

Abstract
The elaboration of the pancreas from epithelial buds to the intricate organ requires complex patterning information that controls fundamental cellular processes such as differentiation and proliferation of pancreatic progenitor cells. During pancreatic organogenesis, endocrine cells are generated from a population of pancreatic progenitor cells. The progenitor cells during the early development simultaneously receive multiple signals, some mitogenic and some inducing differentiation. These extrinsic signals are interpreted through an intrinsic mechanism that either commits the progenitor cell to the mitotic cell cycle or lead to exit from the cell cycle in order to differentiate. The endocrine cells that differentiate from progenitor cells are postmitotic, and direct lineage tracing analyses indicate that a population of progenitor cells persists throughout embryogenesis to allow the differentiation of new endocrine cells. At the end of embryogenesis and early postnatal period is characterized by high rates of beta cell proliferation leading to massive increases in beta cell mass. The beta cell mass expansion considerably slows down in adult animals, though variations in insulin demand due to physiological and pathological states such as pregnancy and obesity can lead to adaptive changes in the beta cells that include hyperplasia, hypertrophy, and increased insulin synthesis and secretion. Deciphering the mechanisms that regulate the plasticity of beta cell mass can be important steps in developing effective strategies to treat diabetes
Introduction
The pancreas is a specialized derivative of the primitive gut endoderm with both endocrine and exocrine function, These two functions of the pancreas are carried out by two distinct populations of cells: the exocrine acinar cells secrete digestive enzymes through the duct system into the gut and the endocrine islet cells secrete hormones into the bloodstream. The hormones produced by the endocrine cells regulate nutrient metabolism: in particular, beta cells produce insulin that is required to maintain glucose homeostasis. Diabetes results from an inadequate mass of functional beta cells. Such inadequacy could result from loss of beta cells due to an immune assault (Type 1) or the lack of compensation to overcome insulin resistance (Type 2). Thus, mechanisms that regulate the number of beta cells will be key to understanding both the pathogenesis of diabetes and for developing therapies.
Cell-cell signaling and refinement of the pancreatic field
Relatively little is known about the molecular mechanisms that underlie the early specification of gut tube to form the pancreas bud [1–4]. The endoderm at this stage receives inductive signals from the adjacent germ layers, the ectoderm and the mesoderm, to form the primitive gut tube, over the next two days. The pancreas derives from the two distinct buds, thickenings or anlagen, on the foregut. One anlage arises dorsally in the upper, duodenal part of the foregut, directly posterior to the developing stomach, while the other is formed ventral to the hepatic endoderm. Proper A–P patterning of the endoderm is necessary for the pancreatic development to proceed, as it makes available the endoderm that can competently receive the inductive signals and has been extensively reviewed recently [5]. A number of these studies to date suggest that signaling from the notochord and endothelium specifies the location of the pancreatic epithelium, while the inductive signals from mesenchyme regulate the size of the pancreatic field.
The elaboration of the pancreas from epithelial buds to the intricate organ requires complex patterning information that coordinates fundamental cellular processes such as differentiation and proliferation of pancreatic progenitor cells. The expansion and differentiation of progenitor cells has been shown to involve Notch signaling, which plays a critical role in specification of individual cell types within the pancreatic domain. Notch signaling is a key pathway in binary cell fate decisions and does so by a process termed “lateral inhibition” [6]. Lateral inhibition is also active in the developing pancreas and is involved in maintaining the progenitor cell population. In context of the pancreatic development, the Notch signaling regulates the decision as to which cells would proliferate versus which cells would differentiate. Loss of function studies using various Notch pathway genes such as the Delta like ligand 1(Dll1), the intracellular mediators RBP-Jk and Hes1 result in depletion of progenitors due to precocious differentiation, Ngn3 upregulation and consequent increase in endocrine formation [7,8]. The similarity of phenotypes of mice defective in different components of the Notch signaling pathway emphasized that Notch signaling via Hes1 regulates self-renewal of pancreatic progenitors in early development. Gain-of-function experiments to activate Notch in the progenitor cells prevent endocrine differentiation, further strengthening this view [9,10]. Thus Notch signaling blocks the differentiation of pancreatic progenitors and forces them to remain undifferentiated.
The near complete conversion of pancreatic progenitors at E9–E10 in the Hes1 null mice, along with the fact that Notch1, Notch2 and Hes1 are expressed in epithelia of the early pancreas, including cells not neighboring a laterally signaling cell, suggests the possibility of existence of Notch independent functions for Hes1 or an alternate mechanism that activates Notch/Hes1 ubiquitously in pancreatic progenitors [8]. The second hypothesis is supported by fact that Hnf6 mutant mice express Notch and Hes1 at normal levels despite the near absence of Ngn3 and loss of endocrine cells [11]. Norgaard et al. [12] choose to refer to this mechanism as “Suppressive maintenance”, where by Hes1 is activated by a process different that lateral inhibition. A major difference between these two mechanisms is the ubiquitous expression of the ligand and receptor in suppressive maintenance, unlike the typical speckled expression of a lateral inhibitory ligand, such as Dll1 [13]. Jagged1 and Jagged2 exhibit an ubiquitous pattern of expression[7,12,14], indicating that they might be the appropriate ligands for suppressive maintenance pathways.
FGF10 signal from the mesenchyme, as suggested before, plays an important role in maintaining the Pdx1+ epithelial progenitor population in the pancreas [15]. FGF10 ectopic expression not only causes abrogation of pancreatic cell differentiation but a simultaneous increased proliferation of pancreatic progenitors as well. These two effects of FGF10 ectopic expression are independent of each other, as the proliferation slows down over time, while the differentiation status is maintained. Here, the FGF10 positive pancreatic cells express Notch1 and Notch2, the Notch-ligands Jagged-1 and Jagged-2, as well as the Notch target Hes1, and exemplify the suppressive maintenance phenomenon. This suggests that FGF10 integrates cell proliferation and terminal differentiation, the latter operating through Notch signaling [12].
What, then, are the regulators of the Notch signaling? The transcription factor Sox9 has recently been shown to be the first specific marker and maintenance factor for multipotent pancreatic progenitors. Pancreatic Sox9 expression is limited to the dividing, Notch responsive, Pdx1 positive subset of multipotent progenitors, and is absent in differentiated cells. Targeted disruption of Sox9 leads to severe pancreatic hypoplasia due to progenitor depletion, a phenotype similar to Notch mutations. Sox9 is involved in the maintenance of the pancreatic progenitor pool and does so by stimulating their proliferation and maintaining them in an undifferentiated state [16]. The authors also demonstrate that Sox9 deficient progenitors have reduced expression of Hes1, indicative of a regulatory effect on Notch signaling. These findings, therefore, suggest that Sox9 potentially maintains the progenitor pool in pancreas by regulating the Notch pathway to However, given their observations that Sox9 deficient mice show more severe pancreatic hypoplasia compared to the Hes1 deficient mice, the authors suggest that Sox9 might have some Notch independent effect on the maintenance of progenitor pool. The Notch pathway has been shown to inhibit Ptf1a function and acinar differentiation in the developing mouse pancreas [17]. Besides, Notch signaling is also essential for region-appropriate pancreas specification in the developing foregut endoderm via regulation of Ptf1a [18]. One of the major targets of the Notch pathways is Neurogenin 3 (Ngn3), a bHLH transcription factor. All endocrine lineages arise from Ngn3+ progenitors [19]. Sox9 has recently been shown to activate the expression of Ngn3 [20]. Further, Sox 9 also regulates the expression of transcription factors Tcf2, Onecut1 and Foxa2, which are critical to the progenitors. Foxa2 and Tcf1 in turn regulate Sox9 expression setting up a feedback loop [20]. These observations suggest that Sox9 not only maintains the transcriptional networks in the progenitor cells but also regulates the programs by which progenitors differentiate into distinct lineages.
