

Abstract
Patients with the genomic instability syndrome Fanconi anemia (FA) commonly develop progressive bone marrow failure and have a high risk of cancer. The prominent role of the FA protein family involves DNA damage response and/or repair. Oxidative stress, defined as an imbalance between the production of reactive oxygen species and antioxidant defense, is considered to be an important pathogenic factor in leukemia-prone bone marrow diseases such as FA. Cellular responses inducing resistance to oxidative stress are important for cellular survival, organism lifespan, and cancer prevention, but until recently, mammalian factors regulating resistance to oxidative stress have not been well characterized. Significant evidence supports excessive apoptosis of hematopoietic stem/progenitor cells, induced by stresses, most significantly oxidative stress, as a critical factor in the pathogenesis of bone marrow failure and leukemia progression in FA. In this brief review, we discuss the functional link between FA proteins and oxidative DNA damage response/repair, with emphasis on the implication of oxidative stress in the pathophysiology and abnormal hematopoiesis in FA. Antioxid. Redox Signal. 10, 1909–1921.
Introduction
Fanconi anemia (FA) is a genetic disorder associated with bone marrow (BM) failure, clonal proliferation of hematopoietic stem cells, and transformation to leukemia and other cancers (2, 5, 8, 12, 21, 40, 92). Somatic cell fusion studies show FA is genetically heterogeneous, with at least 13 complementation groups identified thus far (8, 38, 40, 47). The genes encoding the groups A (FANCA), B (FANCB), C (FANCC), D1 (FANCD1/BRCA2), D2 (FANCD2), E (FANCE), F (FANCF), G (FANCG), -I (FANCI/KIAA1794), J (FANCJ/BRIP1), L (FANCL), M (FANCM), and -N (FANCN/PALB2) have been cloned (15–17, 35, 39, 48, 49, 56, 66–68, 80, 89, 91, 94, 104) (Table 1). The biological function of these FA proteins has been the subject of intense investigation in recent years. Work from Alan D'Andrea and others (9, 13, 16, 66, 89, 95, 100) in the field have established the existence of an FA pathway, in which eight of the FA proteins (namely, FANCA, B, C, E, F, G, L, and M) form a nuclear complex. It is believed this complex functions as an ubiquitin ligase. In response to DNA damage or DNA replication stress, the multimeric FA complex monoubiquitinates two downstream FA proteins, FANCD2 and FANCI, which then recruit other downstream FA proteins including FANCD1 (which is the breast cancer protein BRCA2), the recently identified FANCJ and FANCN, to nuclear loci containing damaged DNA and consequently influence important cellular processes such as DNA replication, cell-cycle control, and DNA damage repair (Fig. 1). It is known that mutations in any of the 13 genes lead to an FA phenotype manifested by developmental abnormalities, BM failure, and cancer (Fig. 2).
Table 1.
Complementation Groups of Fanconi Anemia
| Subtype | FA gene | Chromosome location | Protein (kD) | Patients with FA, estimated (%) | Main function of the protein, etc. |
|---|---|---|---|---|---|
| A | FANCA | 16q24.3 | 163 | 57 | FA core complex |
| B | FANCB (FAAP95) | Xp22.2 | 95 | 0.3 | FA core complex |
| C | FANCC | 9q22.3 | 63 | 15 | FA core complex |
| D1 | FANCD1 | 13q12.3 | 380 | 4 | RAD51 recruitment |
| D2 | FANCD2 | 3p26 | 155, 162 | 3 | Monoubiquitylated protein |
| E | FANCE | 6p22-p21 | 60 | 1 | FA core complex |
| F | FANCF | 11p15 | 42 | 2 | FA core complex |
| G | FANCG/XRCC9 | 9p13 | 68 | 9 | FA core complex |
| I | Unknown | 15q26.1 | ? | Rare | ? |
| J | FANCJ/BACH1/BRIP1 | 17q22-q24 | 130 | 1.6 | 5′ > 3′ DNA helicase/ATPase |
| L | FANCL/PHF9/POG (FAAP43) | 17q22-q24 | 43 | 0.1 | FA core complex, ubiquitin ligase |
| M | FANCM | 14q21.3 | 250 | Rare | FA core complex, ATPase/translocase |
| N | FANCN/PALB2 | 16p12.1 | 130 | 1 | Regulation of BRCA2 location |
FIG. 1.
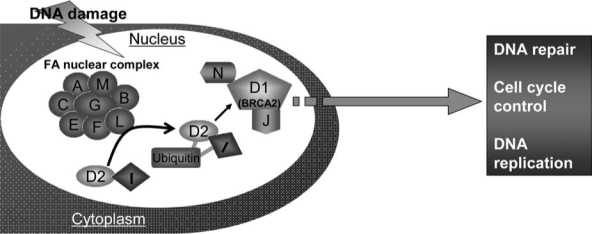
Function of the FA pathway. Eight FA proteins form a nuclear complex, which acts as ubiquitin ligase. In response to DNA damage or replication stress, it monoubiquitinates two other FA proteins, FANCD2 and FANCI, which then recruit other downstream FA proteins FANCD1, FANCJ, and FANCN to damaged DNA and influence DNA replication, cell-cycle control, and DNA repair processes
FIG. 2.
