

Abstract
Growing evidence suggests that pharmacological inhibition of Na/H exchange and Na/HCO3 transport provides protection against damage or injury in cardiac ischemia. In this study, we examined the contribution of the sodium/bicarbonate cotransporter NBCn1 (slc4a7) to cytotoxicity in cultured hippocampal neurons of rats. In neurons exposed to extracellular pH (pHo) ranging from 6.2 to 8.3, NBCn1 protein expression increased by fivefold at pH < 6.5 compared to the expression at pHo 7.4. At pHo 6.5, the intracellular pH of neurons was ~1 unit lower than that at pH 7.4. Immunochemistry showed a marked increase in NBCn1 immunofluorescence in plasma membranes and cytosol of the soma as well as in dendrites, at pHo 6.5. NBCn1 expression also increased by 40% in a prolonged Mg2+-free incubation at normal pHo. Knockdown of NBCn1 in neurons had negligible effect on cell viability. The effect of NBCn1 knockdown on cytotoxicity was then determined by exposing neurons to 0.5 mM glutamate for 10 min and measuring lactate dehydrogenase (LDH) release from neurons. Compared to normal incubation (pHo 7.2 for 6 h) after glutamate exposure, acidic incubation (pHo 6.3 for 6 h) reduced cytotoxicity by 75% for control neurons and 78% for NBCn1-knockdown neurons. Thus, both controls and knockdown neurons showed acidic protection from cytotoxicity. However, in Mg2+-free incubation after glutamate exposure, NBCn1 knockdown progressively attenuated cytotoxicity. This attenuation was unaffected by acidic preincubation before glutamate exposure. We conclude that NBCn1 has a dynamic upregulation in low pHo and Mg2+ depletion. NBCn1 is not required for acidic protection, but increases cytotoxicity in Mg2+-free conditions.
Keywords: acid, ischemia, Na/HCO3 transporter, pH, SLC4A7
Introduction
The acid–base transporters involving Na/H exchange and Na/HCO3 transport are known to affect damage and injury in non-neuronal tissues. In the heart, inhibition or knockout of Na/H exchange protects myocytes against ischemia (Karmazyn, 1999; Wang et al., 2003). Inhibition of Na/HCO3 transport reduces myocardial ischemia–reperfusion injury (Khandoudi et al., 2001). In astrocytes, inhibition of Na/HCO3 transport reduces cell vulnerability to acid injury (Giffard et al., 2000). Thus, inhibition of these proteins reduces cell damage in cardiac tissues and astrocytes.
Na/H exchangers and Na/HCO3 transporters are also present in neurons (Chesler, 2003). However, it is currently unclear whether these proteins are involved in neuronal damage. The primary function of Na/H exchangers and Na/HCO3 transporters is to extrude acid equivalents from the cytosol and recover intracellular pH (pHi) from acidification (Romero et al., 2004; Bobulescu et al., 2005). Intracellular acidification, together with extracellular acidification, occurs in global and focal ischemia (Lipton, 1999). Severe acidification is deleterious to neurons (Nedergaard et al., 1991). However, moderate to mild acidification has been shown to protect neurons from damage in vitro (Lipton, 1999). Thus, neurons can either undergo or be protected from ischemia-induced damage depending upon the magnitude of acidification.
Ionic changes other than acidification occur during ischemia and, among those changes, an aberrant Mg2+ homeostasis is of particular interest. Mg2+ serves as a cofactor for ATP, a modulator of ion channels and receptors, and a cofactor for enzymatic reactions. Low Mg2+ concentrations in the cerebrospinal fluid and serum are found during the early hours after ischemic stroke (McKee et al., 2005). The magnitude of Mg2+ decrease reflects the severity of neuronal damage, and there is a close relationship between low Mg2+ levels and viability of neurons under pathological states.
Acid extrusion involving Na/HCO3 cotransport in neurons is known to be governed by the Na+-driven Cl/HCO3 exchanger, which moves Na+ and HCO3− into the cytosol in exchange for cytosolic Cl− (Schwiening & Boron, 1994; Baxter & Church, 1996). Recent molecular and cellular studies suggests that some neurons also express the Na/HCO3 cotransporter, which moves HCO3− into the cytosol together with Na+ (Cooper et al., 2005; Rickmann et al., 2007; Chen et al., 2008). In prenatal and natal hippocampal neurons, the electroneutral Na/HCO3 cotransporter (NBCn1), encoded by solute carrier family transporter 4A (SLC4A)7, is predominantly localized in the dendrites and soma (Cooper et al., 2005) whereas the Na/HCO3 cotransporter NBCn2 (SLC4A10) and the Na+-driven Cl/HCO3 exchanger NDCBE (SLC4A8) are mostly localized in the soma (Grichtchenko et al., 2001; Giffard et al., 2003; Boedtkjer et al., 2008). In the present study, we examined NBCn1 expression in primary cultures of embryonic rat hippocampal neurons in acidic pH and Mg2+-free incubation. We also examined the contribution of NBCn1 to cytotoxicity of neurons under this aberrant ion homeostasis. We focused our research on NBCn1 in this study, as the transporter is extremely sensitive to cellular and systemic pH changes (Kwon et al., 2002; Jakobsen et al., 2004).
Materials and methods
All experiments were conducted under the NIH guidelines for research on animals, and experimental protocols were approved by the Institutional Animal Care and Use Committee at Emory University.
Primary cultures of embryonic hippocampal neurons
Primary cultures of hippocampal neurons were prepared from fetal E19 Sprague Dawley rats (300 g; Harlan Laboratories, Indianapolis, IN, USA). Pregnant rats were fully anesthetized with isoflurane and quickly decapitated, and fetuses were surgically removed. Fetal brains were removed after decapitation and placed in sterile ice-cold dissecting solution (Eagle’s balanced salt solution; Invitrogen, Carlsbad, CA, USA) with 10mM HEPES and 1mM sodium pyruvate. Hippocampi were removed under a dissecting microscope and then subjected to enzymatic digestion with papain (Worthington Biochemical Corporation; Lakewood, NJ, USA) as well as mechanical digestion. Neurons were plated in Dulbecco’s modification of Eagle’s medium (DMEM) for acidification experiments, or Neurobasal medium (Invitrogen) for knockdown experiments and immunocytochemistry. Neurons were incubated in a 5% CO2 humidified chamber at 37°C for 10–14 days. For acidic (or alkaline) incubation, neurons were incubated for 1 h in media of a series of pH ranging from 6.2 to 8.3. The media contained DMEM (no HCO3−) supplemented with 5% fetal bovine serum, 25 U/mL of combined penicillin and streptomycin, 0.6% dextrose, 0.1% MITO (BD Biosciences, San Jose, CA) serum extender and 5 μMcytosine arabinoside. The pH was adjusted by varying HCO3− levels according to the Henderson–Hasselbalch equation with the solubility coefficient of 0.03 mmol CO2/mmHg and PCO2 bubbled at 35.65 mmHg for pH 6.2–7.7 or 17.83 mmHg for pH 8.0–8.3.