Ngn3 expression predisposes the cells to endocrine fate program, as indicated by the Ngn3 transgenic mice, which express Ngn3 under the control of Pdx1 promoter. These mice show massive conversion of pancreatic cells into glucagons positive endocrine cells [7]. Ngn3+ endocrine progenitor cells have limited mitotic potential and Ngn3 is not expressed in differentiated endocrine cells [7,14,21–23]. Misexpression of Ngn3 induces endocrine program in the endoderm outside the pancreas domain, in the developing chicken gut, with majority of the cells produced being alpha cells [24]. Gradwohl et al. [21] show that absence of Ngn3 leads to complete loss of all pancreatic cell types. Pancreatic progenitors go through different states of competence to allow for different lineages, as demonstrated by the time dependent activation of Ngn3 in transgenic mice [25]. Notch signaling maintains the progenitor status of the cells by inhibiting Ngn3 expression and consequently endocrine differentiation.
Ngn3 in turn positively regulates the insulin expression enhancing bHLH transcription factor NeuroD [26]. NeuroD forms the tissue specific part of a hetero-dimeric complex, along with the ubiquitous E2A [27–29]. NeuroD is expressed in all differentiated and post-mitotic pancreatic endocrine cells. NeuroD null mice generate all all islet cell types, suggesting that its not absolutely required for endocrine differentiation. However, the islet number is greatly reduced and beta cell undergo apoptosis, in these animals [30,31]. The expression of NeuroD precedes that of endocrine postmitotic markers, such as Pax6 and Isl1 [14]. Thus NeuroD may be involved in promoting cell cycle exit. All these observation, put together, underscore the importance of cell-cell signaling in maintaining a balance between progenitor proliferation and differentiation of endocrine cells. The role of transcription factors as intrinsic regulators of endocrine differentiation has been extensively reviewed [32–34].
Coordinating progenitor self-renewal with differentiation to generate proper numbers of beta cells
Very little information exists on how patterning information imparted to progenitors is integrated with cellular processes of differentiation and self-renewal during the process of pancreatic organogenesis. While Notch signaling plays a crucial patterning role by selecting progenitor cells to either differentiate or self-renew, although the intrinsic mechanism by which Notch signaling is interpreted by the progenitor cells during this binary decision process was unknown. The proliferation of multipotent progenitor cells must be precisely regulated as the different endocrine and exocrine cells types emerge at different times during development. For example, if progenitor cells exited the cell cycle too early, cell types that differentiate later in development will be reduced in the mature organ. Thus appropriate terminal cell-cycle exit accompanied by differentiation is important for the size and the cellular make-up of the pancreas.
The decision for progenitors to differentiate is made at the G1 phase of the cell cycle where D-type cyclins and cyclin-dependent kinases inhibitors (CKIs) play an important role in regulating cell cycle exit. The CKIs regulate cell cycle progression by blocking phosphorylation of the retinoblastoma protein. Two groups of CKIs have been described [35]. These include the Ink4 family members that specifically inhibit cyclin D-Cdk4/6 activity, and the CIP/KIP family that includes p21, p27 and p57 which exhibit promiscuous CDK-inhibitory activity. The connection between Notch signaling and cell cycle regulation became clear after the observation that p57 was a target of transcriptional repression by the Notch effector, Hes1. Hes1 is a known transcriptional repressor and has been shown to prevent differentiation by suppressing expression of transcriptional activators of cell differentiation such as Ngn3 [36]. Thus, Hes1 activation in pancreatic progenitors can suppress the expression of both Ngn3 and p57 as well as simultaneously prevent cell cycle exit and differentiation. The inactivation of Hes1, on the other hand, leads to increased number of progenitors expressing p57, leading to their precocious exit from the cell-cycle [37]. These results suggest that Notch signaling coordinates with cell cycle exit with differentiation to regulate the size of the pancreatic progenitor pool.
Differentiation of beta cells from pancreatic progenitors accounts for the bulk of the beta cell mass established before birth [38]. The newly differentiated cells that arise from pancreatic progenitor are mitotically quiescent and do not proliferate until very late in embryogenesis [39–41]. What maintains the postmitotic state of newly differentiated beta cells? Recent work has shown that mitotically quiescent beta cells accumulate another member of CKI family, p27. In the absence of p27, the differentiated cells are no longer quiescent and reenter the cell cycle to proliferate. This proliferation of beta cells in p27−/− mice during embryogenesis results in increased beta cell mass at birth when compared to wild-type littermates. Accumulation of p27 in beta cells that differentiate during embryogenesis, prevents reentry into the cell cycle. Thus, p27 functions to maintain the quiescent state of the newly differentiated beta cells during embryogenesis and regulate the total number of beta cell at birth [42]. Stanger et al. [43] have examined the effect of perturbations in progenitor pool size on the determination of final organ size in mouse models. These authors have used cell ablation with targeted and controlled expression of Diptheria Toxin A (DTA) and complementation of Pdx1 null embryos with wild type embryonic stem cells, to analyze the effect of progenitor population size during early pancreatic and liver development. Reduction of the pancreatic progenitor pool led to a smaller pancreas indicating compensatory mechanism for organ size are limited, thus underlining the importance of elucidating the mechanisms that regulate the numbers of pancreatic progenitors during embryogenesis.
Postnatal mechanism that regulate beta cell growth and development
The postnatal period in rodent life between birth and weaning is characterized by a massive expansion in beta cell mass to align proper insulin secretion with increasing with the physiological demands. During postnatal development, the number of endocrine cells is governed by balancing endocrine cell growth and endocrine cell death. The increase in the number of beta cells is a result of increased proliferation of beta cells and possible contributions from differentiation. Early work involving the use of 3H thymidine incorporation to study cell proliferation suggested that endocrine cells in the adult pancreas could be maintained by the proliferation of differentiated cells [44,45]. The proliferation of differentiated beta cells can be as high as 7% in neonatal rats [46] and mice [47]. The expansion of beta cell mass during the neonatal period is balanced by waves of apoptosis during pancreatic remodeling [46,48]. A transient wave of beta cell apoptosis occurs at weaning, which is thought to be associated with islet remodeling and/or changes in beta cell maturation [49,50]. The rate of beta cell mass expansion is, therefore, governed by the relative rates of new beta cell formation and beta cell loss. It has been proposed that the neonatal beta cell apoptosis may function as a possible trigger for autoimmune diabetes by exposing potential autoantigens [51].