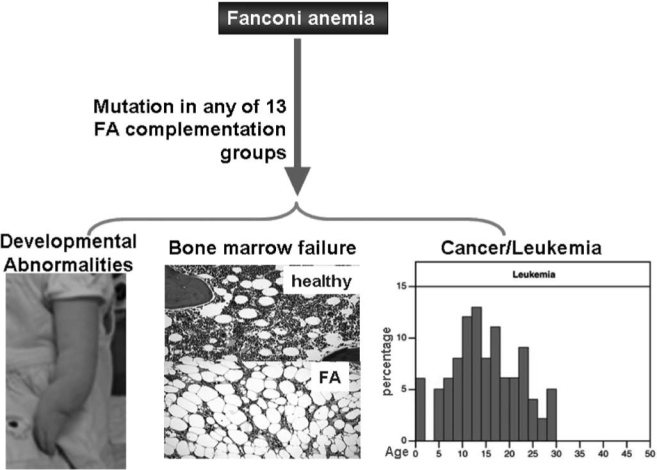
Consequence of FA defects. Mutations in any of the 13 genes lead to the FA phenotype such as developmental abnormalities, bone marrow failure, and cancer in FA patients
Since patients with FA uniformly develop BM failure and have high risk of progression to leukemia, FA proteins likely play specific roles in hematopoiesis by governing responses of hematopoietic cells to both genotoxic and cytotoxic stresses. Extensive studies indicate that chronic cytotoxic or genotoxic stresses differentially affect FA hematopoiesis by causing excessive apoptosis of hematopoietic stem cells and progenitors (HSC/P) (11, 12, 21, 31–33, 42, 51, 52, 61, 71, 74–76, 78, 79, 88, 100, 101, 103). Thus, the maintenance of HSC/P cells in bone marrow may require different functions of the FA proteins. This seems to be consistent with clinical outcomes and evolution of the disease: loss of FA functions causes excessive apoptosis of HSC/P in the early stage of FA, leading to BM failure. As the disease progresses, both apoptosis and genomic instability impose a selective pressure on FA HSC/P cells and promote the development of mutant clones leading to leukemia (Fig. 3).
FIG. 3.
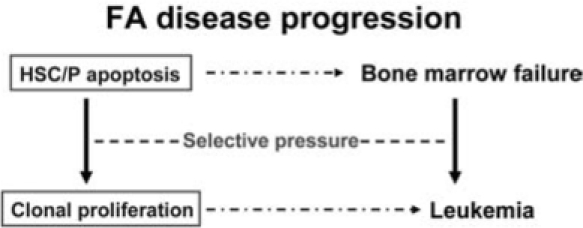
A model of FA disease progression. FA HSC/P cells are highly susceptible to stress-induced apoptosis, which may lead to HSC depletion and bone marrow failure. The combination of apoptosis and genomic instability provides recrudescent selective sweeps that purge hematopoietic tissues of all but the selected or adapted neoplastic stem cells
FA Hematopoiesis
The most important clinical features of FA are hematological. Children with FA often develop pancytopenia during the first few years of life. Complications of BM failure are the major causes of morbidity and mortality of FA, and 80% of FA patients die from BM failure (3, 6, 22, 45, 47, 53). In addition, FA patients have increased susceptibility of developing myelodysplasia (MDS) or acute myeloblastic leukemia (AML) (3, 6, 13, 22, 41, 95). They have also been known to frequently develop clonal chromosomal abnormalities in the BM progenitor cells in the later stage of the disease (3, 47, 95). In fact, certain clonal cytogenetic abnormalities, such as monosomy 5 and monosomy 7, are common in MDS and AML occurring secondary to treatment with alkylating agents and in children with FA who have evolved to MDS and AML (58, 83, 102). Thus, FA has been proposed as a genetic model system for studying these hematological malignancies (3, 13, 95).
Increasing evidence indicates progressive BM failure in children with FA results from excessive apoptosis and subsequent failure of the HSC compartment. The first evidence that FA BM cells demonstrate increased apoptosis was from a report that demonstrated CD34+ cells from children with FA expressed high levels of the death receptor Fas (11, 72). Subsequently, many laboratories have reported FA BM cells are hypersensitive to a variety of extracellular biological apoptotic cues, including interferon-γ (IFN-γ) and tumor necrosis factor α (TNF-α) (19, 21, 31, 42, 51, 52, 71, 74–76, 78–81, 86, 88, 101, 103). The most compelling evidence comes from studies of the functions of FANCC in apoptotic responses of hematopoietic cells, largely because FANCC was the first FA gene cloned and the Fancc knockout mouse was the first murine model of FA generated. For example, suppression of FANCC expression represses clonal growth of normal erythroid and granulocyte-macrophage progenitor cells and disruption of the Fancc gene in mice renders hematopoietic progenitor cells hypersensitive to the pro-apoptotic effects of IFN-γ and TNF-α (21, 31, 42, 51, 71, 74–76, 78, 79, 88, 101, 103). Conversely, overexpression of FANCC suppresses apoptosis in human hematopoietic progenitor cell lines (27, 28), in CD34+ cells from FA patients with FANCC mutations (101) and in HSC/P cells from Fancc knock-out mice (79, 103). These data suggest deregulation of apoptotic responses in hematopoietic cells may account at least in part for the nearly universal development of BM failure in children with inactivating FA mutations. However, the mechanism by which FA proteins modulate apoptotic responses and the downstream effector molecules involved are mostly unknown.