Immunoblots
The anti-NBCn1 antibody (provided by Walter Boron, Case Western Reserve University, USA) was generated by immunizing a guinea pig with a GST-fusion protein containing the C-terminal 87 amino-acid residues of rat NBCn1-B (Chen et al., 2007). The characterization of this antigen shows no crossreactivity to other neuronal Na/HCO3 transporters (Chen et al., 2007). Neurons were scraped in ice-cold homogenization buffer containing 300 mM mannitol, 5 mM HEPES (pH 7.2), 0.1 mg/mL phenylmethanesulphonyl fluoride and 1× protease inhibitor cocktail I (Calbiochem, San Diego, CA, USA). Cells were homogenized with a 26-gauge needle and centrifuged at 810 g at 4°C for 10 min. Supernatants were collected and assayed for the determination of protein concentration using the Bradford reagents (Sigma–Aldrich, St Louis, MO, USA). Immunoblot analyses were carried out using equal amounts of protein from the samples. Samples were separated on 4–15% sodium dodecylsulfate (SDS) polyacrylamide gels and blotted onto polyvinylidene fluoride (PVDF) membrane. Blots were incubated with the guinea pig anti-NBCn1 antibody in phosphate-buffered saline (PBS) containing 0.05% Tween 20 and 5% nonfat dry milk (dilution 1:500) for 2 h. Blots were washed four times for 10 min and then incubated with horseradish peroxidase (HRP)-conjugated goat antiguinea pig IgG (Millipore, Billerica, MA, USA) for 1 h. Blots were washed and visualized by ECL chemiluminescence (GE Healthcare, Chicago, IL, USA). For β-actin, blots were stripped with 62.6 mM Tris-HCl (pH 6.7), 2% SDS and 0.7% β-mercaptoethanol at 50°C for 10 min, and reprobed with the mouse β-actin antibody (Millipore). Densitometric measurements of NBCn1 and β-actin signals were carried out using IMAGEQUANT image analysis software (Molecular Dynamics, Sunnyvale, CA, USA) according to the manufacturer’s protocol. Mean pixel intensities of NBCn1 and β-actin were measured by positioning boxes around protein bands.
Immunofluorescence immunocytochemistry
After enzymatic and mechanical digestion of the hippocampus as described above, neurons (1–2 × 105 cells) were plated on 60-mm dishes containing poly-D-lysine-coated coverslips and incubated for 10–14 days. Cells were fixed with 4% paraformaldehyde, permeabilized with PBS containing 0.1% Triton X-100 (PBT), and blocked in PBT containing 5% normal goat serum (NGS) for 30 min. After washes, coverslips were incubated with the anti-NBCn1 antibody (1:250) in PBS containing 0.025% Triton X-100 and 2.5% NGS at 4°C overnight. Coverslips were rinsed with PBT twice for 20 min and incubated with the goat HRP-conjugated antiguinea pig IgG (1:400; GE Healthcare) for 30 min. Coverslips were then incubated with TSA + tetramethylrhodamine (1:300; Perkin Elmer Life Sciences, Boston, MA, USA). Coverslips were mounted on slides with Vectashield (Vector Laboratories, Burlingame, CA, USA) and examined with a Zeiss Axiovert 100M confocal microscope (Oberkochem, Germany) using a Plan Neofluar 100 × lens (numerical aperture 1.3; oil immersion). Immunofluorescence images were acquired using the LSM510 confocal acquisition software. Images were obtained from a single optical section (130 × 130 × 1.49 μm) with a 543-nm fluorescence filter. A single optical section was achieved by stacking images at an interval of 0.5 μm for 12 stacks. Exposure time was 6.29 s for sections. The pinhole setting was 186 μm. Captured images were analyzed using IMAGEQUANT image analysis software. The tetramethylrhodamine immunofluorescence image was converted to a gray scale and inverted. Fluorescence intensity was calibrated by setting the gray value range from 0 (white) to 255 units (black). The fluorescence intensity of tetramethylrhodamine was in the linear range. Mean pixel intensity of immunofluorescence was determined by positioning boxes (700 square pixels) at different locations around the soma and dendrites. Background intensity was determined by positioning boxes in the areas where there was no cell.
Measurements of pHi
The steady-state pHi of neurons was determined according to the protocol of Wang et al. (2006) with a slight modification. Briefly, neurons on a coverslip were loaded with 6.5 μm of 2′,7′-bis- (2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECFAM) for 10 min and then mounted in a closed-bath confocal imaging chamber RC-30 (Warner Instruments, Hamden, CT, USA) affixed on the stage of a Nikon TE200 inverted microscope. The microscope was equipped with a Lambda 10–2 filter wheel controller and a multi-wavelength filter set. The dye was alternately excited with 500 and 440 nm light, and the emission light at 530 nm was captured. The ratio of the emission from 500 nm (I500) to the emission from 440 nm (I440) was calculated after the subtraction of backgrounds (i.e. neurons without dye) from total I500 and I440. Calibration of the fluorescence signal was performed using the K+/H+ ionophore nigericin. The steady-state pHi was calculated by determining linear least-squares analysis over a minimum of 20 s. Data were acquired using METAflUOR software (Universal Imaging, Downingtown, PA, USA). All experiments were performed at 37°C.
Production of small interference RNA (siRNA)/NBCn1 lentiviral particles
Viral vectors for siRNA/NBCn1-knockdown and siRNA/ns control were made by the custom design and construction service in Cellogenetics (Ijamsville, MD, USA). The nucleotides corresponding to position 589–607, 1641–1659, and 2739–2757 of rat NBCn1 (Genbank accession number: NM_058211) were selected as the best target sequences for siRNA knockdown according to the company’s design algorism. The nucleotides were separately inserted downstream of the human U6 promoter in the lentiviral vector, pLenti-h6BX. The control vector pLenti-hsNS containing 19 non-specific nucleotides GTTCTCCGAACGTGTCACG was purchased from Cellogenetics. The vector contains enhanced green fluorescence protein (eGFP), which is used to detect infection with the green fluorescence filter. Viral particles were produced by transfecting human embryonic kidney HEK293-FT cells with pLenti-h6BX/NBCn1 and pLenti-h6BX/ns vectors. The day before transfection, the HEK293-FT cells (5 × 106 cells) were plated on a 100-mm plate in the culture medium (DMEM with 10% fetal bovine serum, 25 mM HEPES and 120 μg/mL penicillin and streptomycin, pH 7.4). Next day, the medium was replaced with fresh culture medium without antibiotics. Cells (90–95% confluent) were transfected with 5 μg lentiviral vectors, 2.5 μg pVSVG (encoding G envelope protein), 3.75 μg pCM?8.9 (encoding Gag and Pol proteins) and 36 μL Lipofectamine 2000 (Invitrogen) in 1.5 mL of Opti-MEM medium according to the manufacturer’s protocol. Cells were incubated at 37°C overnight, and the medium was replaced with a culture medium containing 2 mM L-glutamine and 0.5 mg/mL Geneticin. The medium was collected 72 h post-transfection and centrifuged at 500 g for 10 min. The supernatant containing viral particles was filtered through a membrane (0.45 μm pore size) and stored at −80°C.