A number of observations, including prevalence of hormone-positive ductal cells as well as the close proximity of islets to ducts, have led to the idea that some endocrine cell differentiation occurs in the ducts. Ductal cell differentiation from putative precursor cells, sometimes referred to as “neogenesis” [46,48], has been suggested to occur in the neonatal animals [49,52] and there have been studies indicating that the observational incidence of ductal cell differentiation increases in experimental models of pancreatic injury [53,54]. Melton and colleagues have questioned the significance of ductal cell differentiation contributing to the generation of new beta cells and shown that the majority of beta cells originate from pre-existing beta cells [55]. In contrast to studies that invoke the idea of ductal cell differentiation based on histological observations, more recent work using detailed lineage tracing experiments provided strong evidence that implicates beta cell proliferation to be the main source of adult beta cell mass expansion [56]. In such a scenario, the beta cells could be a) a homogenous population, with all beta cells contributing equally to maintenance of beta cell mass, b) a heterogeneous population, with some highly replicative cells functioning as unipotent adult stem cells, or c) a beta cell subpopulation could maintain the beta cell mass by reversible de-differentiating into replicating cells [57]. More recently, there has been increasing evidence to indicate that all beta cells contribute equally to islet growth and maintenance. [57,58] Thus, endocrine pancreas provide unique model, where differentiated beta cells are capable of self-renewal to regulate beta cell mass in adults.
Elucidating the role of cell cycle regulators in beta cell proliferation has lent support to the concept that self-renewal of beta cells is the main driving force of postnatal beta cell growth. These studies indicate that the balance between cyclin D2-Cdk4 complexes that form in response to mitotic signals, and cyclin kinase inhibitors that block the activity of cyclinE-Cdk2 complex regulates beta cell proliferation. Cdk4 is essential for adult beta cell proliferation, as the cdk4−/− mice develop severe diabetes due to hypoplastic islets within 10 weeks after birth [59,60]. In contrast, transgenic mice expressing either a cdk4 allele insensitive to the Ink4 class of cell cycle inhibitors or cdk4 mutant develop massive islet hyperplasia [59,61,62]. The D-type cyclins levels are controlled by mitogens and upon induction, these cyclins pair with the Cdk4/6 to drive the cell into S phase from G1 phase [63,64]. In the beta cell, cyclin D2 is uniquely required for beta cell replication and proper expansion of beta cell mass during postnatal development [47] and that other D-type cyclins are unable to adequately compensate for the absence of cyclin D2 [65].
Whether a beta cell proliferates depends on the balance between cyclin D2-Cdk4 complex that forms in response to mitotic signals, and cyclin kinase inhibitors that block the activity of cyclinE-Cdk2 complex. One cyclin kinase inhibitor, p27 accumulates in quiescent beta cells and degradation of p27 was shown to be essential for beta cell proliferation. Hence, cellular abundance of p27 protein governs the decision of a beta cell to either divide or remain quiescent by attenuating the activity of the cyclin-cdk complexes that are the mitotic sensors for cell cycle progression. Skp2 and Cks1 (which is also a part of the SCF complex) are also regulated by the transcription factor FoxM1. Targeted deletion of FoxM1 in the mouse pancreas leads to postnatal deficits in beta cell mass, progressive glucose intolerance and diabetes [66]. The transcriptional co-activator Menin (product of Men1 locus, which is mutated in familial multiple endocrine neoplasia type 1: MEN1; [67]) also regulates the growth of pancreatic islets by promoting histone methylation and the expression from p27 and p18 loci encoding CKIs. Menin directly associates with the promoter regions of these two loci and increases the methylation of lysine-4 in histone H3, thus modulating the transcrption of these CKIs [68].
Plasticity of beta cell mass and coping with metabolic demand
The beta cell mass expansion considerably slows down in adult animals, though some expansion continues at very slow rates through the life of the animal. While beta cell mass expands primarily through replication during neonatal growth, the replication rate becomes slower postnatally and in adulthood. Finegood et al. [69] have previously suggested that nearly 2% of adult beta cells undergo replication. However, long term BrdU labeling studies by Teta et al. [70] suggest a much slower rate of beta cell replication in the adult mice and extremely low beta cell turnover. Besides maintaining the beta cell mass under normal circumstances, animals adapt beta cell mass to cope with changing metabolic demands for insulin. Variations in insulin demand due to physiological and pathological states such as pregnancy and obesity can lead to adaptive changes in the beta cells that include hyperplasia, hypertrophy, and increased insulin synthesis and secretion [48,71–73]. Under such circumstances, blood glucose levels are maintained by regulated increases in insulin secretion, which is accompanied by an expansion of pancreatic beta cell mass. However, the inability of the endocrine pancreas to adapt to the changing insulin demand can result in hyperglycemia and development of diabetes mellitus [74–76]. In humans, this is typified by increased insulin resistance in obese type II diabetics and by the onset of gestational diabetes during pregnancy. Thus, deciphering the mechanisms that regulate the plasticity of beta cell mass can be important step in developing effective strategies to treat diabetes.
A linear correlation has been shown to exist between beta-cell mass and body weight [77]. Diet induced obesity and the consequent insulin resistance correlates with a compensatory increase in beta cell mass. C57Bl/6 mice demonstrate a 2 fold increase in their beta cell mass after four months of high fat diet regimen [78]. Cell cycle molecules play an important role in diet induced adaptive expansion of beta cell mass. Increased insulin resistance due to diet-induced obesity leads to the Skp2−/− mice becoming overtly diabetic, as beta cell growth in the absence of cell division can not compensate for increased metabolic pressure. This suggests that regulation of p27 turnover is needed to respond to increased metabolic demand associated with insulin resistance (Zhong et al., unpublished).
Beta cell compensation is witnessed in naturally occurring genetic models of obesity and insulin resistance. The db/db mice, which lack a functional leptin receptor, show a twofold increment in beta cell mass by 8 weeks of age [79]. This timing correlates with the onset of diabetes, and is preceded by glucose intolerance. Zucker diabetic fatty (ZDF) rats, another model of leptin receptor deficiency, display increased beta cell mass and proliferation before the onset of diabetes. However, increased apoptosis after the onset of diabetes results in a decreased beta cell mass unable to compensate for increased insulin demand. This is in sharp contrast to the non-diabetic, Zucker fatty (ZF) rats, which are also obese but able to adequately expand beta cell mass to respond to increased metabolic demand for insulin due to insulin resistance [80]. Islet hyperplasia also accompanies insulin resistance in case of the hyperglycemic obese ob/ob mice [81].
