produce ions that are oppositely charged to those of the absorbing partner [Fig. 1 C and F and supporting information (SI) Fig. S1]. To explore this concept, we investigated ion pairs of 4 aromatic bases with aliphatic acids monitoring the formation of anions using TOF mass spectrometry. The first experiments were performed with a “proton sponge” (15, 16), which is a strong base [pKa of its conjugate acids 12.5 (15)], 1,8-bis(dimethylamino)naphthalene (DMAN), never previously used as a matrix. Analytes (fatty acids, fatty acid–amino acid conjugates and other anionic species) were mixed in a 1:1 molar ratio with DMAN in ethanol and allowed to dry on a MALDI metallic target. Under both vacuum and atmospheric pressure, gas-phase anions were formed upon UV-laser irradiation (337 or 355 nm) and detected at physiologically relevant concentrations (Fig. S2). Negligible fragmentation was observed except in polyhydroxylated compounds such as water losses in prostaglandin E1 (Fig. S2G). In contrast to common MALDI matrices, no matrix ions were detected. The absence of matrix ions in mass spectra affords a crucial advantage over existing matrices. To ensure that we were observing a matrix-assisted process and not laser desorption/ionization (LDI), mixtures of an aliphatic base (triethylamine) and an inorganic base (KOH) with strong aliphatic acids (e.g., trifluoroacetic acid) were made and tested for the presence of anion signals. No analyte signal was observed even at ablative laser fluences, thus confirming the importance of the UV-absorbing partner in the ionization process.
Fig. 1.

Comparison of mass spectra of 1,8-bis(dimethylamino)naphthalene (DMAN) with conventional matrices; 4-hydroxy-α-cyanocinnamic acid (α-CHCA) and 2,5-dihydroxybenzoic acid (DHB) when mixed with stearic acid (SA) in 1:1 molar ratios. Positive mode analysis. (A) Spectra for α-CHCA + SA. (B) Spectra for DHB + SA. (C) Spectra for DMAN + SA. Spectra in A and B are dominated by copious matrix ions with no signal for [M+H]+ of SA at m/z 285 (transparent blue bars); clearly, none of the 3 matrices shows the protonated analyte. C shows DMAN [M+H]+ at m/z 215 resulting from DMAN protonated with SA. Negative mode analysis: (D) Spectra for α-CHCA + SA. (B) Spectra for DHB + SA. (F) Spectra for DMAN + SA. Spectra in D and E show only copious matrix clusters with no signal for the deprotonated stearate ion at m/z 283 (transparent red bar); single ion at m/z 283 corresponding to stearate anion is observed, with no additional matrix peaks (F).
Potential of Ionless Matrices for Targeted Metabolomics Was Demonstrated on Plant, Insect, and Blood Samples.
In situ profiling of a damaged Arabidopsis thaliana leaf spotted with DMAN solution provided rich negative ion-mode mass spectra (108 monoisotopic peaks; Fig. 2A) with the mass accuracy suitable for identifying at least 46 metabolites by using the KNApsAcK database (17) (Table 1), which exceeds the number of metabolites identified from a single biological tissue by the existing state-of-the-art LAESI-MS method (18). Most of the intermediates of important plant metabolic pathways (Krebs' cycle, fatty acid biosynthesis and glucosinolate biosynthesis) were detected (Fig. 2A Inset and Table 1). In addition to primary metabolites, secondary metabolites such as kaempherol glycosides were identified (Table 1). The analysis of individual or pooled extracts of Drosophila melanogaster males and virgin females showed distinct sex-related profiles (Fig. 2B, Table S1) composed of short-, medium-, and long-chain fatty acids, many more than were detected using GC/MS (Table S1). Direct profiling of male D. melanogaster's body (Fig. 2C Upper) and pea aphid's (Acyrthosiphon pisum) wing (Fig. 2C Lower) showed clear ion signals, presumably lipids, thus documenting the potential of our method for tissue profiling/imaging. Clear spectra were also obtained from submicroliter volumes of human blood (Fig. 2D), thus highlighting the potential of the matrices for clinical diagnostics by direct analysis of biofluids. Overall, our approach has a wide range of applications—high-throughput metabolomic screening, tissue profiling/imaging, direct biofluids analysis—which can be further developed for biomedical studies.