Results from BM culture assays have also demonstrated defective hematopoiesis in FA (3, 22, 95). Bone marrow cells from FA patients also show altered expression of certain growth factors and cytokines, such as reduced expression of interleukin-6 (IL-6) and granulocyte-macrophage colony-stimulating factor (GM-CSF) but increased secretion of mitotic inhibitor TNF-α (14, 19, 81, 82, 86, 90). These alterations may change the BM microenvironment (for instance, leading to factor deprivation or constant exposure to mitogenic inhibitors) and cause deregulation of cellular homeostasis. Studies of FA patients have demonstrated that BM from FA patients has decreased frequency of colony-forming progenitor cells, as well as a reduction in colony size (18, 29). These data suggest loss of FA gene function results in reduced survival or proliferation of lineage restricted progenitors and ultimately injury to the progenitor cell compartment. Using a murine model of FA complementation group C (Fancc), Haneline et al. (32, 33) convincingly demonstrated that Fancc−/− BM cells had a profound decrease in repopulating ability, and complementation with a retroviral vector encoding the normal human FANCC could restore Fancc−/− mouse BM stem and progenitor cell growth and proliferation in vivo. These data are consistent with damage to the stem cell compartment in FA.
FA Oxidant Hypersensitivity
Like other somatic stem cells, hematopoietic stem cells (HSCs) maintain life-long hematopoiesis in the bone marrow via their ability to self-renew and to differentiate into all blood lineages (Fig. 4). These HSCs are essentially required for the hematopoietic homeostasis. During differentiation, long-term hematopoietic stem cells (LT-HSCs) transit through short-term (ST)–HSCs and committed progenitor stages, which are characterized by restricted lineage potential and reduced self-renewal capacity. This is followed by ultimate differentiation into mature myeloid, erythroid, or lymphoid lineages. Molecular mechanisms controlling self-renewal and cell-fate decisions within the hematopoietic system remain poorly defined. In this context, HSC do not only need to replenish peripheral blood cells of all lineages, but also have to keep their pool relatively constant. There is good evidence that certain extrinsic cues provided in a special environment, the HSC-niches, essentially take part in regulating the HSC pool in vivo and might also be involved in leukemogenesis. Hematopoietic cells are exposed to various reactive oxygen species (ROS), which are routinely generated during metabolic or inflammatory process. ROS stimulation induces a variety of responses in hematopoietic cells, including cellular proliferation and growth inhibition (50, 70, 87, 108). Like cells from other tissues, hematopoietic cells have developed several mechanisms to prevent the damage induced by oxidative stress. Antioxidant enzymes, including superoxide dismutases (SODs), catalase, glutathione peroxidases, and peroxiredoxins, can directly eliminate ROS. Other cellular enzymes can function to repair DNA damage induced by ROS in hematopoietic tissues.
FIG. 4.
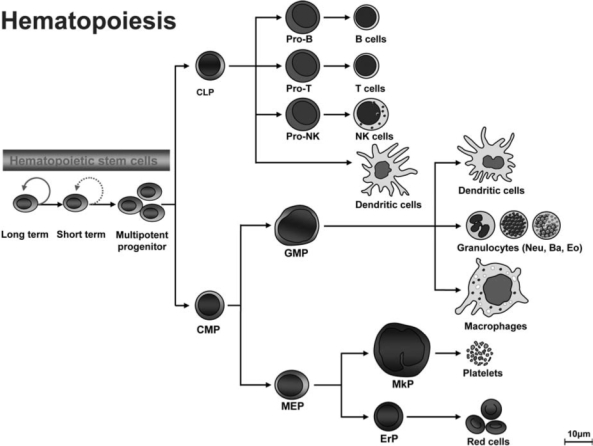
Normal hematopoiesis. Long-term hematopoietic stem cells (LT-HSCs) maintain life-long hematopoiesis in the bone marrow (BM) via their ability to self-renew and to differentiate into all blood lineages. During differentiation, LT-HSCs transit through short-term (ST)–HSCs and committed progenitor stages (characterized by restricted lineage potential and reduced self-renewal capacity) before differentiating into mature myeloid, erythroid, or lymphoid lineages
Studies on pathophysiological mechanisms of oxidative stress responses in stem cell diseases such as aplastic anemia (AA) have been very instructive and provide insights into the function of normal hematopoietic stem cells and their self-renewal capacity. One of the well-studied AA disease models is FA. There is strong evidence that FA cells are intolerant of oxidative stress. This was first suggested by the observation that cultured FA cells are vulnerable to oxygen-induced chromosomal aberrations (38). The finding was later confirmed by two other groups that showed FA fibroblasts and primary bone marrow cells grow better under hypoxic conditions than in ambient air (8, 85). Over the last decade, FA oxidant hypersensitivity has been documented in many studies using primary and immortalized cell cultures as well as ex vivo materials from patients (5, 8, 11, 27, 30, 43, 73, 77, 84).