Titration of viral stocks and infection of hippocampal neurons
Titration of viral stocks was done by transducing HEK293 FT cells or HEK cells that stably express NBCn1 (Cooper et al., 2005). The day before titration, cells were plated in a six-well plate (1.8–2 × 105 cells per well) and incubated in the culture medium overnight. On the day of infection, 10-fold serial dilutions (10−2 to 10−6) of the viral supernatants were added to 1 mL of fresh culture medium and the medium in the wells was replaced. Cells were incubated at 37°C for 2 days. The infection was monitored by counting green fluorescent cells examined via an inverted microscope Axiovert 135 (Zeiss) with a GFP filter. The titer in transducing units (TU) was calculated by counting GFP-positive cells in two consecutive diluted plates. The titer was typically 1 × 105 TU/mL or higher. To concentrate the viral stocks, the supernatants were centrifuged at 72,100 g at 16°C for 90 min and pellets were resuspended in PBS and stored at −80°C. The titer of the concentrated viral stocks was typically ~1 × 107 TU/mL. Hippocampal neurons were then infected with virus at a multiplicity of infection (MOI) of 0.36–1.1. The infection was monitored on a daily basis for green fluorescence. At 72-h post infection, neurons were incubated in the culture medium at pH 6.3 or 7.2 for 6 h and then lysed with the lysis buffer for immunoblot to determine the efficacy of NBCn1 knockdown.
Cell damage and cytotoxicity
Cell damage or cytotoxicity of neurons was determined by the quantitative analysis of lactate dehydrogenase (LDH) released from neurons (Koh & Choi, 1987). Following enzymatic and mechanical digestion of hippocampi, neurons were split in a 96-well plate (1.8 × 104 cells/well) and incubated in Neurobasal medium at 37°C for 10–14 days. Cytotoxicity was induced by treating neurons with 0 or 500 μM glutamate for 10 min in the exposure medium (in mM: NaCl, 119; KCl, 5; CaCl2, 2; MgCl2, 1.8; glucose, 10; HEPES, 10; glycine, 0.05; and NaHCO3, 28; pH 7.2) and then incubating neurons in pH 6.3 or 7.2 media without glutamate for 6 h. MgCl2 was omitted from Mg2+- free medium. For knockdown experiments, neurons were infected with siRNA/NBCn1 or siRNA/ns lentiviral particles in normal culture medium and then incubated for 72 h before glutamate exposure. Cell damage or cytotoxicity was determined using the LDH-Cytotoxicity Assay kit (BioVision, Mountain View, CA, USA). The maximum amount of LDH release was achieved by breaking neurons with 1% Triton X-100. The media (no cells) with or without Triton X-100 served as backgrounds. The amount of formazan produced in culture supernatants was determined by measuring the absorbance of samples at 490 nm with the microtiter plate reader MRX (Dynex, Chantilly, VA, USA). Cell damage or cytotoxicity was expressed as the percentage of the total LDH release from cell lysis after subtracting backgrounds.
Statistical analysis
The n-value in the manuscript refers to the number of samples that were independently prepared from three to five experiments. Data are reported as mean ± SE. Level of significance was assessed using: (i) an unpaired, two-tailed Student’s t-test to analyze immunofluorescence between neurons at normal vs. acidic pH; (ii) a paired, two-tailed Student’s t-test to analyze immunoblot and LDH release of paired groups of neurons (control vs. knockdown) subjected simultaneously to normal and acidic pH; (iii) a one-way ANOVA with the Bonferroni post-test to analyze NBCn1 protein expression and pHi in a series of pH; and (iv) a two-way ANOVA with the Bonferroni post-test to analyze LDH release from control and knockdown neurons at different time in Mg2+-free medium. P < 0.05 was considered significant. Data were analyzed using ORIGIN 8.1 software (OriginLab Corporation; Northampton, MA, USA).
Results
Characterization of the new NBCn1 antibody
We validated the new guinea pig polyclonal antibody against the C-terminal 87 amino acids of rat NBCn1 by immunoblot. These residues correspond to most of the predicted cytoplasmic C-terminal domain of a rat NBCn1-B splice variant (Choi et al., 2000). The characterization of this antigen shows no cross-reactivity with other neuronal Na/HCO3 transporters (Chen et al., 2007). Figure 1A shows an immunoblot of a membrane preparation from the primary cultures of embryonic rat hippocampal neurons. The antibody recognized a band at ~150 kDa, similar to NBCn1 protein in cerebral cortex and hippocampus of mouse as detected with a rabbit polyclonal antibody against the same antigenic domain (Chen et al., 2007). The immunoreactive band disappeared when the primary antibody was preabsorbed with the GST/NBCn1 fusion protein containing the C-terminal 87 amino acids of rat NBCn1-B. The specificity of the antibody was also tested by immunoblot of a membrane preparation from Xenopus oocytes expressing rat NBCn1 or injected with water (Fig. 1B). The antibody recognized a band at ~150 kDa in oocytes expressing the transporter, but not in control oocytes.
Fig. 1.

Characterization of the NBCn1 antibody. (A) Immunoblot of crude plasma membranes isolated from the primary cultures of embryonic rat hippocampal neurons. Immunoblot was performed with the guinea pig NBCn1 antibody against the C-terminal 87 amino acids of rat NBCn1-B. For preabsorption, the antibody was preabsorbed with GST/NBCn1 fusion protein containing the immunogen (50 μg/mL) for 1 h before probing the membrane. (B) Immunoblot of membrane preparation from Xenopus oocytes. Oocytes were injected with cRNA encoding rat NBCn1-E or water.
NBCn1 in cultured hippocampal neurons was upregulated at low pH
Significant decreases in pHi and extracellular pH (pHo) occur during cerebral ischemia (Lipton, 1999). The magnitude of the decrease varies between 0.6 and 1.2 pH units for global and focal ischemia. The decrease occurs within minutes of global ischemia whereas for focal ischemia the decrease occurs within minutes to hours (core region) and hours (penumbra). To examine the effect of acidic pH on NBCn1, we first determined NBCn1 protein expression in primary cultures of rat hippocampal neurons that were incubated in media at pH ranging from 6.2 to 7.4 [established by adjusting (HCO3−) at fixed 5% CO2] for 1 h. Neurons were also incubated in media at pH ranging from 7.4 to 8.3 [established by increasing (HCO3−) and lowering PCO2] to examine the transporter expression at alkaline pH. Crude plasma membranes were isolated from neurons and assessed for immunoblot. Figure 2A shows a marked increase in NBCn1 protein expression at pHo < 6.5. Changes in NBCn1 expression were negligible at pHo ranging from 6.8 to 8.3. The signals for β-actin, an internal control, remained unaffected at pHo ranges we tested. Analyzed by densitometric measurements of NBCn1 relative to β-actin (Fig. 2B), the expression of NBCn1 at pHo < 6.5 was significantly stronger than those at pHo 6.8–8.3 (F7,22 = 6.3, P < 0.001). The expression at pHo 6.5 was five times higher than that at pHo 7.4. A similar increase at pHo 6.2 was also observed when neurons were incubated for 30 min (data not shown).