Genetic models of insulin resistance have proven to be powerful tools in understanding the molecular mechanisms that regulate beta cell plasticity. The Insulin and Insulin like Growth Factor (IGF) signaling pathways are critical for proper maintenance and functioning of beta cells. Genetic studies reveal that these pathways actively mediate peripheral insulin action and beta cell function [82–85]. Both Insulin and IGF pathways signal through Insulin Receptor Substrate proteins (IRS), and of particular interest are IRS1/2. IRS1 plays a role in compensatory beta cell expansion in response to insulin resistance, while IRS2 plays an important role in beta cell growth and survival [84,86–88]. Because of the drastic differences in phenotypes of the different IRS1/2 null mice, it has become clear that IRS1 and IRS2 have distinct, non-redundant functions in insulin/IGF signaling. Further, it has been shown that an upregulation of IRS2 leads to the prevention of diabetes in mice [89].
Combinations of IR/IRS models of insulin resistance with Pdx/Foxo haploinsufficient models have elucidated the important role of Pdx1 expression and beta cell proliferation. Recent work from Accili lab showed that Foxo1 overexpression in liver and/or beta cells restricts the compensatory increase of beta cell mass in response to insulin resistance via restricting proliferation in the beta cells of Insulin receptor deficient mice [90]. Similarly, it has been shown that Pdx1 haploinsufficency can restrict the proliferation-based compensatory increase of beta cell mass in response to insulin resistance in liver-specific insulin receptor and IR-IRS1 double haploinsufficient animals [91]. Conversely, overexpression of Pdx1 in IRS2-null animals has been shown to prevent diabetes and allow for adaptive expansion of beta cell mass via increased proliferation of beta cells [92]. The clear involvement of Pdx1 action in the adaptive expansion of beta cell mass begs further investigation into how Pdx1 influences cell cycle machinery to allow for beta cell proliferation.
Pregnancy also leads to increased metabolic demand for insulin, resulting in compensatory increase of beta cell mass. Rats show 2–2.5 fold increase in beta cell mass during pregnancy, attained mainly by an increased proliferation of beta cells. This increase in the beta cell mass is a combined effect of increased beta cell proliferation and beta cell hypertrophy (an increase in cell size) [50]. Beta cell hyperplasia and hypertrophy has also been observed in pregnant humans [93]. After delivery, the beta cell mass returns to normal, through a decrease in beta cell proliferation and size and an increase in apoptosis [50]. It has been shown that exposure to prolactin and placental lactogen, both lactogenic hormones, come into play to accommodate the increased insulin demand during pregrnancy, and do so by increasing proliferation [94–97]. Non pregnant mice which ectopically express placental lactogen 1(PL1) in beta cells (through Rat Insulin Promoter-RIP) exhibit hypoglycemia and hyperinsulinemia, resulting from a doubling of beta cell mass. This expansion of beta cell mass is due to increased proliferation as well as hypertrophy [98]. The absence of progesterone, another model of the hormonal changes that occur during pregnancy, results in increased beta cell mass due to increases in beta cell proliferation [99].
The regenerative capacity of beta cells
In adult life, beta cell loss and renewal are balanced to maintain homeostasis. Rapid renewing organs such as hair follicle, blood and gut utilize dedicated stem cells to constantly renew themselves throughout life. In contrast, organs that exhibit slow turnover such as the pancreas and liver rely on self-renewal of beta cell and hepatocytes respectively [100]. Regeneration of pancreatic beta-cell mass following either toxin or autoimmune-mediated destruction is possible in the young rodent, but the extent of the recovery decreases with age and is incomplete in adult life. Treatment with incretins, such as glucagon-like peptide 1 and its long lasting homolog exendin-4, results in increased beta cell proliferation. The proliferative effects of GLP1 are dependent on PDX1 expression [101] and concurrent nuclear exclusion of Foxo1 [102]. Even though histological observations serve as evidence of ductal cell differentiation expanding the beta cell population after injury such as partial pancreatectomy, pancreatic duct ligation, or EGF and gastrin treatment following drug induced diabetes [103], lineage tracing of genetically marked beta cells shows that no new beta cells are formed from non-insulin-expressing stem cells or progenitor cells following 70% pancreatectomy [56]. The experiments cited have been performed on young mice and some data exists to suggest that the regenerative capacity of the endocrine pancreas declines with age. In one-month old rats, the insulin content of the residual pancreas after 90% pancreatectomy increased 3-fold but no such increase was observed in five and fifteen-month old rats. Consistent with increased insulin content, plasma glucose levels of one-month old rats after 90% pancreatectomy increased for two weeks before declining while no such decline was observed in five and fifteen-month old rats [104].
The decline in beta cell proliferation correlates with increased expression of p16 in islet cells [105]. p16, another CyclinD-cdk4 inhibitor and a mediator of cellular senescence, is known to accumulate in many tissue with ageing [106–108]. p16 RNA expression is enriched in the murine pancreatic islets and shows a significant increase with ageing. Overexpression of p16 in mice results in decreased islet proliferation. The loss of p16 expression, on the other hand, shows an increased proliferation in the old mice while not affecting the proliferation in younger mice. Further, the p16 knockout mice are better able to survive STZ mediated islet ablation. These findings suggest that ageing related increase in p16 expression limits the regenerative potential of beta cells [105]. Thus, p16 is an important regulator of adult pancreatic beta cell survival and regenerative potential.
Recently, studies have linked the expression of other key cell cycle regulators with established roles in beta cell proliferation to key signaling pathways important to establishing and maintaining a adaptively responsive beta cell mass. Previously, it has been reported that mice with a conditional ablation of LRP5, a co-receptor in the Wnt signaling pathway, mouse islets resulted in reduced beta cell function and poor glucose tolerance in adult mice that were challenged with a high fat diet, indicating that wnt signaling may play a role in the adaptive response of beta cells to obesity and insulin resistance [109]. Though there were no mechanistic explanations as to how wnt signaling was involved in the adaptive response to high fat diet, recently it has been reported that conditional expression of Axin, an inhibitor of Wnt pathway, reduces the expression of Cyclin D2 and Pitx2, leading to reduced neonatal beta cell mass and glucose intolerance. Conversely, Wnt3a promotes the expression of Pitx2, a direct target of Wnt signaling, and that Pitx2 binds to the Cyclin D2 promoter, upregulates cyclin D2 mRNA levels, and subsequently increase proliferation in MIN6 cells. To mimic these in vitro experiments in vivo, mice were bred that conditionally expressed constitutively active beta-catenin (the downstream effector of canonical Wnt pathway), and showed the same effects in vivo, resulting in expansion of beta cell mass and insulin production. These studies suggest that Wnt signaling is essential for beta cell proliferation [110]. TGF-beta is also an important regulator of islet development. Conditional expression of Smad7, a potent inhibitor of TGF-beta pathway in the embryonic Pdx1 positive cells leads to severe beta cell hypoplasia and neonatal lethality. Smad7 expression in the adult Pdx1 positive cells reduces the expression of beta cell regulatory factors such as menin and MafA. This also leads to diabetes, which can be reversed by inducing TGF-beta signaling again [111]. Thus, there is a whole set of different signaling pathways and molecules that can be used as potential targets to allow in vitro and in vivo proliferation of beta cells or the islets.
Figure 1.