Fig. 2.

Sections of negative-mode mass spectra obtained on MAILD-TOF/MS measurements from diverse biological materials. Matrix [1,8-bis(dimethylamino)naphthalene; DMAN] dissolved in chloroform/methanol (2/1) mixture was applied either on extracts or directly on tissues. (A) Spectral profile of A. thaliana Col-0, 4-week-old leaf scratched with a scalpel and with applied DMAN showing 108 monoisotopic peaks (signal/noise ratio 5/1) in m/z range 50–800 Th The Inset shows the Krebs' cycle with the identified intermediates in red. The identified compounds are listed in Table 1. (B) Overlaid mass spectra measured from extracts of D. melanogaster males and females show distinct sex-related peaks. Fatty acids identified are reported in Table S1. (C) Mass spectra obtained from D. melanogaster's body (Upper) and A. pisum wing (Lower). (D) A drop of human blood deposited on the target covered with the matrix solution provided a rich spectrum of blood-associated fatty acids.
Table 1.
The 46 metabolites identified in negative ion MAILD-TOF/MS profiling of A. thaliana Col-0 4-week-old leaf damaged with a scalpel and spotted with DMAN [10 mg/mL in 2:1 CHCl3:CH3OH)] matrix
| Observed m/z | Analyte identified* | Metabolic pathway† or function |
|---|---|---|
| 87.00 | Pyruvic acid | Super pathway of glycolysis, pyruvate dehydrogenase, TCA, and glyoxylate bypass |
| 96.97 | Phosphoric acid | — |
| 115.00 | Fumaric acid | Superpathway of glyoxalate cycle and TCA cycle |
| 117.01 | Succinic acid | TCA cycle, flavanoid biosynthesis, glyoxylate cycle, fatty acid oxidation |
| 131.00 | Oxaloacetic acid | TCA cycle, glyoxylate cycle |
| 133.01 | Malic acid | TCA cycle, glyoxylate cycle, fatty acid oxidation |
| 137.01 | Salicylic acid | SA biosynthesis, volatile benzenoid ester biosynthesis, plant hormone |
| 145.02 | α-Ketoglutaric acid | TCA cycle, flavanoid biosynthesis |
| 147.08 | Mevalonic acid | Mevalonate pathway |
| 163.04 | Phenylpyruvic acid | Phenyalanine biosynthesis |
| 175.02 | Ascorbic acid | Ascorbate biosynthesis |
| 179.06 | Hexose sugars | Glycolysis |
| 180.05 | Tyrosine | Tyrosine, tryptophan and phenylalanine biosynthesis |
| 185.17 | Undecanoic acid | Fatty acid pathway |
| 191.03 | Citric/isocitric acid | TCA cycle |
| 193.08 | Ferulic acid | Phenylpropanoid biosynthesis and suberin biosynthesis |
| 197.04 | Syringic acid | — |
| 199.18 | Lauric acid | Fatty acid pathway |
| 202.10 | Indole-3-butyric acid | Plant hormone |
| 203.07 | Tryptophan | Tryptophan biosynthesis, glucsinolate biosynthesis |
| 211.99 | Aspartyl phosphate | Lysine and homoserine biosynthesis |
| 217.10 | 2-Oxo-9-methylthiononanoic acid | Side-chain elongation of cyclic aliphatic glucosinolates |
| 221.08 | 2-(3′-Ethylthio)propylmalic cid | Side-chain elongation of cyclic aliphatic glucosinolates |
| 229.20 | 2-Hydroxytridecanoic acid | Fatty acid pathway |
| 231.10 | 2-Oxo-10-methylthiodecanoic acid | Side-chain elongation of cyclic aliphatic glucosinolates |
| 232.14 | Hexahomomethionine | Glucosinolate biosynthesis |
| 245.05 | 2-(Indole-3-yl)-4,5-dihydro-1,3-thiazole-4-carboxylic acid | Camalexin biosynthesis |
| 247.09 | 2-(Indole-3-yl)-1,3-thiazolidine-4-carboxylic acid | Camalexin biosynthesis |
| 263.10 | 2-(6′-Methylthio)hexylmalic acid | Side-chain elongation of aliphatic glucosinolates |