Fig. 2.

NBCn1 expression in hippocampal neurons at different pH. (A) Immunoblot of NBCn1. Cultured hippocampal neurons of rats were incubated in media at pH ranging from 6.2 to 8.3 for 1 h. Immunoblot of crude plasma membranes isolated from neurons was performed with an anti-NBCn1 antibody. The blot was striped and reprobed with the anti-β-actin antibody. One of four experiments is shown. (B) Densitometric measurement of NBCn1. The pixel intensity of the NBCn1-immunoreactive band was measured and normalized to that of β-actin (n = 4); *P < 0.05. (C) Steady-state pHi of neurons incubated at different pHo for 1 h. The pHi was measured using the ratiometric pH-sensitive dye BCECF-AM. Significant decreases in pHi were observed at pHo < 6.5. The pHi values at pHo 6.5–8.0 were similar (P > 0.05, two-way ANOVA). Measurements were made with four to nine preparations at each pHo; error bars are SEM.
The steady-state pHi of neurons at different pHo was measured using the ratiometric pH-sensitive dye BCECF-AM. Large decreases in pHi were observed at pHo < 6.5 (F7,30 = 62.6, P < 0.001; Fig. 2C). At pHo 6.5, the pHi was 6.32 ± 0.04 (n = 8), significantly lower than 7.55 ± 0.02 (n = 6) at pHo 7.4 (F1,12 = 509.6, P < 0.001). Changes in pHi at pHo 6.5–8.0 were relatively small. These data show a close correlation of the pHi profile with the NBCn1 expression profile: lower pHi with greater NBCn1 expression.
Figure 3 shows NBCn1 immunocytochemistry of cultured neurons that were incubated at pHo 6.5 or 7.4 for 1–3 h (n = 3 for each). Tetramethylrhodamine-amplified immunofluorescence was localized in puncta in somatodendrites, consistent with the previous observation of NBCn1 localization in these neurons (Cooper et al., 2005). Acidic incubation markedly increased NBCn1 immunofluorescence (Fig. 3A and D). The increase occurred in the intensity and number of the punctuate fluorescence in soma and in dendrites at 1 h and remained for 3 h. Controls without the primary antibody had no labeling. Immunofluorescence was quantitated by measuring mean pixel intensity at different locations around single neurons (see Materials and methods). An increase in NBCn1 labeling was detected at low pHo in membranes (t80 = −20.1, P = 0.004) and cytosol of soma (t60 = −20.7, P = 0.005) as well as in dendrites (t189 = −18.4, P < 0.001; Fig. 3H). Neurons had negligible dendritic blebbing or swelling, indicating that cells were not significantly damaged at pHo 6.5. The negligible cell damage is compatible with low values of LDH release in these neurons at acidic pHo as described below.
Fig. 3.

Immunocytochemistry of NBCn1 in hippocampal neurons. (A–E) NBCn1 immunofluorescence in neurons incubated at pH 6.5 or 7.4 for 1–3 h. The labeling was amplified with tetramethylrhodamine (see Materials and methods). Control was neurons without the primary antibody. One of three immunofluorescence experiments is shown. (F and G) NBCn1 immunofluorescence in dendrites of neurons incubated at pH 6.5 or 7.4 for 1 h. The whole cell images are shown in insets. (H) Quantitation of NBCn1 immunofluorescence. The immunofluorescence images were converted to a gray scale and inverted (0 units as white and 255 units as black). Mean pixel intensity of NBCn1 immunofluorescence was determined by positioning boxes (700 square pixels) at different locations around plasma membrane, cytosol and dendrites. Numbers in parentheses are numbers of counts, and error bars are SEM. Scale bars, 50 μm. *P < 0.05.
NBCn1 was upregulated in Mg2+-free medium
In addition to acidification, other ionic changes occur during ischemia and one of these changes is a decrease in the extracellular Mg2+ level; for review see McKee et al., 2005). To examine NBCn1 protein expression under these conditions, we incubated cultured hippocampal neurons in Mg2+-free medium and determined NBCn1 expression by immunoblot. Figure 4A shows a representative immunoblot of membrane preparation from neurons that were incubated in Mg2+-free medium at pH 7.2 for 3–6 h. NBCn1 protein expression was upregulated under this condition, while β-actin expression remained unaffected. The densitometric measurements of NBCn1 relative to β-actin showed a 35–40% increase during these incubations (n = 3 for each, t2 = −5.8, P = 0.03; Fig. 4B). This magnitude was relatively small compared to the fivefold increase at pHo 6.2–6.5 in Fig. 2, but we obtained consistent results in four independent experiments. The negligible change in NBCn1 expression at 1 h is not shown.
Fig. 4.

NBCn1 expression in Mg2+-free medium. (A) Immunoblot of NBCn1. Cultured hippocampal neurons were incubated in Mg2+-free medium (pH 7.2) for 3–6 h. Immunoblots of crude plasma membrane preparation from neurons were probed with the anti-NBCn1 antibody. The blot was striped and reprobed with the anti-β-actin antibody. (B) Densitometric measurements of NBCn1. The pixel intensity of NBCn1 was measured and normalized to that of β-actin (n = 4 for each). Error bars are SEM. *P < 0.05.
Neurons with NBCn1 knockdown maintained viability
In studying the effect of NBCn1 on cell damage or cytotoxicity, it is not possible to pharmacologically inhibit NBCn1 because no specific blockers are available. For this reason, we developed a lentivirus-mediated siRNA knockdown approach by which NBCn1 transcripts, and subsequent proteins, are degraded in neurons. The nucleotide sequences corresponding to positions 589–607, 1641–1659, and 2739–2757 of rat NBCn1 were selected and separately introduced to a lentiviral vector containing eGFP. The control was a non-specific 19 nucleotide sequence (see Materials and methods). The efficacy of NBCn1 knockdown was tested by infecting HEK cells that stably express the transporter (Cooper et al., 2005) with siRNA/NBCn1 knockdown and siRNA/ns control lentivirus (data not shown). Hippocampal neurons were then infected with knockdown or control lentivirus (1.8–5.4 × 105 viruses per 5 × 105 neurons) and 72 h post-infection the medium was switched to an acidic medium of pH 6.3 for 6 h. Immunoblot showed a marked reduction in NBCn1 expression mediated by siRNA/NBCn1 knockdown (Fig. 5A). Densitometric measurement indicated a reduction of 90% on average (n = 3 for each; t2 = 11.3, P = 0.008; Fig. 5B).
Fig. 5.