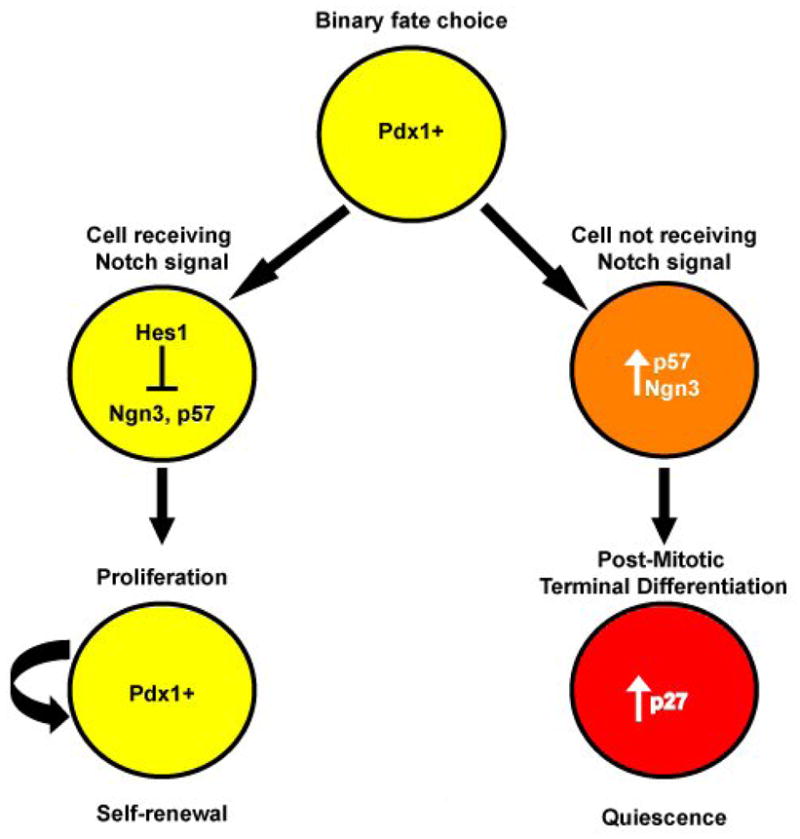
The decision to proliferate or differentiate. Pdx1+ progenitor cells face a binary decision to either self-renew to expand the progenitor pool, or to undergo cell cycle arrest and begin the differentiation process. Progenitor cells that receive the Notch signal pathway via downstream effector Hes1 represses not only genes specific to differentiation but also the cell cycle arrest mediator, p57, thereby allowing the progenitor cells to continue to self-renew. Cells that do not receive the Notch signal upregulate p57 and exit the cell cycle to allow the differentiation process. The quiescent state of differentiated cell type is maintained by accumulation of cell cycle inhibitor p27.
Figure 2.

The proliferative capacity of beta cells changes during the progression from embryogenesis through adulthood. During early embryogenesis, beta cell umbers are established by direct differentiation from Pdx1+ progenitor cells. In late gestation through the neonatal stages of life, the beta cell population is expanded through high rates of proliferation in existing beta cells. In adulthood, low levels of beta cell replication maintain a constant set of beta cells ready to provide insulin to meet metabolic demand.
Figure 3.

The inability of beta cell mass to adaptively expand in order to compensate for increased insulin demand leads to diabetes. When either pregnancy- or obesity–related insulin resistance creates an increase in metabolic demand for insulin, healthy beta cells are able to expand in number and size to compensate for increased metabolic need for insulin, thereby maintaining glucose homeostasis. When beta cells to expand and adapt to changing metabolic demand, the reduced insulin secretion results in hyperglycemia.
Acknowledgments
We are grateful to Peter Butler, Ulupi Jhala, Aleksey Matveyenko and other members of the Larry Hillblom center for helpful discussions. The Bhushan lab is supported by grants from NIH R01 DK-068763 and the Larry Hillblom Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Edlund H. Pancreas: how to get there from the gut? Curr Opin Cell Biol. 1999;11:663–668. doi: 10.1016/s0955-0674(99)00033-2. [DOI] [PubMed] [Google Scholar]
- 2.Slack JM. Developmental biology of the pancreas. Development. 1995;121:1569–1580. doi: 10.1242/dev.121.6.1569. [DOI] [PubMed] [Google Scholar]
- 3.Wells JM, Melton DA. Vertebrate endoderm development. Annu Rev Cell Dev Biol. 1999;15:393–410. doi: 10.1146/annurev.cellbio.15.1.393. [DOI] [PubMed] [Google Scholar]
- 4.Kim SK, MacDonald RJ. Signaling and transcriptional control of pancreatic organogenesis. Curr Opin Genet Dev. 2002;12:540–547. doi: 10.1016/s0959-437x(02)00338-6. [DOI] [PubMed] [Google Scholar]
- 5.Zorn AM, Wells JM. Molecular basis of vertebrate endoderm development. Int Rev Cytol. 2007;259:49–111. doi: 10.1016/S0074-7696(06)59002-3. [DOI] [PubMed] [Google Scholar]
- 6.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 7.Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, Hrabe de Angelis M, Lendahl U, Edlund H. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400:877–881. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 8.Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P, Madsen OD. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24:36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- 9.Hald J, Hjorth JP, German MS, Madsen OD, Serup P, Jensen J. Activated Notch1 prevents differentiation of pancreatic acinar cells and attenuate endocrine development. Dev Biol. 2003;260:426–437. doi: 10.1016/s0012-1606(03)00326-9. [DOI] [PubMed] [Google Scholar]
- 10.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100:14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacquemin P, Durviaux SM, Jensen J, Godfraind C, Gradwohl G, Guillemot F, Madsen OD, Carmeliet P, Dewerchin M, Collen D, et al. Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol Cell Biol. 2000;20:4445–4454. doi: 10.1128/mcb.20.12.4445-4454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Norgaard GA, Jensen JN, Jensen J. FGF10 signaling maintains the pancreatic progenitor cell state revealing a novel role of Notch in organ development. Dev Biol. 2003;264:323–338. doi: 10.1016/j.ydbio.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 13.Lammert E, Brown J, Melton DA. Notch gene expression during pancreatic organogenesis. Mech Dev. 2000;94:199–203. doi: 10.1016/s0925-4773(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 14.Jensen J, Heller RS, Funder-Nielsen T, Pedersen EE, Lindsell C, Weinmaster G, Madsen OD, Serup P. Independent development of pancreatic alpha- and beta-cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes. 2000;49:163–176. doi: 10.2337/diabetes.49.2.163. [DOI] [PubMed] [Google Scholar]
- 15.Bhushan A, Itoh N, Kato S, Thiery JP, Czernichow P, Bellusci S, Scharfmann R. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development. 2001;128:5109–5117. doi: 10.1242/dev.128.24.5109. [DOI] [PubMed] [Google Scholar]
- 16.Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, Scherer G, Sander M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A. 2007;104:1865–1870. doi: 10.1073/pnas.0609217104. **The authors show that Sox9 maintains pancreatic progenitor population in an undiffernetiated state and also increases their proliferation. Very importantly, this study demonstrates that these effects of Sox 9 are due to its effects on Hes1 levels and Notch pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esni F, Ghosh B, Biankin AV, Lin JW, Albert MA, Yu X, MacDonald RJ, Civin CI, Real FX, Pack MA, et al. Notch inhibits Ptf1 function and acinar cell differentiation in developing mouse and zebrafish pancreas. Development. 2004;131:4213–4224. doi: 10.1242/dev.01280. [DOI] [PubMed] [Google Scholar]
- 18.Fukuda A, Kawaguchi Y, Furuyama K, Kodama S, Horiguchi M, Kuhara T, Koizumi M, Boyer DF, Fujimoto K, Doi R, et al. Ectopic pancreas formation in Hes1 -knockout mice reveals plasticity of endodermal progenitors of the gut, bile duct, and pancreas. J Clin Invest. 2006;116:1484–1493. doi: 10.1172/JCI27704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 20.Lynn FC, Smith SB, Wilson ME, Yang KY, Nekrep N, German MS. Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc Natl Acad Sci U S A. 2007;104:10500–10505. doi: 10.1073/pnas.0704054104. *This study reveals a role for Sox9 in the regulation and coordination of the pro-endocrine transcrptional program, especially via Ngn3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maestro MA, Boj SF, Luco RF, Pierreux CE, Cabedo J, Servitja JM, German MS, Rousseau GG, Lemaigre FP, Ferrer J. Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum Mol Genet. 2003;12:3307–3314. doi: 10.1093/hmg/ddg355. [DOI] [PubMed] [Google Scholar]