While FA murine models do not recapitulate some of the major FA clinical manifestations such as BM failure and leukemia, hematopoietic cells from FA knockout mice exhibit extreme oxidant sensitivity. Saadatzadeh et al. (84) show that primary hematopoietic progenitors and murine embryonic fibroblasts (MEFs) isolated from Fancc−/− mice exhibit hypersensitivity to oxidative stress generated by hydrogen peroxide (H2O2). Furthermore, pretreatment with antioxidants selenomethionine or N-acetylcysteine (NAC) preferentially enhanced the survival of Fancc−/− cells. Mechanistically, the authors found that H2O2 induced overactivation of the serine-threonine kinase apoptosis signal-regulating kinase 1 (ASK1) in Fancc−/− cells, which was correlated with elevated H2O2-induced apoptosis in these mutant cells. Interestingly, ASK1 has been shown to be an important kinase involved in oxidant- and TNF-α-induced apoptosis (36), and Fancc−/− cells are uniquely hypersensitive to TNF-α (31, 78, 87, 107). It is therefore a plausible hypothesis that the TNF-α-oxidant–ASK1 pathways may play an important role in the observed functional deficits of Fancc−/− hematopoietic stem/progenitor cells, including reduced reconstitutional ability and proliferative potential, diminished self-renewal, and increased apoptosis. In addition, mice with combined deficiencies of the antioxidative enzyme, Cu/Zn superoxide dismutase, and Fancc genes demonstrated a defective hematopoiesis (30). Although not displaying development defects or increased chromosomal aberrations typical of FA, Fancc−/− Sod−/− mice had a phenotype of bone marrow hypocellularity, which is not detected in single mutant mice. Furthermore, hepatocytes from Fancc−/− Sod−/− mice exhibited a zonal pattern of microvesicular steatosis. All of the observations indicated the altered redox state of the hematopoietic progenitors in these mice was responsible for an impairment of cell proliferation or survival.
To dissect the relationship between oxidative damage and BM failure in FA, we have recently demonstrated that oxidative stress generated by repeated cycles of hypoxia-reoxygenation leads to excessive oxidative DNA damage and premature senescence in Fancc−/− bone marrow hematopoietic stem/progenitor cells (106, 107). These studies suggest that stress-induced senescence may be a novel mechanism underlying hematopoietic stem cell depletion in bone marrow failure diseases, including FA. More recently, we showed that ROS induce hematopoietic suppression in Fancc−/− mice exposed to bacterial toxin lipopolysaccharide or pro-inflammatory cytokine TNF-α (4, 65, 87, 108). TNF-α-induced senescence correlates with the accumulation of ROS and oxidative DNA damage, and pretreatment of TNF-α-injected Fancc−/− mice with a ROS scavenger significantly reduces oxidative base damage, DNA strand breaks, and senescence. Furthermore, Fancc−/− hematopoietic stem/ progenitor cells show increased chromosomal aberrations and have a lagging oxidative DNA damage repair. These results point to a potential link between oxidative DNA damage and hematopoietic stem cell defect in FA. The observed inefficient repair of oxidative damage in Fancc−/− hematopoietic stem cells may lead to decrease of stem cell quality (self-renewal capacity), which may ultimately cause premature exhaustion of the hematopoietic stem cell pool leading to BM failure.
One important issue concerns oxidative stress as a pathological factor in FA disease progression. Numerous reports indicate that FA cells, including HSC/P cells, are hypersensitive to oxidative stress (5, 8, 11, 27, 30, 38, 43, 73, 77, 84, 85, 106, 107). A number of hypotheses regarding the effect of oxidative stress in FA have been suggested, including the proposal that ROS could damage DNA and inability of FA hematopoietic stem/progenitor cells to repair such damage would result in exacerbated genomic instability leading to apoptosis and malignant transformation (Fig. 5).
FIG. 5.
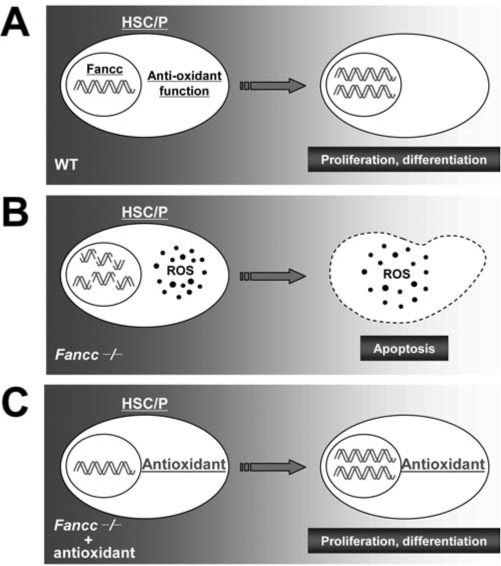
The effects of oxidative stress on HSC function. (A) Wild-type (WT) hematopoiesis, in which FA proteins regulate antioxidant response in HSC/P cells. The WT cells then recover from oxidative damage, survive, and proliferate normally. (B) Loss of FA function in HSC/P cells results in an increase in ROS, which ultimately results in increased accumulation of DNA damage and apoptotic death. (C) Reversal of this detrimental effect on HSC/P cells by administration of exogenous antioxidant, such as N-acetyl-L-cysteine (NAC) to the Fancc−/− mice, most likely due to the decrease in ROS
Redox-Sensitive FA Proteins
Because FA hematopoietic progenitor and stem cells have high rates of stress-induced apoptosis and reduced repopulating ability, FA proteins are subsequently believed to play an important role in the maintenance of normal hematopoiesis during oxidant metabolism. Consistent with the observations that cells derived from FA patients are intolerant of oxidative stress, an extensive body of evidence suggests that FA proteins play crucial role in oxidative stress signaling in variety of cell types including hematopoietic stem/progenitor cells.