Effect of NBCn1 knockdown on viability of neurons. (A) Immunoblots of NBCn1 in neurons treated with control and knockdown lentivirus. (B) Densitometric measurements of NBCn1. The intensity of NBCn1 was normalized to that of β-actin (n = 3). (C) LDH release from neurons infected with siRNA/ns control lentivirus. Neurons were incubated in media at pH 6.3 or 7.2 for 6 h and LDH release was determined by measuring the medium absorbance at 490 nm. The absorbance value from cell lysis represents the total LDH release from neurons lysed with 1% Triton X-100, and the value from medium is the background without cells. (D) LDH release from neurons infected with siRNA/NBCn1 knockdown lentivirus (n = 6). Error bars are SEM. *P < 0.05.
To examine the effect of NBCn1 knockdown on cell viability, we infected neurons with siRNA/ns or siRNA/NBCn1 lentivirus and 72 h post-infection switched medium to a bath pH of 6.3 or 7.2 for 6 h. pH 6.3 was chosen to ensure NBCn1 upregulation in acidic incubation (i.e. within pH 6.2–6.5). The toxic effect of NBCn1 knockdown was then tested by measuring LDH release from neurons. LDH release from cell lysis represents the total LDH release from neurons treated with 1% Triton X-100, while the release from medium only (no cells) refers to background. Both total and basal releases were unaffected by changes in pH, thus validating the analysis of cell damage at different pH values. Cell damage is presented as the percentage of LDH release normalized to total release after subtracting backgrounds. In neurons with siRNA/ns control (Fig. 5C), the mean cell damage was 5.8 ± 0.1% at acidic pH incubation and 6.3 ± 0.7% at normal pH incubation (n = 6 for each). These values were not significantly different from each other (t5 = −2.5, P = 0.06). Low LDH release at pHo 6.3 is consistent with the finding by others (Yermolaieva et al., 2004) that cell damage is minimal in cultured hippocampal neurons of mouse at pHo > 6.0. Similarly, in neurons with siRNA/NBCn1 knockdown (Fig. 5D), the mean cell damage was 9.0 ± 0.5% at acidic pH incubation and 9.2 ± 0.6% at normal pH incubation (n = 6 for each), not significantly different from each other (t5 = −2.5, P = 0.53). LDH release from knockdown neurons remained small at both acidic and normal pH. Thus, despite NBCn1 expression being severely reduced by knockdown, neurons maintained their viability at both pHo 6.3 and 7.2.
NBCn1 knockdown did not affect acidic protection of neurons from glutamate cytotoxicity
The above results indicate that NBCn1 had negligible effect on the viability of neurons maintained at low pHo although the transporter is upregulated in acidic environments. We then examined whether NBCn1 was linked to acidic protection from cytotoxicity. Cytotoxicity was induced by applying 500 μM glutamate (50 μM glycine and 0 Mg2+, pHo 7.2) for 10 min (Choi et al., 1987). Neurons were infected with control and knockdown virus and, at 72 h post-infection, neurons were exposed to glutamate and then switched to medium at pH 6.3 or 7.2 for 6 h without the added glutamate. In control neurons (Fig. 6, left bar graphs), glutamate exposure induced cell damage at pHo 7.2 incubation (31 ± 2%, n = 5). Acidic incubation significantly reduced damage (7.6 ± 1.0%, n = 5; t4 = −10.6, P < 0.001). This low value was similar to the basal values (5.8%) for neurons that were incubated at acidic pH without glutamate exposure (Fig. 5A). The decrease following acidic incubation was 75% on average. In NBCn1-knockdown neurons (Fig. 6, right bar graphs), glutamate exposure caused cell damage at pHo 7.2 (30 ± 2%, n = 5). The damage was significantly reduced at pHo 6.3 (6.5 ± 1.0%, n = 5; t4 = −8.4, P = 0.001), showing a decrease by 78% on average. This decrease was not different from the decrease for control neurons (F1,17 = 1.1, P = 0.31). Thus, neurons were protected from glutamate cytotoxicity by acidic pHo regardless of NBCn1 knockdown.
Fig. 6.
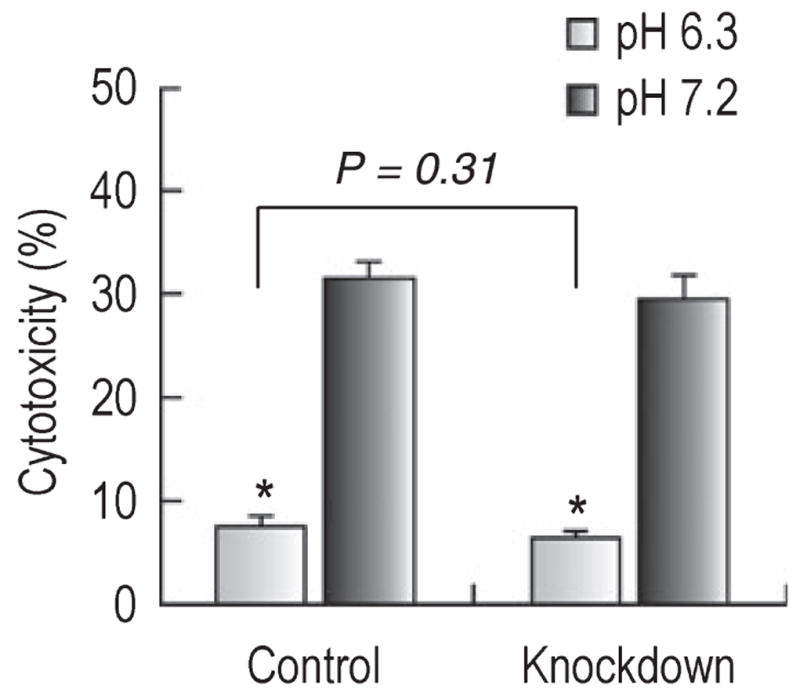
Glutamate cytotoxicity in neurons at acidic and normal pH. Neurons with siRNA control and NBCn1 knockdown lentivirus (72 h post-infection) were exposed to 500 μM glutamate (50 μM glycine and 0 Mg2+, pH 7.2) for 10 min and then switched to medium at pH 6.3 or 7.2 for 6 h without the added glutamate. Cytotoxicity was determined as the percentage of LDH released from neurons relative to the total release after background subtraction (n = 5 for each groups of neurons). Error bars are SEM. *P < 0.05.