- 23.Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD, German MS. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development. 2000;127:3533–3542. doi: 10.1242/dev.127.16.3533. [DOI] [PubMed] [Google Scholar]
- 24.Grapin-Botton A, Majithia AR, Melton DA. Key events of pancreas formation are triggered in gut endoderm by ectopic expression of pancreatic regulatory genes. Genes Dev. 2001;15:444–454. doi: 10.1101/gad.846001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johansson KA, Dursun U, Jordan N, Gu G, Beermann F, Gradwohl G, Grapin-Botton A. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev Cell. 2007;12:457–465. doi: 10.1016/j.devcel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Huang HP, Liu M, El-Hodiri HM, Chu K, Jamrich M, Tsai MJ. Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol Cell Biol. 2000;20:3292–3307. doi: 10.1128/mcb.20.9.3292-3307.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- 28.Naya FJ, Stellrecht CM, Tsai MJ. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev. 1995;9:1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- 29.Nelson C, Shen LP, Meister A, Fodor E, Rutter WJ. Pan: a transcriptional regulator that binds chymotrypsin, insulin, and AP-4 enhancer motifs. Genes Dev. 1990;4:1035–1043. doi: 10.1101/gad.4.6.1035. [DOI] [PubMed] [Google Scholar]
- 30.Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson ME, Scheel D, German MS. Gene expression cascades in pancreatic development. Mech Dev. 2003;120:65–80. doi: 10.1016/s0925-4773(02)00333-7. [DOI] [PubMed] [Google Scholar]
- 33.Servitja JM, Ferrer J. Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia. 2004;47:597–613. doi: 10.1007/s00125-004-1368-9. [DOI] [PubMed] [Google Scholar]
- 34.Gradwohl G. Development of the endocrine pancreas. Diabetes Metab. 2006;32:532–533. doi: 10.1016/s1262-3636(06)72807-5. [DOI] [PubMed] [Google Scholar]
- 35.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 36.Lee JC, Smith SB, Watada H, Lin J, Scheel D, Wang J, Mirmira RG, German MS. Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes. 2001;50:928–936. doi: 10.2337/diabetes.50.5.928. [DOI] [PubMed] [Google Scholar]
- 37.Georgia S, Soliz R, Li M, Zhang P, Bhushan A. p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev Biol. 2006;298:22–31. doi: 10.1016/j.ydbio.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 38.McEvoy RC, Madson KL. Pancreatic insulikn-, glucagon-, and somatostatin-positive islet cell populatins during the perinatal development of the rat. I. Morphometric quantitation. Biol Neonate. 1980;38:248–254. doi: 10.1159/000241372. [DOI] [PubMed] [Google Scholar]
- 39.Bernard-Kargar C, Kassis N, Berthault MF, Pralong W, Ktorza A. Sialylated form of the neural cell adhesion molecule (NCAM): a new tool for the identification and sorting of beta-cell subpopulations with different functional activity. Diabetes. 2001;50 (Suppl 1):S125–130. doi: 10.2337/diabetes.50.2007.s125. [DOI] [PubMed] [Google Scholar]
- 40.Hellerstrom C, Swenne I. Functional maturation and proliferation of fetal pancreatic beta-cells. Diabetes. 1991;40 (Suppl 2):89–93. doi: 10.2337/diab.40.2.s89. [DOI] [PubMed] [Google Scholar]
- 41.Swenne I. Effects of cyclic AMP on DNA replication and protein biosynthesis in fetal rat islets of Langerhans maintained in tissue culture. Biosci Rep. 1982;2:867–876. doi: 10.1007/BF01114892. [DOI] [PubMed] [Google Scholar]
- 42.Georgia S, Bhushan A. p27 Regulates the transition of beta-cells from quiescence to proliferation. Diabetes. 2006;55:2950–2956. doi: 10.2337/db06-0249. [DOI] [PubMed] [Google Scholar]
- 43.Stanger BZ, Tanaka AJ, Melton DA. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature. 2007;445:886–891. doi: 10.1038/nature05537. [DOI] [PubMed] [Google Scholar]
- 44.Messier B, Leblond CP. Cell proliferation and migration as revealed by radioautography after injection of thymidine-H3 into male rats and mice. Am J Anat. 1960;106:247–285. doi: 10.1002/aja.1001060305. [DOI] [PubMed] [Google Scholar]
- 45.Tsubouchi S, Kano E, Suzuki H. Demonstration of expanding cell populations in mouse pancreatic acini and islets. Anat Rec. 1987;218:111–115. doi: 10.1002/ar.1092180203. [DOI] [PubMed] [Google Scholar]
- 46.Bonner-Weir S. Life and death of the pancreatic beta cells. Trends Endocrinol Metab. 2000;11:375–378. doi: 10.1016/s1043-2760(00)00305-2. [DOI] [PubMed] [Google Scholar]
- 47.Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonner-Weir S. Islet growth and development in the adult. J Mol Endocrinol. 2000;24:297–302. doi: 10.1677/jme.0.0240297. [DOI] [PubMed] [Google Scholar]
- 49.Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes. 1995;44:249–256. doi: 10.2337/diab.44.3.249. [DOI] [PubMed] [Google Scholar]
- 50.Scaglia L, Smith FE, Bonner-Weir S. Apoptosis contributes to the involution of beta cell mass in the post partum rat pancreas. Endocrinology. 1995;136:5461–5468. doi: 10.1210/endo.136.12.7588296. [DOI] [PubMed] [Google Scholar]
- 51.Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Neonatal beta-cell apoptosis: a trigger for autoimmune diabetes? Diabetes. 2000;49:1–7. doi: 10.2337/diabetes.49.1.1. [DOI] [PubMed] [Google Scholar]
- 52.Hellerstrom C, Andersson A, Groth CG, Sandler S, Jansson L, Korsgren O, Swenne I, Petersson B, Tollemar J, Tyden G. Experimental pancreatic transplantation in diabetes. Diabetes Care. 1988;11 (Suppl 1):45–53. [PubMed] [Google Scholar]
- 53.Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes. 1993;42:1715–1720. doi: 10.2337/diab.42.12.1715. [DOI] [PubMed] [Google Scholar]
- 54.Gu D, Sarvetnick N. Epithelial cell proliferation and islet neogenesis in IFN-g transgenic mice. Development. 1993;118:33–46. doi: 10.1242/dev.118.1.33. [DOI] [PubMed] [Google Scholar]
- 55.Dor Y, Melton DA. How important are adult stem cells for tissue maintenance? Cell Cycle. 2004;3:1104–1106. [PubMed] [Google Scholar]