Three major FA proteins, FANCA, FANCC, and FANCG, as parts of the FA protein complex, are found to associate with a variety of cellular factors that primarily function in redox-related processes (Table 2). For example, the FANCC protein interacts with NADPH cytochrome P450 reductase and glutathione S-transferase P1-1 (11, 43), two enzymes involved in either triggering or detoxifying reactive intermediates including ROS. Fancc−/− mice with deficiency in the antioxidative enzyme Cu/Zn superoxide dismutase demonstrated a defective hematopoiesis (30). Another FA protein, FANCG, interacts with cytochrome P450 2E1, a member of the P450 superfamily that is associated with the production of reactive oxygen intermediates, and mitochondrial antioxidant enzyme peroxiredoxin-3 (27, 69), suggesting a possible role of FANCG in protection against oxidative DNA damage. Significantly, Saadatzadeh et al. (84) recently showed oxidant hypersensitivity of Fancc−/− cells was due to an altered redox regulation and hyperactivation of ASK1, a serine-threonine kinase that plays an important role in redox apoptotic signaling. Moreover, oxidative stress induces complex formation by two major FA proteins, FANCA and FANCG (77).
Table 2.
Fanconi Anemia Proteins in Redox Signaling
| FA proteins | Interacting factors | References |
|---|---|---|
| FANCA | FANCG | Park et al., 2004 |
| FANCC | NADPH cytochrome P450 (RED) | Kruyt et al., 1998 |
| Glutathine S-transferase P1-1 (GSTP1) | Cumming et al., 2001 | |
| Cu/Zn superoxide dismutase (SOD) | Hadjur et al., 2001 | |
| Apoptosis signal-regulating kinase 1 (ASK1) | Saadatzadeh et al., 2003 | |
| FANCG | Cytochrome P450 2E1 (CYP2E1) | Futaki et al., 2002 |
| Mitochondrail anti-oxidant enzyme peroxiredoxin-3 | Mukhopadhyay et al., 2006 |
Oxidative Stress Response in FA Hematopoietic Cells: A p53 Connection
The tumor suppressor p53 is a key transcription factor that activates vital damage containment procedures to restrict aberrant cell growth in response to DNA damage, oncogene activation, and loss of normal cell contacts (28, 55). By eliciting cell cycle arrest and DNA damage response to oxidative DNA damage and oncogenic stress, p53 restricts cellular growth by inducing senescence, growth inhibition, or apoptosis that maintain genomic stability (37). Thus, p53 plays a major role in the prevention of cancer. Consistent with this, emerging evidence suggest that p53 deficiency may increase cancer development in patients with FA and FA mice. For example, studies have found a higher proportion of human papillomavirus–positive squamous cell carcinomas (SCC) in patients with FA than in healthy controls. Furthermore, SCC in FA patients is probably associated with the inactivation of p53 by HPV-associated oncoproteins rather than by direct mutagenesis, indicating that loss of functional p53 facilitated the tumor development in these cases (46, 57). Mice deficient for Fancd1 or Fancd2 have accelerated tumor development in Trp53-deficient background (34, 40). In addition, Fancc deficiency accelerates the development of certain blood and solid tumors in mice heterozygous at Trp53 (24). Moreover, these studies demonstrate that FA proteins and p53 cooperate in apoptosis and cell-cycle checkpoint control following DNA damage (25, 34, 54). Thus, p53 may function to prevent the propagation of damaged DNA through apoptosis. In FA patients, this leads to stem cell depletion, which may cause congenital abnormalities and bone marrow failure. Loss of p53 function may predispose to cancer by allowing premalignant cells to survive (41, 93).
Primary cells from FA patients and knockout mice are uniquely hypersensitive to oxidative stress-induced DNA damage and growth arrest. This suggests FA proteins may interplay with p53 in oxidative stress response. Encouraged by recent reports that p53 deficiency increases cancer development in patients with FA and FA knockout mice (24, 34, 40, 46, 57), we formally tested the hypothesis that FA proteins may functionally interact with the p53-activating signals in response to oxidative stress. Our unpublished results suggest that two major FA proteins, Fanca and Fancc, may coordinate with p53 in the regulation of oxidative stress response. This notion is supported by (i) hypersensitive response to oxidative stress in tissues in vivo and in cells in vitro derived from Fanca−/− or Fancc−/− mice is correlated with a persistent p53 overactivation; (ii) manipulation of p53 signaling alters H2O2-induced cell-cycle checkpoint (Fig. 6) and DNA damage response in primary Fanca−/− cells; and (iii) the functional status of p53 dictates the kinetics and persistence of response to oxidative stress in FA cells.
FIG. 6.