NBCn1 knockdown reduced cytotoxicity in a prolonged Mg2+-free incubation
NBCn1 was upregulated in Mg2+-free medium (Fig. 4). To examine whether this upregulation was linked to cytotoxicity, we exposed neurons to glutamate as described above and incubated them in Mg2+-free medium for 1–6 h (pH 7.2). Figure 7A shows that glutamate cytotoxicity progressively increased with time in both control neurons and NBCn1-knockdown neurons. This development of cytotoxicity is consistent with the previous report (Sun et al., 2001) that a prolonged Mg2+ depletion induces spontaneous epileptiform-like activity and causes cell death in hippocampal neurons. However, glutamate cytotoxicity in a prolonged Mg2+-free incubation was attenuated in NBCn1-knockdown neurons (F1,54 = 11.9, P = 0.001). The difference between the two groups of neurons was progressively more prominent with time. Thus, at 6 h incubation, NBCn1-knockdown neurons had ~30% less cytotoxicity than controls. A similar Mg2+-free incubation without glutamate exposure had negligible cytotoxicity in both groups of neurons (Fig. 7B), thus indicating that Mg2+-free incubation itself is not sufficient to cause cytotoxicity. To test whether the reduction in cytotoxicity by NBCn1 knockdown was dependent upon pH, we incubated control neurons and NBCn1-knckdown neurons in acidic medium without Mg2+ after glutamate exposure. However, this approach did not provide valuable information as both groups of neurons had a reduction in cytotoxicity due to the protective effect of acidity (data not shown). As an alternative approach, we incubated neurons at pH 6.3 for 1 h before glutamate exposure (Fig. 7C). Following incubation at acidic pH, NBCn1-knockdown neurons had lower glutamate cytotoxicity than control neurons (t9 = 4.0, P = 0.003). The decrease was ~16% on average, not significantly different from 23% glutamate cytotoxicity on average at pHo 7.2 preincubation (F1,29 = 2.7, P = 0.11). Therefore, NBCn1 knockdown reduced glutamate cytotoxicity regardless of acidic preconditioning.
Fig. 7.

Effects of NBCn1 knockdown on cytotoxicity in Mg2+-free medium. (A) Glutamate cytotoxicity in Mg2+-free medium. Neurons with siRNA control and knockdown were incubated for 1–6 h in Mg2+-free medium (pH 7.2) after glutamate exposure and assessed for the cytotoxicity assay (n = 5 for each). (B) Cytotoxicity in Mg2+-free medium without glutamate exposure. The experimental procedure was similar to the above except that neurons were exposed to 0 mM glutamate (n = 5 for each). (C) Preincubation of neurons at pH 6.3 before glutamate exposure. Neurons were incubated at pH 6.3 or 7.2 before glutamate exposure and then incubated in Mg2+-free medium (pH 7.2) for 2.5 h (n = 10 for pH 6.3 and n = 5 for pH 7.2). Error bars are SEM. *P < 0.05.
In conclusion, our data show that NBCn1 in cultured hippocampal neurons was upregulated at low pH and in Mg2+-free medium. The upregulation had minimal effect on acidic protection from glutamate cytotoxicity. In the absence of Mg2+, however, the upregulation could make neurons more vulnerable to glutamate cytotoxicity. The significance of our findings is discussed below.
Discussion
In this study, we provide the first evidence for the association of NBCn1 with cytotoxicity in cultured hippocampal neurons. Using molecular and cellular approaches, we determined the effects of acidity and Mg2+ on NBCn1 expression, knocked down endogenous NBCn1 expression in neurons, and examined the effect of NBCn1 knockdown on glutamate cytotoxicity under conditions where the transporter was upregulated. The major finding is that NBCn1 in hippocampal neurons had a dynamic upregulation in low pH as well as Mg2+-free incubation, but its contribution to glutamate cytotoxicity varied depending upon the availability of acidity and Mg2+. Our data could be a valuable starting point for future studies aimed at determining the pathological role of NBCn1 in cerebral ischemia.
Upregulation of NBCn1
A dramatic increase in NBCn1 protein expression occurred when neurons were incubated at pH 6.2–6.5 (Fig. 2). The expression remained unaffected at pHo 6.5–8.3, which is in the range of ~1 pH unit from the physiological pH 7.2–7.4. NBCn1 is known to have a dynamic response to acidic environments in some tissues. In the kidney, the expression and activity of NBCn1 is upregulated by chronic metabolic acidosis (Kwon et al., 2002; Jakobsen et al., 2004). Chronic metabolic acidosis increases NH4+ absorption from the lumen in the medullary thick ascending limb of the kidney. The increase in NH4+ absorption overloads intracellular H+ in the tubules as NH4+ dissociates into NH3 and H+ (Knepper et al., 1989). The thick ascending limb cells compensate intracellular H+ overloads by upregulating NBCn1 in chronic metabolic acidosis. Whether a similar NBCn1-dependent compensatory mechanism exists in neurons is unclear, but our study shows a close correlation between low pHi and NBCn1 upregulation in primary cultures of hippocampal neurons. The pHi of cultured neurons markedly decreased at pHo < 6.5 (Fig. 2C) and this decrease was similar to NBCn1 upregulation at these pHo ranges. In adjusting pHo, we varied (HCO3−) and PCO2 in the medium. The (HCO3−) in the medium of pH 6.5 is eight times less than that at pHo 7.4 (3.25 vs. 26 mM). Despite this low (HCO3−) in the medium, NBCn1 was markedly upregulated at pHo 6.5. Conversely, the (HCO3−) in the medium of pH 8.3 was four times higher (104 vs. 26 mM). Despite this, NBCn1 expression remained unaffected at pHo 8.3. Therefore, it is unlikely that HCO3− in the medium determined NBCn1 expression.
Negligible role of NBCn1 in acidic protection
With the observation of NBCn1 upregulation in acidic environments, we hypothesized that the transporter would play a role in protecting neurons from cytotoxicity at low pH. We instead found both control neurons and NBCn1-knockdown neurons having a similar reduction in glutamate toxicity at low pHo (Fig. 6), thus indicating a negligible effect of NBCn1 knockdown on acidic protection. In our experiments, the glutamate exposure condition was identical in both control neurons and knockdown neurons before they were placed in either acidic or normal medium. This suggests that the protection of neurons at acidic pHo is not caused by the process of glutamate exposure but instead is directly related to the acidity of media or in the cell. It is not clear how the acidity of media or in the cell reduces cytotoxicity mediated by glutamate exposure, but the mechanism should involve inhibition of a pH-dependent process downstream of glutamate receptor activation.
Effect of NBCn1 on glutamate cytotoxicity caused by Mg 2+-free incubation
Despite the negligible role of NBCn1 in acidic protection, the transporter can affect neuronal damage. NBCn1-knockdown neurons had an attenuation of glutamate cytotoxicity in a prolonged Mg2+-free incubation (Fig. 7A). This attenuation did not occur when cytotoxicity was examined in the presence of Mg2+ (Fig. 6), thus indicating that the effect of NBCn1 on neuronal damage was dependent upon the availability of Mg2+ in the medium. In our experiments, ‘Mg2+-free’ refers to the nominal removal of Mg2+ from the medium but does not imply a complete lack of Mg2+, as discussed by others (Sombati & DeLorenzo, 1995). Mg2+ may be released from the cell via Mg2+ transporters (Schmitz et al., 2007). The exact concentration of Mg2+ in our Mg2+-free medium is not determined, but it would probably be lower than that in vivo during pathological states. The Mg2+ level is reduced to 0.3–0.48 mM within 8 h after the onset of ischemia in human (Altura et al., 1997). It remains to be determined whether the effect of NBCn1 on damage of hippocampal neurons can occur at such Mg2+ levels.