| 269.26 | Heptadecanoic acid | Fatty acid oxidation |
| 275.02 | 6-Phospho-d-gluconate | Pentose pathway and gluconate degradation |
| 277.24 | Linolenic acid | Fatty acid pathway |
| 279.24 | Linoleic acid | Fatty acid pathway |
| 283.29 | Stearic acid | Fatty acid pathway |
| 285.21 | 1,16-hexadecanedioic acid | Suberin biosynthesis |
| 294.26 | 18-hydroxy-9-octadecanoic acid | Suberin biosynthesis |
| 311.20 | Octadeca-9-ene-1,18-dioic acid | Suberin biosynthesis |
| 325.23 | Nonadecanoic acid | Fatty acid pathway |
| 339.33 | Behenic acid | Fatty acid pathway |
| 367.38 | Lignoceric acid | Fatty acid pathway |
| 420.08 | 4-Methylthiobutyl-glucosinolate | Glucosinolate biosynthesis |
| 436.08 | 4-Methylsulfinylbutyl-glucosinolate | Glucosinolate biosynthesis |
| 447.09 | 3-Indolylmethylglucosinolate | Glucosinolate biosynthesis |
| 577.19 | Kaempferol-3,7-dirhamnoside | Kaempferol biosynthesis |
| 593.18 | Kaempferol-3-galatoside-7-rhamnoside | Kaempferol glucoside biosynthesis |
| 755.22 | Kaempferol-3-gentobioside-7-rhamnoside | Kaempferol glucoside biosynthesis |
Data obtained on externally calibrated instrument were internally recalibrated; the mass accuracies for identified metabolites are within ±20 mDa. TCA, tricarboxylic acid; cycle, Krebs cycle.
*Identified as [M-H]− ions.
†Using KNApsAcK database (http://prime.psc.riken.jp/?action=metabolites_index ).
Acid–Base Model Was Further Validated by a Combination of MS, NMR, and X-ray Experiments and Supported by Density Functional Quantum Chemical Calculations.
To study the effects of matrix structure and basicity, additional bases structurally similar to DMAN were selected, and stearic acid (SA) as a representative acid (pKa = 10.15), was analyzed. A strong dependence of the stearate ion abundance on the pKb values of the studied bases was observed (Fig. 3). As in DMAN, no matrix ions were observed for N,N-dimethylaniline (DMA) (Fig. 3B) and aniline (Fig. 3D). The negative-ion TOF mass spectrum obtained by using 1,8-diaminonaphthalene shows, in addition to the expected m/z 283 of stearate, copious matrix cluster ions obscuring both high- and low-mass regions (Fig. 3C). It seems that the gas-phase basicity of the matrix is important and that the suitable matrix in the negative-ion mode must not tend toward deprotonation. The gas-phase pKb of all 4 bases used here were calculated and the obtained values correlated well with the observed matrix ion formations (Table S2). Given the results of these preliminary experiments, we suggest a mechanistic model (Eq. 1) to explain our observations.
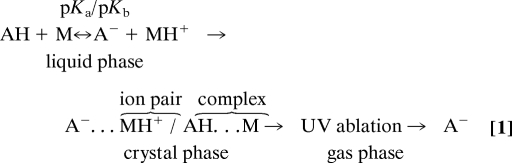 |
The equilibrium between the ion pair and the associated base–acid complex in the liquid phase is characterized by pKa and pKb values of base or acid in the solution in terms of the Brønsted–Lowry concept (14). This equilibrium is reflected in the crystal phase, and the amounts of observed ions are manifested by the ion pair/complex equilibrium. We are not considering gas-phase ionization/reionization (19). According to the proposed model, the ion yield should depend on both the matrix/analyte ratio and the pKa (Eq. 1). Based on the suggested mechanisms we are proposing to name it as matrix-assisted ionization/laser desorption, abbreviated as MAILD, mass spectrometry.
Fig. 3.