Manipulation of p53 signaling alters H2O2-induced cell-cycle checkpoint in Fanca −/− bone marrow cells. Note that Fanca−/− cells expressing the p53 inhibitor (p53DD) evade from H2O2-induced G2/M arrest but suffer apoptosis; whereas forced activation of p53 by Nutlin-3a does not induce G2/M arrest in Fanca−/− cells with a functional p53, as evidenced by induction of G0/G1 arrest
The mechanistic link between p53 signaling and FA has not been well defined. The involvement of p53 in FA pathophysiology has been highlighted by recent studies that show mice deficient for Fancd1, Fancd2, or Fancc have accelerated tumor development in Trp53-deficient background (24, 34, 40). Furthermore, developmental defects and increased apoptosis in Fancd2-deficient zebrafish could be corrected by knockdown of p53, suggesting p53-dependent apoptosis may be an underlying mechanism for developmental defect in the
Fancd2−/− fish (54). Some reports suggested that the activation of p53 leads to an increase in ROS that, perhaps by interfering with mitochondrial function and/or integrity, contributes to cell death. In addition, the higher levels of ROS appear to be part of the feedback loop that stabilizes p53 resulting in more p53 activity (26). Cellular stresses may increase mitochondrial ROS generation and increase p53 protein levels in some cell lines, whereas antioxidant NAC and the Cu/Zn SOD inhibitor can abolish the stress-induced increase in ROS and p53 levels (7). While these studies have not formally conducted in FA cells, the difference in redox status may result in different levels of p53 activation in WT and FA cells. In WT cells, ROS attack chromosomal DNA and generate oxidative DNA damage, mainly in the form of 8-oxo-deoxyguanosine (8-oxo-dG). The damage can induce the activation of p53 by phosphorylation (for example, phospho-p53 at Ser20–p53Ser20), leading to the repair of the oxidative DNA damage (Fig. 7). Loss of FA function leads to increased level of ROS or reduced repair of the oxidative DNA damage. Consequently, FA cells accumulate higher level of oxidative DNA damage, leading higher level of p53 activation. Additional investigations into functional interaction between the p53 and FA pathways in oxidative DNA damage stress response may aid us in better understanding how cells can bypass the normal checkpoints and continue to proliferate in the presence of damaged DNA and oncogenic activation. In the context of FA, new insights on the role of FA proteins in oxidative DNA damage response/repair can suggest new pathways and proteins to target for therapeutic prevention of cancer progression of the disease.
FIG. 7.
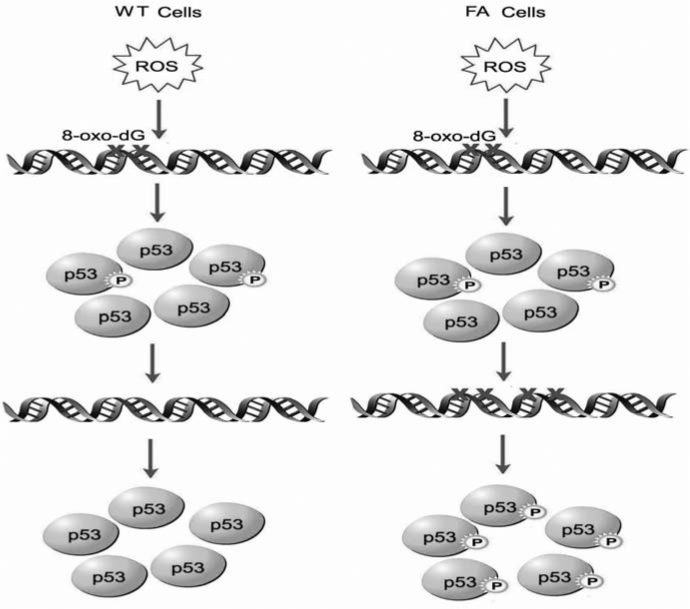
The difference in redox status may result in different levels of p53 activation in WT and FA cells. In WT cells, ROS attach chromosomal DNA and generate oxidative DNA damage, mainly in the form of 8-oxo-deoxyguanosine (8-oxo-dG). The damage can induce the activation of p53 by phosphorylation (for example, phospho-p53 at Ser20–p53Ser20), leading to the repair of the oxidative DNA damage. Loss of FA function leads to increased level of ROS or reduced repair of the oxidative DNA damage. Consequently, FA cells accumulate higher level of oxidative DNA damage, leading higher level of p53 activation
The Link Between Inflammatory ROS and FA Leukemogenesis
Certain chronic inflammatory conditions have long been known to link to cancer. There is compelling evidence that chronic inflammation increases the risk of human cancers such as hepatocellular carcinoma, colon and bladder cancers, B cell lymphomas, and visceral malignancies (44, 62, 92, 97). Chronic inflammation in the intestinal or bronchial epithelia also promotes carcinomas of the colon and lung (20). Other inflammatory diseases like Barrett's syndrome and Crohn's disease are linked to the development of esophageal cancer and bowel cancer, respectively (3, 9). In these pathological conditions, unresolved inflammation provokes cell turnover coupled with ROS generated at sites of inflammation, leading to chromosomal DNA mutations and malignant transformation of the cells (2, 10).