By what mechanism does NBCn1 affect cytotoxicity caused by Mg2+ depletion? The development of cytotoxicity requires glutamate (Fig. 7B) and this led us to speculate that Mg2+-sensitive N-methyl-D-aspartate (NMDA) receptors may be involved in cytotoxicity under Mg2+-free conditions. The receptors mediate a large intracellular Ca2+ load that initiates apoptotic cellular events leading to cell death (Dingledine et al., 1999). The receptors are blocked by Mg2+ in a voltage-sensitive and noncompetitive manner. Taken together, it is possible that Mg2+ depletion from the glutamate-containing medium causes NMDA receptor activation, which results in cytotoxicity.
Based on the data in the present study, we propose a model for the pathological effect of NBCn1 on cytotoxicity in hippocampal neurons. The pHi of neurons falls under acidic conditions and the acid-extruder NBCn1 is upregulated to compensate for intracellular acidification. The upregulation of NBCn1 may cause neuronal damage, but this does not occur at low pHi due to the protective effect of acidity. The transporter can also be upregulated at normal pHo when Mg2+ is less available. Under this low-Mg2+ condition, the upregulation of the transporter facilitates deleterious effects on neurons and renders neurons more vulnerable to cytotoxicity. Thus, the cytotoxicity potentially mediated by the transporter is normally protected but can be uncovered by low Mg2+ in the extracellular fluid of the brain. NBCn1 could be one of the proteins to facilitate neuronal injury in hypomagnesemia.
Comparison with Na/HCO3 transporter-mediated damage in astrocytes
The electrogenic Na/HCO3 cotransporter (NBCe1; SLC4A4) is predominantly expressed in astrocytes (Rickmann et al., 2007; Majumdar et al., 2008). Inhibition of this transporter reduces cell vulnerability to acid injury (Giffard et al., 2000). The data in our study are thus consistent with the report by Giffard et al. (2000) in that inhibition or knockdown of Na/HCO3 transporters reduces cell damage. Despite this similarity, several differences are recognizable. First, hippocampal neurons are viable at pH 6.3 incubation whereas astrocytes die at pH 6.8 incubation. This difference is consistent with the previous reports that neurons are more resistant than astrocytes to acidic injury. Second, NBCn1 knockdown causes negligible effects on neuronal viability at low pHo whereas NBCe1 inhibition increases glial viability at low pHo. Third, NBCn1 contributes to glutamate cytotoxicity in Mg2+-free conditions whereas NBCe1 appears to have a limited effect on acidic injury. Apparently, both NBCn1 and NBCe1 affect cytotoxicity or cell death, but the mechanisms underlying their effects are distinct.
Maintenance of cell viability at low pH
In addition to the finding of a pathological role of NBCn1, we also observed that cultured hippocampal neurons maintained their viability at pHo 6.3 (Fig. 5). The threshold pHo for acidic cell death depends on both time and degree of intracellular acidification. In cortical cultures of neurons, the threshold to induce 50% cell death is pHo 6.4 for 6 h acid exposure in the absence of CO2/HCO3− buffer (Nedergaard et al., 1991). In cultured hippocampal neurons of mouse, LDH release at pHo > 6.0 is minimal (Yermo- laieva et al., 2004). A prolong exposure to an acidic environment can induce the acid-activated ion channels (ASIC) in central neurons (Johnson et al., 2001; Xiong et al., 2004), and mediates Ca2+ influx via ASIC, causing acidic injury in neurons. However, these channels are mostly activated at pHo < 6.0 (Johnson et al., 2001; Xiong et al., 2004). Consistent with this, our data show low LDH release from hippocampal neurons at pHo 6.3.
Summary
We found that NBCn1 knockdown reduces cytotoxicity of hippocampal neurons subjected to Mg2+ depletion from the medium. This finding is consistent with the previous reports that inhibition of Na/H exchange or Na/HCO3 cotransport protects cells against damage or injury in cardiac cells. Nevertheless, in our system, the mechanism for NBCn1 contribution to cytotoxicity appears to be different from those in cardiac cells, and further investigation is needed. In particular, it will be interesting to investigate what mechanisms ameliorate the cytotoxicity that would be predicted with the upregulation of NBCn1.
Acknowledgments
The authors acknowledge Walter Boron (Case Western Reserve) for providing the guinea pig polyclonal NBCn1 antibody. The authors acknowledge Ronald Abercrombie (Emory) for helpful discussion. This work was supported by the American Heart Association Southeast Affiliate and the URC grant at Emory (I.C.), and NIH DK61418 (C.C.Y).
Abbreviations
- BCECF-AM
2′,7′-bis-(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester
- DMEM
Dulbecco’s modification of Eagle’s medium
- eGFP
enhanced green fluorescence protein
- HEPES
4-(2-hydroxyethyl)-1-piperazineethane-sulfonic acid
- HRP
horseradish peroxidase
- LDH
lactate dehydrogenase
- MOI
multiplicity of infection
- NBCe1
electrogenic Na/bicarbonate cotransporter 1
- NBCn1
electroneutral Na/bicarbonate cotransporter 1
- NGS
normal goat serum
- PBS
phosphate-buffered saline
- pHi
intracellular pH
- pHo
extracellular pH
- PVDF
polyvinylidene fluoride
- siRNA
small interference RNA
- SLC4A
solute carrier family transporter 4A
- TU
transducing unit
References
- Altura BT, Memon ZI, Zhang A, Cheng TP, Silverman R, Cracco RQ, Altura BM. Low levels of serum ionized magnesium are found in patients early after stroke which result in rapid elevation in cytosolic free calcium and spasm in cerebral vascular muscle cells. Neurosci Lett. 1997;230:37–40. doi: 10.1016/s0304-3940(97)00471-0. [DOI] [PubMed] [Google Scholar]
- Baxter KA, Church J. Characterization of acid extrusion mechanisms in cultured fetal rat hippocampal neurones. J Physiol. 1996;493:457–470. doi: 10.1113/jphysiol.1996.sp021396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobulescu IA, Di Sole F, Moe OW. Na+/H+ exchangers: physiology and link to hypertension and organ ischemia. Curr Opin Nephrol Hypertens. 2005;14:485–494. doi: 10.1097/01.mnh.0000174146.52915.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boedtkjer E, Praetorius J, Fuchtbauer EM, Aalkjaer C. Antibody-independent localization of the electroneutral Na+-HCO3− co-transporter NBCn1 (slc4a7) in mice. Am J Physiol Cell Physiol. 2008;294:C591–C603. doi: 10.1152/ajpcell.00281.2007. [DOI] [PubMed] [Google Scholar]