Averaged (30 scans with 20 laser shots per scan) MAILD TOF/MS negative-ion spectra for 2.5 nmol of stearic acid using: 1,8-bis(dimethylamino)naphthalene (A), N,N-dimethylaniline (B), 1,8-diaminonaphthalene (C), aniline (D) as MAILD matrices. Insets in each section show the mass spectra in the region 0–200 Th. Peaks marked “A” correspond to stearate anion at m/z 283.2. Peaks in C marked as “M” correspond to matrix peaks. First peak at 313.1 corresponds to [2M-3H]−, and second peak at 336.0 corresponds to [2M-3H+Na]−. (E) Comparison of the stearate monoisotopic signal for 2.5 nmol of stearic acid when mixed with 2.5 nmol of the 4 matrices. Red peak corresponds to the signal obtained with DMAN as the matrix, green peak with N,N-dimethylaniline as the matrix, pink peak with 1,8-diaminonaphthalene as the matrix, and blue peak with aniline as the matrix.
In additional experiments, this model was tested using: (i) different matrix/analyte molar ratios and (ii) diverse ion pairs from bases and acids with different pKa values. The effect of increasing amounts of DMAN and DMA cocrystallized with 1 molar equivalent of trifluoroacetic acid (TFA, Fig. 4A) or SA (Fig. 4B) on trifluoroacetate and stearate intensity, respectively, is summarized in Fig. 4. As predicted by the model, the ion intensity increased from a 0–1 molar equivalent of bases, reaching a maximum at equimolar proportions. Upon further increase of the amount of base, signals for both anions decreased, probably due to the gas-phase ion reneutralization. DFT/B3LYP calculations show high stability of the AH…M complex with respect to its dissociation into A− and MH+ species in the gas phase (106 kcal·mol−1 for a simple acetic acid and DMAN model), preventing gas-phase reionization. A similar trend was observed for the much weaker base, DMA (Fig. 4 A and B), suggesting that these phenomena might be rather general. This observation differs from the widely accepted idea that excess matrix/analyte ratio has beneficial effects. Here, it must be noted that for the analysis of large biomolecules, excess matrix may act as an energy receptacle or a buffer zone preventing analyte degradation. Furthermore, the observed maximum in Fig. 4 A and B at equimolar proportions of base and acid could be used to quantify acids/bases using a series of matrix–analyte mixtures with known amounts of the matrix. A matrix-assisted method for absolute analyte quantification has not previously been used.
Fig. 4.

Dependence of ion signal intensities on analyte/matrix ratios. (A and B) Plot of relative intensity of trifluoroacetate (A) and stearate (B) anions versus MAILD matrices (DMAN, solid lines; DMA, dashed lines), concentrations plotted for clarity as log2 [matrix]. The amount of analytes was kept constant at 250 pmol, and the matrix concentrations were increased to have the following matrix-to-analyte molar ratios: 0.02:1. 0.05:1. 0.1:1. 0.2:1. 0.5:1. 1:1. 2:1. 5:1. 10:1 and 100:1, for which MAILD-TOF/MS measurements were made; 100% = 40,000 counts (A); 100% = 35,000 counts (B). (C) A plot of pKa versus limit-of-detection (LOD) for 4 different acids, namely TFA, TCA, PA, and SA. The solid line represents the LOD studies with DMAN as matrix (TFA 300 fmol; TCA 750 fmol; PA 7.8 pmol; SA 15.6 pmol). The dashed line represents the LOD studies with N,N-dimethylaniline as the matrix (TFA 1 pmol; TCA 2.5 pmol; PA 10 pmol; SA 20 pmol). Inset shows the same curve for just 2 acids, TFA and TCA to highlight the difference in the LOD obtained for the 2 acids. Error bars represent SEM (n = 5, 20 scans with 20 laser shots per scan).
The effect of pKa/pKb on the ion yield was investigated by using 2 bases as matrices and 4 acids with a pKa range of 0.5–10.2. The limit of detection (LOD) at signal-to-noise (S/N) ratio 5 was determined for studied pairs and plotted on a pKa scale (Fig. 4C). An unambiguous dependence of LOD values on decreasing pKa was observed. This agrees with the Brønsted–Lowry concept, which predicts increased ion pair stability with decreasing pKa − pKb difference. This trend can be supported by DFT calculations. Only two halogenated acids are strong enough to form A−…MH+ pair in ethanol solutions of the studied acids and DMAN. The Gibbs free-energy difference for the ionization process as described in Eq. 1 favors a strong acid (Table S3).