Oxidative stress is considered to be an important pathogenic factor in leukemia-prone bone marrow diseases like FA (5, 8, 11, 27, 30, 38, 43, 69, 73, 77, 84, 85, 106, 107). The expression of inflammatory mediators, particularly the pro-inflammatory cytokines tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), and IL-6 in these patients is often associated with increased production of ROS either as a component of their immune response or as a consequence of increased metabolism (59, 63, 64, 96). Thus, the presence of pro-inflammatory cytokines and increased oxidative stress in these patients may account for profound physiologic changes, including the development of BM failure and progression to leukemia. Many studies (24, 51, 81, 108) have shown a correlation between elevated circulating pro-inflammatory cytokines and anemia in patients with leukemia-related BM diseases but direct evidence for the mechanistic link between inflammation and leukemia is lacking.
The inflammatory cytokine TNF-α is considered as one important pathological factor involved in the abnormal hematopoiesis In FA. Studies from our laboratory and others have suggested excessive apoptosis of FA hematopoietic cells induced by TNF-α, which is overproduced in FA patients,?? may contribute to the pathophysiology of BM failure frequently occurring in FA children. The recent pioneer work from the laboratories of Wade Clapp and Laura Haneline (31–33, 51, 88) has demonstrated that ex vivo culture of Fancc−/− BM cells leads to an increase in cytogenetic abnormalities and myeloid malignancies that are associated with an acquired resistance to TNF-α, suggesting FA hematopoietic cells are prone to clonal hematopoiesis and malignancy. It is well established that TNF-α-induced ROS production involves the c-JUN NH2-terminal kinase (JNK) and nuclear factor-kappa B (NF-κB) pathways (71, 98). TNF-α-induced ROS activate the JNK kinase, which in turn leads to more ROS production, and sustained JNK activation in NF-κB-deficient cells was suggested to depend on ROS. Studies have shown that the production of ROS by TNF-α at inflammatory sites causes DNA damage (1, 92, 99). The persistent high levels of oxidative DNA damage observed in HSC/progenitor cells from TNF-α-injected Fancc−/− mice suggest that a deficiency in the FA pathway renders chromosomal DNA susceptible to ROS attack, thereby increasing oxidative DNA damage (87, 108). Our recent studies have also indicated that TNF-α not only is a pro-apoptotic signal suppressing FA hematopoietic progenitor activity, but also promotes leukemic transformation of FA hematopoietic stem/progenitor cells (50). Specifically, we tested the leukemia-promoting effects of TNF-α in Fancc−/− stem cells in vitro. We found that TNF-α exposure initially inhibited the growth of Fancc−/− bone marrow hematopoietic stem/progenitor cells, but longer term exposure of these cells promoted the outgrowth of TNF-α-resistant cytogenetically abnormal clones that upon transplantation into congenic wild-type mice led to acute myelogenous leukemia. TNF-α induced ROS-dependent genetic instability in Fancc−/− but not in WT cells. The leukemic clones were TNF-α-resistant but retained their characteristic hypersensitivity to mitomycin C, and exhibited high levels of chromosomal instability. Expression of FANCC cDNA in Fancc−/− stem/progenitor cells protected them from TNF-α-induced clonal evolution. The molecular etiology of FA leukemogenesis remains unknown. We hypothesize that FA disease progression to leukemia is governed not only by genetic changes intrinsic to the FA cells, but also by epigenetic and environmental factors and that TNF-α-mediated inflammation is one of the most important epigenetic and environmental factors contributing to FA leukemogenesis. Our studies on the role of TNF-α in FA leukemogenesis suggest that TNF-α exposure creates an environment in which somatically mutated preleukemic stem cell clones are generated and selected for. Our results thus provide direct confirmation of the importance of selective pressure in the evolution of leukemic clones in FA. These studies also suggest a model, in which mutations in the FA genes can cause genomic instability and overproduction of TNF-α, which induces apoptosis through upregulation of ROS and JNK/p38 kinases. Patients with excessive apoptosis of BM cells develop bone marrow failure. Chronic exposure of FA BM cells to proinflammatory cytokine TNF-α selects for progenitor cells that are apoptosis-resistant and acquire proliferative advantage. Patients with these TNF-α-resistant BM cells advance to myelodysplasia (MDS) and acute myelogenous leukemia (AML) via a mechanism involving genomic instability, coupled with inflammation driven by high NF-κB transcriptional activity (Fig. 8).
FIG. 8.

The pro-inflammatory cytokine TNF-α and its potential role in FA pathophysiology. Overproduced TNF-α plays role in not only pro-apoptotic signal suppressing FA hematopoietic progenitor activity, but also promoting leukemic transformation of FA hematopoietic stem/progenitor cells, which lead to typical phenotype of FA patients
While the role of FA proteins in the regulation of TNF-α-induced ROS production remains to be elucidated, it is likely FA proteins can disrupt downstream ROS signaling by protecting chromosomal DNA from ROS attack or facilitating the repair of oxidative DNA damage. Recently, the Grompe group reported that treatment of Fancd2−/−;Trp53 +/− mice with the antioxidant tempol delayed solid tumor development, possibly through a mechanism involving the enhanced repair of oxidative DNA damage by the antioxidant (105). However, it is also possible FA proteins can influence the expression of antioxidant enzymes (such as glutathione S-transferases and catalase) or the biosynthesis of ROS metabolic molecules such as glutathione. So far, there is no direct evidence for any of these assumptions. Another potential target is the redox-sensitive transcription factor NF-κB whose activation is known to enhance inflammation and promote cancer (10, 23, 60). Indeed, TNF-α-resistant BM hematopoietic stem/progenitor cells advance to acute myelogenous leukemia via a mechanism involving genomic instability coupled with inflammation driven by high NF-κB transcriptional activity (50).