- 56.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 57.Brennand K, Huangfu D, Melton D. All beta Cells Contribute Equally to Islet Growth and Maintenance. PLoS Biol. 2007;5:e163. doi: 10.1371/journal.pbio.0050163. *Using extensive lineage tracing, this study highlights and provides further evidence for the proposal that adult beta cells give rise to all new beta cells and do so with equal potential. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. 2007;12:817–826. doi: 10.1016/j.devcel.2007.04.011. *The authors here use multiple labeling of beta cells and lineage tracing for long periods and further strengthen the notion that all new beta cells arise from existing pool of beta cells in the adults. [DOI] [PubMed] [Google Scholar]
- 59.Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 60.Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, Kiyokawa H. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol. 1999;19:7011–7019. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hino S, Yamaoka T, Yamashita Y, Yamada T, Hata J, Itakura M. In vivo proliferation of differentiated pancreatic islet beta cells in transgenic mice expressing mutated cyclin-dependent kinase 4. Diabetologia. 2004;47:1819–1830. doi: 10.1007/s00125-004-1522-4. [DOI] [PubMed] [Google Scholar]
- 62.Marzo N, Mora C, Fabregat ME, Martin J, Usac EF, Franco C, Barbacid M, Gomis R. Pancreatic islets from cyclin-dependent kinase 4/R24C (Cdk4) knockin mice have significantly increased beta cell mass and are physiologically functional, indicating that Cdk4 is a potential target for pancreatic beta cell mass regeneration in Type 1 diabetes. Diabetologia. 2004;47:686–694. doi: 10.1007/s00125-004-1372-0. [DOI] [PubMed] [Google Scholar]
- 63.Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bates S, Parry D, Bonetta L, Vousden K, Dickson C, Peters G. Absence of cyclin D/cdk complexes in cells lacking functional retinoblastoma protein. Oncogene. 1994;9:1633–1640. [PubMed] [Google Scholar]
- 65.Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang H, Ackermann AM, Gusarova GA, Lowe D, Feng X, Kopsombut UG, Costa RH, Gannon M. The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol Endocrinol. 2006;20:1853–1866. doi: 10.1210/me.2006-0056. [DOI] [PubMed] [Google Scholar]
- 67.Crabtree JS, Scacheri PC, Ward JM, McNally SR, Swain GP, Montagna C, Hager JH, Hanahan D, Edlund H, Magnuson MA, et al. Of mice and MEN1: Insulinomas in a conditional mouse knockout. Mol Cell Biol. 2003;23:6075–6085. doi: 10.1128/MCB.23.17.6075-6085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. *This study delineates an epigenetic mechanism of action for the tumor supressor menin, mutations in which lead to multiple endocrine neaoplasia. The authors demonstrate that Menin regulates the expression of key cell cycle regulators such as p18 and p27 by modulating their chromatin structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finegood DT, Tzur D. Reduced glucose effectiveness associated with reduced insulin release: an artifact of the minimal-model method. Am J Physiol. 1996;271:E485–495. doi: 10.1152/ajpendo.1996.271.3.E485. [DOI] [PubMed] [Google Scholar]
- 70.Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 71.Accili D, Kido Y, Nakae J, Lauro D, Park BC. Genetics of type 2 diabetes: insight from targeted mouse mutants. Curr Mol Med. 2001;1:9–23. doi: 10.2174/1566524013364040. [DOI] [PubMed] [Google Scholar]
- 72.Bruning JC, Winnay J, Cheatham B, Kahn CR. Differential signaling by insulin receptor substrate 1 (IRS-1) and IRS-2 in IRS-1-deficient cells. Mol Cell Biol. 1997;17:1513–1521. doi: 10.1128/mcb.17.3.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lingohr MK, Buettner R, Rhodes CJ. Pancreatic beta-cell growth and survival--a role in obesity-linked type 2 diabetes? Trends Mol Med. 2002;8:375–384. doi: 10.1016/s1471-4914(02)02377-8. [DOI] [PubMed] [Google Scholar]
- 74.Bonner-Weir S. Perspective: Postnatal pancreatic beta cell growth. Endocrinology. 2000;141:1926–1929. doi: 10.1210/endo.141.6.7567. [DOI] [PubMed] [Google Scholar]
- 75.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 76.Flier SN, Kulkarni RN, Kahn CR. Evidence for a circulating islet cell growth factor in insulin-resistant states. Proc Natl Acad Sci U S A. 2001;98:7475–7480. doi: 10.1073/pnas.131192998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Montanya E, Nacher V, Biarnes M, Soler J. Linear correlation between beta-cell mass and body weight throughout the lifespan in Lewis rats: role of beta-cell hyperplasia and hypertrophy. Diabetes. 2000;49:1341–1346. doi: 10.2337/diabetes.49.8.1341. [DOI] [PubMed] [Google Scholar]
- 78.Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia. 2005;48:58–67. doi: 10.1007/s00125-004-1605-2. [DOI] [PubMed] [Google Scholar]
- 79.Wang Q, Brubaker PL. Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia. 2002;45:1263–1273. doi: 10.1007/s00125-002-0828-3. [DOI] [PubMed] [Google Scholar]
- 80.Pick A, Clark J, Kubstrup C, Levisetti M, Pugh W, Bonner-Weir S, Polonsky KS. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes. 1998;47:358–364. doi: 10.2337/diabetes.47.3.358. [DOI] [PubMed] [Google Scholar]
- 81.Tomita T, Doull V, Pollock HG, Krizsan D. Pancreatic islets of obese hyperglycemic mice (ob/ob) Pancreas. 1992;7:367–375. doi: 10.1097/00006676-199205000-00015. [DOI] [PubMed] [Google Scholar]
- 82.Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, Accili D. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest. 2000;105:199–205. doi: 10.1172/JCI7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96:329–339. doi: 10.1016/s0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- 84.Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 85.Xuan S, Kitamura T, Nakae J, Politi K, Kido Y, Fisher PE, Morroni M, Cinti S, White MF, Herrera PL, et al. Defective insulin secretion in pancreatic beta cells lacking type 1 IGF receptor. J Clin Invest. 2002;110:1011–1019. doi: 10.1172/JCI15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Araki E, Lipes MA, Patti ME, Bruning JC, Haag B, 3rd, Johnson RS, Kahn CR. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372:186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 87.Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, Terauchi Y, Ueki K, Kaburagi Y, Satoh S, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994;372:182–186. doi: 10.1038/372182a0. [DOI] [PubMed] [Google Scholar]