For 2 base–acid systems (DMAN plus SA and DMAN plus TFA), the conjugated acid–base was examined more carefully by using crystallization, X-ray diffraction, and melting point experiments. The crystallization of equimolar DMAN mixtures with weak acids (acetic and stearic) did not provide salts with sharp melting points (Fig. S3 E and F). True salt of DMAN with TFA exhibited a strong fluorescence and a sharp melting point (188 °C), which was much higher than that of DMAN (65 °C) (Fig. S3 C and D). The NMR measurements on isolated crystals of DMAN plus TFA salt confirmed the equimolar proportions of CF3COO− and DMAN plus H+. Critical data obtained by using X-ray crystallography (deposited in the Cambridge Structural Database under FO3376) further confirmed the salt nature of the crystal, which is in agreement with the previous literature (20) for DMAN salts. Further confirmation was acquired by DFT calculations (Table S3 and Fig. S1). Excellent surface coverage was observed for the matrix crystals alone and when mixed with analytes, limiting the “sweet spot” phenomena of other matrices like DHB (Fig. S4).
Inspired by the successful use of strong bases for MAILD analysis of acidic compounds, we tested the suitability of several sulfonic acids as MAILD matrices for the positive-ion mode. 2-Naphthalenesulfonic acid was successfully applied for MAILD analysis of simple amines (Fig. 5). Clear spectra with no interfering matrix signals were obtained. Again, stronger bases gave higher ion count manifested in the low LOD observed for diaza(1,3)bicyclo[5.4.0] undecane (Fig. 5E). The protonation of 2-naphthalenesulfonic acid in the gas phase is negligible and supports the calculated pKa value (Table S2).
Fig. 5.

Positive ion MAILD analysis of amines. (A–D) MAILD TOF/MS positive-ion spectra for 250 pmol of triethylamine (A), diisopropylamine (B), ethyldiisopropylamine (C), 1,8-diazabicyclo[5.4.0]undec-7-ene (D). (E) Calibration curves for DBU [solid line; limit of detection (LOD) = 7.8 pmol] and triethylamine (dashed line; LOD = 31.3 pmol).
Conclusions
An ionization mechanism termed MAILD is introduced. The matrices developed herein are ionless, in other words they produce no matrix-related interfering ions, solving the problem of conventional matrices and allowing the detection of small molecules (0–1,000 Da). Furthermore, the enormous applicability of our matrices in targeted metabolomic studies is illustrated. The analysis of biological tissues/extracts/biofluids shown here clearly demonstrates numerous possible metabolomic applications; a range of biomedical applications, namely high-throughput clinical diagnostics and drug distribution/imaging studies, are likely to be developed. Our model, which reflects situations in equi- and subequimolar amounts of a matrix, is a crucial addition to photoionization/protonation (21–23) and “lucky survivor” (24) MALDI models. Several important physicochemical characteristics for the rational design of matrices are illustrated: negative ion-mode matrices should have an absorption maximum matching the laser frequency, high basicity (pKa > 10), and no acidic protons, as ours does. Furthermore, high acidity with minimal protonation in the gas phase seems to be an important characteristic of matrices designed for positive ion-mode analysis. Such simple matrix attributes can be calculated by using DFT methods, which opens the door for rational in silico MAILD matrix design. Our concept represents another level in rational design of matrices initiated by Karas and coworkers (25).
Materials and Methods
Sample Preparation.
Stock solutions of all matrices and analytes were prepared in ethanol at appropriate concentrations and 1-μL aliquots each of the matrix and analyte solutions were mixed before MAILD measurements. One microliter of the resulting mixture was spotted on a circular metallic area with 2.5-mm diameter. DMAN-trifluoroacetate crystals, suitable for X-ray crystallography (Tables S4 and S5), were obtained by crystallization of an equimolar mixture of TFA and DMAN from benzene (melting point 188 °C). The pKa values used: DMAN, pKa(conjugate-acid) = 12.5 (16); stearic acid, pKa = 10.15 (26); trifluoroacetic acid, pKa = 0.52; trichloroacetic acid, pKa = 0.81; pivalic acid, pKa = 4.98 (all, refs.27 and 28).
Mass Spectrometry.