- Chen LM, Choi I, Haddad GG, Boron WF. Chronic continuous hypoxia decreases the expression of SLC4A7 (NBCn1) and SLC4A10 (NCBE) in mouse brain. Am J Physiol Regul Integr Comp Physiol. 2007;293:R2412–R2420. doi: 10.1152/ajpregu.00497.2007. [DOI] [PubMed] [Google Scholar]
- Chen LM, Kelly ML, Rojas jd, Parker MD, Gill HS, Davis BA, Boron WF. Use of a new polyclonal antibody to study the distribution and glycosylation of the sodium-coupled bicarbonate transporter NCBE in rodent brain. Neuroscience. 2008;151:374–385. doi: 10.1016/j.neuroscience.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I, Aalkjaer C, Boulpaep EL, Boron WF. An electroneutral sodium/bicarbonate cotransporter NBCn1 and associated sodium channel. Nature. 2000;405:571–575. doi: 10.1038/35014615. [DOI] [PubMed] [Google Scholar]
- Cooper DS, Saxena NC, Yang HS, Lee HJ, Moring AG, Lee A, Choi I. Molecular and functional characterization of the electroneutral Na/HCO3 cotransporter NBCn1 in rat hippocampal neurons. J Biol Chem. 2005;280:17823–17830. doi: 10.1074/jbc.M408646200. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Giffard RG, Papadopoulos MC, van Hooft JA, Xu L, Giuffrida R, Monyer H. The electrogenic sodium bicarbonate cotransporter: developmental expression in rat brain and possible role in acid vulnerability. J Neurosci. 2000;20:1001–1008. doi: 10.1523/JNEUROSCI.20-03-01001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giffard RG, Lee YS, Ouyang YB, Murphy SL, Monyer H. Two variants of the rat brain sodium-driven chloride bicarbonate exchanger (NCBE): developmental expression and addition of a PDZ motif. Eur J Neurosci. 2003;18:2935–2945. doi: 10.1046/j.1460-9568.2003.03053.x. [DOI] [PubMed] [Google Scholar]
- Grichtchenko II, Risso-Bradley S, Rojas jd, Richerson GB, Boron WF. Localization of the Na+-driven Cl-HCO3 exchanger protein (NDCBE1) in rat brain. Soc Neurosci Abstr. 2001:A527, 12. [Google Scholar]
- Jakobsen JK, Odgaard E, Wang W, Elkjaer ML, Nielsen S, Aalkjaer C, Leipziger J. Functional up-regulation of basolateral Na+-dependent HCO3− transporter NBCn1 in medullary thick ascending limb of K+-depleted rats. Pflügers Arch. 2004;448:571–578. doi: 10.1007/s00424-004-1303-4. [DOI] [PubMed] [Google Scholar]
- Johnson MB, Jin K, Minami M, Chen D, Simon RP. Global ischemia induces expression of acid-sensing ion channel 2a in rat brain. J Cereb Blood Flow Metab. 2001;21:734–740. doi: 10.1097/00004647-200106000-00011. [DOI] [PubMed] [Google Scholar]
- Karmazyn M. The role of the myocardial sodium-hydrogen exchanger in mediating ischemic and reperfusion injury. From amiloride to cariporide. Ann NY Acad Sci. 1999;874:326–334. doi: 10.1111/j.1749-6632.1999.tb09248.x. [DOI] [PubMed] [Google Scholar]
- Khandoudi N, Albadine J, Robert P, Krief S, Berrebi-Bertrand I, Martin X, Bevensee MO, Boron WF, Bril A. Inhibition of the cardiac electrogenic sodium bicarbonate cotransporter reduces ischemic injury. Cardiovasc Res. 2001;52:387–396. doi: 10.1016/s0008-6363(01)00430-8. [DOI] [PubMed] [Google Scholar]
- Knepper MA, Packer R, Good DW. Ammonium transport in the kidney. Physiol Rev. 1989;69:179–249. doi: 10.1152/physrev.1989.69.1.179. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Kwon TH, Fulton C, Wang W, Kurtz I, Frokiaer J, Aalkjaer C, Nielsen S. Chronic metabolic acidosis upregulates rat kidney NaHCO3 cotransporters NBCn1 and NBC3 but not NBC1. Am J Physiol Renal Physiol. 2002;282:F341–F351. doi: 10.1152/ajprenal.00104.2001. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Majumdar D, Maunsbach AB, Shacka JJ, Williams JB, Berger UV, Schultz KP, Harkins LE, Boron WF, Roth KA, Bevensee MO. Localization of electrogenic Na/bicarbonate cotransporter NBCe1 variants in rat brain. Neuroscience. 2008;155:818–832. doi: 10.1016/j.neuroscience.2008.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee JA, Brewer RP, Macy GE, Borel CO, Reynolds JD, Warner DS. Magnesium neuroprotection is limited in humans with acute brain injury. Neurocrit Care. 2005;2:342–351. doi: 10.1385/NCC:2:3:342. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Goldman SA, Desai S, Pulsinelli WA. Acid-induced death in neurons and glia. J Neurosci. 1991;11:2489–2497. doi: 10.1523/JNEUROSCI.11-08-02489.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickmann M, Orlowski B, Heupel K, Roussa E. Distinct expression and subcellular localization patterns of Na+/HCO3− cotransporter (SLC 4A4) variants NBCe1-A and NBCe1-B in mouse brain. Neuroscience. 2007;146:1220–1231. doi: 10.1016/j.neuroscience.2007.02.061. [DOI] [PubMed] [Google Scholar]
- Romero MF, Fulton CM, Boron WF. The SLC4 family of HCO3− transporters. Pflügers Arch. 2004;447:495–509. doi: 10.1007/s00424-003-1180-2. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Deason F, Perraud AL. Molecular components of vertebrate Mg2+ -homeostasis regulation. Magnes Res. 2007;20:6–18. [PubMed] [Google Scholar]
- Schwiening CJ, Boron WF. Regulation of intracellular pH in pyramidal neurons from the rat hippocampus by Na+-dependent Cl− -HCO3− exchange. J Physiol. 1994;475:59–67. doi: 10.1113/jphysiol.1994.sp020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sombati S, DeLorenzo RJ. Recurrent spontaneous seizure activity in hippocampal neuronal networks in culture. J Neurophysiol. 1995;73:1706–1711. doi: 10.1152/jn.1995.73.4.1706. [DOI] [PubMed] [Google Scholar]
- Sun DA, Sombati S, DeLorenzo RJ. Glutamate injury-induced epileptogenesis in hippocampal neurons: an in vitro model of stroke-induced “epilepsy”. Stroke. 2001;32:2344–2350. doi: 10.1161/hs1001.097242. [DOI] [PubMed] [Google Scholar]
- Wang Y, Meyer JW, Ashraf M, Shull GE. Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ Res. 2003;93:776–782. doi: 10.1161/01.RES.0000094746.24774.DC. [DOI] [PubMed] [Google Scholar]
- Wang D, Lee HJ, Cooper DS, Cebotaro L, Walden PD, Choi I, Yun CC. Coexpression of MAST205 inhibits the activity of Na+/H+ exchanger NHE3. Am J Physiol Renal Physiol. 2006;290:F428–F437. doi: 10.1152/ajprenal.00161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci USA. 2004;101:6752–6757. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]