- 88.Kubota N, Tobe K, Terauchi Y, Eto K, Yamauchi T, Suzuki R, Tsubamoto Y, Komeda K, Nakano R, Miki H, et al. Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes. 2000;49:1880–1889. doi: 10.2337/diabetes.49.11.1880. [DOI] [PubMed] [Google Scholar]
- 89.Hennige AM, Burks DJ, Ozcan U, Kulkarni RN, Ye J, Park S, Schubert M, Fisher TL, Dow MA, Leshan R, et al. Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. J Clin Invest. 2003;112:1521–1532. doi: 10.1172/JCI18581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Okamoto H, Hribal ML, Lin HV, Bennett WR, Ward A, Accili D. Role of the forkhead protein FoxO1 in beta cell compensation to insulin resistance. J Clin Invest. 2006;116:775–782. doi: 10.1172/JCI24967. *By overexpressing a mutant, constitutively nuclear form of Foxo1, in the ductal and beta cells, the authors show that such mutant Foxo1 prevents beta cell replication in mouse models of beta cell hyperplasia during insulin resistance. Their study demonstrates that nuclear exclusion of Foxo1 is very critical in response of beta cells to insulin resistance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest. 2004;114:828–836. doi: 10.1172/JCI21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kushner JA, Ye J, Schubert M, Burks DJ, Dow MA, Flint CL, Dutta S, Wright CV, Montminy MR, White MF. Pdx1 restores beta cell function in Irs2 knockout mice. J Clin Invest. 2002;109:1193–1201. doi: 10.1172/JCI14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas in human pregnancy. Br J Obstet Gynaecol. 1978;85:818–820. doi: 10.1111/j.1471-0528.1978.tb15835.x. [DOI] [PubMed] [Google Scholar]
- 94.Brelje TC, Scharp DW, Lacy PE, Ogren L, Talamantes F, Robertson M, Friesen HG, Sorenson RL. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology. 1993;132:879–887. doi: 10.1210/endo.132.2.8425500. [DOI] [PubMed] [Google Scholar]
- 95.Brelje TC, Stout LE, Bhagroo NV, Sorenson RL. Distinctive roles for prolactin and growth hormone in the activation of signal transducer and activator of transcription 5 in pancreatic islets of langerhans. Endocrinology. 2004;145:4162–4175. doi: 10.1210/en.2004-0201. [DOI] [PubMed] [Google Scholar]
- 96.Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology. 1992;130:1459–1466. doi: 10.1210/endo.130.3.1537300. [DOI] [PubMed] [Google Scholar]
- 97.Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29:301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- 98.Vasavada RC, Garcia-Ocana A, Zawalich WS, Sorenson RL, Dann P, Syed M, Ogren L, Talamantes F, Stewart AF. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. J Biol Chem. 2000;275:15399–15406. doi: 10.1074/jbc.275.20.15399. [DOI] [PubMed] [Google Scholar]
- 99.Picard F, Wanatabe M, Schoonjans K, Lydon J, O’Malley BW, Auwerx J. Progesterone receptor knockout mice have an improved glucose homeostasis secondary to beta -cell proliferation. Proc Natl Acad Sci U S A. 2002;99:15644–15648. doi: 10.1073/pnas.202612199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rawlins EL, Hogan BL. Epithelial stem cells of the lung: privileged few or opportunities for many? Development. 2006;133:2455–2465. doi: 10.1242/dev.02407. [DOI] [PubMed] [Google Scholar]
- 101.Li Y, Cao X, Li LX, Brubaker PL, Edlund H, Drucker DJ. beta-Cell Pdx1 expression is essential for the glucoregulatory, proliferative, and cytoprotective actions of glucagon-like peptide-1. Diabetes. 2005;54:482–491. doi: 10.2337/diabetes.54.2.482. [DOI] [PubMed] [Google Scholar]
- 102.Buteau J, Spatz ML, Accili D. Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic beta-cell mass. Diabetes. 2006;55:1190–1196. doi: 10.2337/db05-0825. [DOI] [PubMed] [Google Scholar]
- 103.Bouwens L, Rooman I. Regulation of pancreatic beta-cell mass. Physiol Rev. 2005;85:1255–1270. doi: 10.1152/physrev.00025.2004. [DOI] [PubMed] [Google Scholar]
- 104.Tanigawa K, Nakamura S, Kawaguchi M, Xu G, Kin S, Tamura K. Effect of aging on B-cell function and replication in rat pancreas after 90% pancreatectomy. Pancreas. 1997;15:53–59. doi: 10.1097/00006676-199707000-00008. [DOI] [PubMed] [Google Scholar]
- 105.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. **This study unravels the molecular basis for age related decline in the regenerative potential of beta cells and implicates the age related increase in cell cycle regulator p16 in this process. [DOI] [PubMed] [Google Scholar]
- 106.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nielsen GP, Stemmer-Rachamimov AO, Shaw J, Roy JE, Koh J, Louis DN. Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab Invest. 1999;79:1137–1143. [PubMed] [Google Scholar]
- 108.Zindy F, Soares H, Herzog KH, Morgan J, Sherr CJ, Roussel MF. Expression of INK4 inhibitors of cyclin D-dependent kinases during mouse brain development. Cell Growth Differ. 1997;8:1139–1150. [PubMed] [Google Scholar]
- 109.Fujino T, Asaba H, Kang MJ, Ikeda Y, Sone H, Takada S, Kim DH, Ioka RX, Ono M, Tomoyori H, et al. Low-density lipoprotein receptor-related protein 5 (LRP5) is essential for normal cholesterol metabolism and glucose-induced insulin secretion. Proc Natl Acad Sci U S A. 2003;100:229–234. doi: 10.1073/pnas.0133792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rulifson IC, Karnik SK, Heiser PW, ten Berge D, Chen H, Gu X, Taketo MM, Nusse R, Hebrok M, Kim SK. Wnt signaling regulates pancreatic beta cell proliferation. Proc Natl Acad Sci U S A. 2007;104:6247–6252. doi: 10.1073/pnas.0701509104. *This study identifies Wnt signaling as a major regulator of beta cell proliferation and demonstrates that Wnt signaling enhances proliferation through an effect on Cyclin D2 levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Smart NG, Apelqvist AA, Gu X, Harmon EB, Topper JN, MacDonald RJ, Kim SK. Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-beta signaling and induces reversible diabetes mellitus. PLoS Biol. 2006;4:e39. doi: 10.1371/journal.pbio.0040039. *The authors make use of conditional overexpression of Smad7 in Pdx1 positive cells to show that TGF-beta signaling is really critical for the function of mature beta cells. [DOI] [PMC free article] [PubMed] [Google Scholar]