A MALDI micro MX mass spectrometer (Waters/Micromass) fitted with a nitrogen laser (337-nm, 4-ns laser pulse duration, max 330 μJ per laser pulse, max 20-Hz repetition rate) was used in reflectron mode and negative polarity for data acquisition. The instrument operated at 0.7 × 10−7 bars with voltages of 5 kV on the sample plate, 12 kV on the extraction grid, pulse and detector voltages of 1.95 and 2.35 kV, respectively. The laser frequency was set to 5 Hz and energy was optimized for different analytes (fatty acids, other anions at 80 μJ per pulse, and strong volatile acids at 100 μJ per pulse). The extraction delay time was optimized to 150 ns. Polyethylene glycol (PEG) 600 sulfate (Sigma–Aldrich) was used to calibrate the mass spectrometer for an m/z range of 100–1,200 Th in the negative ion mode. For measurements relating to Fig. 3, all mass spectrometric parameters were kept identical as mentioned above. For positive mode calibration, a mixture of PEG 200 and 600 was used. The laser energy for positive-mode measurements of the low-molecular-weight bases as analytes and 2-naphthylsulfonic acid as matrix was optimized to 96 μJ per pulse with remaining parameters identical to those used in negative mode. Details on ion identifications are given in SI Text.
NMR Spectroscopy.
All spectra were measured in CDCl3 (400 MHz for 1H- and 100 MHz for 13C-NMR).
DFT Calculations.
Refer to SI Text for details on DFT calculations.
See Tables S2 and S3 and Fig. S1 for additional information.
Supplementary Material
Acknowledgments.
We gratefully acknowledge Prof. Emeritus Dr. Franz Hillenkamp for reading the manuscript and for his invaluable suggestions, Dr. Bernd Schneider for his help with designing the NMR experiments, Dr. Helmar for performing the X-ray crystallography studies, Dr. Dirk Hölscher for training R.S. to use the laser capture microdissection microscope, Eduardo Hatano (Max Planck Institute for Chemical Ecology, Jena, Germany) and Lars W. Clement (Friedrich Schiller University, Jena, Germany) for providing the aphids and the aphid photograph, and Prof. David Heckel and Mrs. Emily Wheeler for editing the text. This work was supported by the International Max Planck Research School, the Max Planck Society (R.S.), and Ministry of Education of the Czech Republic Grant LC512 (to L.R.).
Footnotes
Conflict of interest statement: The principal part of the article was filled as US patent application.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Cambridge Structural Database (accession no. FO3376).
This article contains supporting information online at www.pnas.org/cgi/content/full/0900914106/DCSupplemental.
References
- 1.Karas M, Bachmann D, Bahr U, Hillenkamp F. Matrix-assisted ultraviolet laser desorption of non-volatile compounds. Int J Mass Spectrom Ion Proc. 1987;78:53–68. [Google Scholar]
- 2.Tanaka K, et al. Protein and polymer analyses up to m/z 100,000 by laser ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1988;2:151–153. [Google Scholar]
- 3.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 4.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 5.Kirpekar F, Berkenkamp S, Hillenkamp F. Detection of double-stranded DNA by IR- and UV-MALDI mass spectrometry. Anal Chem. 1999;71:2334–2339. doi: 10.1021/ac990018w. [DOI] [PubMed] [Google Scholar]
- 6.Caprioli RM, Farmer TB, Gile J. Molecular imaging of biological samples: Localization of peptides and proteins using MALDI-TOF MS. Anal Chem. 1997;69:4751–4760. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- 7.Want EJ, Cravatt BF, Siuzdak G. The expanding role of mass spectrometry in metabolite profiling and characterization. ChemBioChem. 2005;6:1941–1951. doi: 10.1002/cbic.200500151. [DOI] [PubMed] [Google Scholar]
- 8.Strupat K, Karas M, Hillenkamp F. 2,5-Dihydroxybenzoic acid: A new matrix for laser desorption-ionization mass spectrometry. Int J Mass Spectrom Ion Proc. 1991;111:89–102. [Google Scholar]
- 9.Beavis RC, Bridson JN. Epitaxial protein inclusion in sinapic acid crystals. J Phys D. 1993;26:442–447. [Google Scholar]
- 10.Horneffer V. Muenster, Germany: University of Muenster; 2002. Matrix-Analyt-Wechselwirkunen bei der Matrix-Untershutzten Laserdesorption/Ionization Massenspekrometrie (MALDI-MS) PhD dissertation. [Google Scholar]
- 11.Wei J, Buriak JM, Siuzdak G. Desorption-ionization mass spectrometry on porous silicon. Nature. 1999;399:243–246. doi: 10.1038/20400. [DOI] [PubMed] [Google Scholar]
- 12.Northen TR, et al. Clathrate nanostructures for mass spectrometry. Nature. 2007;449:1033–1036. doi: 10.1038/nature06195. [DOI] [PubMed] [Google Scholar]
- 13.Guo Z, He L. A binary matrix for background suppression in MALDI-MS of small molecules. Anal Bioanal Chem. 2007;387:1939–1944. doi: 10.1007/s00216-006-1100-3. [DOI] [PubMed] [Google Scholar]
- 14.Clayden J, Warren S. Organic Chemistry. 1st Ed. Oxford: Oxford Univ Press; 2000. [Google Scholar]
- 15.Staab H, Saupe T. “Proton Sponges” and the geometry of hydrogen bonds: Aromatic nitrogen bases with exceptional basicities. Angew Chem Int Ed. 1988;7:865–879. [Google Scholar]
- 16.Llamas-Saiz A, Faces-Foces C, Elguero J. Proton sponges. J Mol Struct. 1994;328:297. [Google Scholar]
- 17.Akiyama K, et al. PRIMe: A web site that assembles tools for metabolomics and transcriptomics. In Silico Biol. 2008;8:27. [PubMed] [Google Scholar]
- 18.Li Y, Shrestha B, Vertes A. Atmospheric pressure infrared MALDI imaging mass spectrometry for plant metabolomics. Anal Chem. 2008;80:407–420. doi: 10.1021/ac701703f. [DOI] [PubMed] [Google Scholar]
- 19.Knochenmuss R, Zenobi R. MALDI ionization: The role of in-plume processes. Chem Rev. 2003;103:441–452. doi: 10.1021/cr0103773. [DOI] [PubMed] [Google Scholar]
- 20.Grech E, Malarski Z, Sobczyk L. IR Spectroscopic properties of hydrogen bonding in 1:1 salts of 1,8bis(N,N-dimethylamino)naphthalene. J Mol Struct. 1985;129:35–43. [Google Scholar]
- 21.Knochenmuss R. Ion formation mechanisms in UV-MALDI. Analyst. 2006;131:966–986. doi: 10.1039/b605646f. [DOI] [PubMed] [Google Scholar]
- 22.Knochenmuss R. Photoionization pathways and free electrons in UV-MALDI. Anal Chem. 2004;76:3179–3184. doi: 10.1021/ac035501s. [DOI] [PubMed] [Google Scholar]
- 23.Ehring H, Karas M, Hillenkamp F. Role of photoionization and photochemistry in ionization processes of organic molecules and relevance for matrix-assisted laser desorption ionization mass spectrometry. Org Mass Spectrom. 1992;27:472–480. [Google Scholar]
- 24.Karas M, Glückmann M, Schäfer J. Ionization in matrix-assisted laser desorption/ionization: Singly charged molecular ions are the lucky survivors. J Mass Spectrom. 2000;35:1–12. doi: 10.1002/(SICI)1096-9888(200001)35:1<1::AID-JMS904>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 25.Jaskolla TW, Lehmann WD, Karas M. 4-Chloro-alpha-cyanocinnamic acid is an advanced, rationally designed MALDI matrix. Proc Natl Acad Sci USA. 2008;105:12200–12205. doi: 10.1073/pnas.0803056105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanicky JR, Shah DO. Effect of degree, type, and position of unsaturation on the pKa of long-chain fatty acids. J Colloid Interface Sci. 2002;256:201–207. doi: 10.1006/jcis.2001.8009. [DOI] [PubMed] [Google Scholar]
- 27.Serjeant EP, Dempsey B. Oxford: Pergamon Press; 1979. lonisation Constants of Organic Acids in Aqueous Solutions, IUPAC Data, Series No. 23. [Google Scholar]
- 28.Albert A, Serjeant EP. Ionization Constants of Acids and Bases. New York: Wiley; 1962. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.