Conclusion
FA is now considered the only human genomic instability syndrome that is uniquely sensitive to oxidative stress. As a bona-fide hematopoietic stem cell disease, FA represents an excellent disease model for studying oxidative stress response in hematopoietic stem/progenitor cells. Since BM failure and leukemia are rarely found in other known genomic instability syndromes such as ataxia telangiectasia, Nijmegen breakage syndrome, xeroderma pigmentosum, and Werner syndrome, further investigation into the function of FA proteins in oxidative damage response and repair will provide information on whether oxidative stress is a common signal that drives FA disease progression to leukemia. Therefore, understanding the relationship between oxidative stress and FA disease progression provides a unique opportunity to mechanistically comprehend and potentially intervene in these physiologically important processes.
Acknowledgments
The work of the authors is supported by NIH Grants R01 CA109641, R01 HL076712, and a Leukemia and Lymphoma Scholar award. We thank Dr. Keqin Ren for graphic support.
Abbreviations
ASK1, apoptosis signal-regulating kinase 1; AML, acute myeloblastic leukemia; BMF, bone marrow failure; FA, Fanconi anemia; GM-CSF, granulocyte-macrophage colony-stimulating factor; H2O2, hydrogen peroxide; HSC/P, hematopoietic stem cells and progenitors; IFN-γ, interferon-gamma; IL-6, interleukin-6; JNK; c-JUN NH2-terminal kinase; MEFs, murine embryonic fibroblasts; MDS, myelodysplasia; NAC, N-acetyl-L-cysteine; NF-κB, nuclear factor-kappa B; redox, reduction/oxidation reaction; ROS, reactive oxygen species; SCC, squamous cell carcinoma; SODs, superoxide dismutases; TNF-α, Tumor necrosis factor-alpha.
References
- 1.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nature Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 2.Ames BN. Gold LS. Willett WC. The causes and prevention of cancer. Proc Natl Acad Sci USA. 1995;92:5258–5265. doi: 10.1073/pnas.92.12.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagby GC., Jr Genetic basis of Fanconi anemia. Curr Opin Hematol. 2003;10:68–76. doi: 10.1097/00062752-200301000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Balkwill F. Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 5.Bogliolo M. Cabré O. Callén E. Castillo V. Creus A. Marcos R. Surrallés J. The Fanconi anaemia genome stability and tumour suppressor network. Mutagenesis. 2002;17:529–538. doi: 10.1093/mutage/17.6.529. [DOI] [PubMed] [Google Scholar]
- 6.Buchwald M. Moustacchi E. Is Fanconi anemia caused by a defect in the processing of DNA damage? Mutat Res. 1998;408:75–90. doi: 10.1016/s0921-8777(98)00024-x. [DOI] [PubMed] [Google Scholar]
- 7.Chandel NS. Vander Heiden MG. Thompson CB. Schumacker PT. Redox regulation of p53 during hypoxia. Oncogene. 2000;19:3840–3848. doi: 10.1038/sj.onc.1203727. [DOI] [PubMed] [Google Scholar]
- 8.Cohen–Haguenauer O. Pult B. Bauche C. Daniel MI. Casal b. Levy V. Dausset J. Boiron M. Auclair C. Gluckman E. Marty M. In vivo repopulation ability of genetically corrected bone marrow cells from Fanconi anemia patients. Proc Natl Acad Sci USA. 2006;103:2340–2345. doi: 10.1073/pnas.0510613103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins N. Kupfer GM. Molecular pathogenesis of Fanconi anemia. Int J Hemato. 2005;82:176–83. doi: 10.1532/IJH97.05108. [DOI] [PubMed] [Google Scholar]
- 10.Coussens LM. Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cumming RC. Liu JM. Youssoufian H. Buchwald M. Suppression of apoptosis in hematopoietic factor-dependent progenitor cell lines by expression of the FAC gene. Blood. 1996;88:4558–4567. [PubMed] [Google Scholar]
- 12.Cumming RC. Lightfoot J. Beard K. Youssoufian H. O'Brien PJ. Buchwald M. Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat Med. 2001;7:814–820. doi: 10.1038/89937. [DOI] [PubMed] [Google Scholar]
- 13.D'Andrea AD. Grompe M. The Fanconi anaemia/ BRCA pathway. Nat Rev Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 14.de Cremoux P. Gluckman E. Podgorniak MP. Menier C. Thierry D. Calvo F. Socie G. Decreased IL-1 beta and TNF alpha secretion in long-term bone marrow culture supernatant from Fanconi's anaemia patients. Eur J Haematol. 1996;57:202–207. doi: 10.1111/j.1600-0609.1996.tb01364.x. [DOI] [PubMed] [Google Scholar]
- 15.de Winter JP. Waisfisz Q. Rooimans MA. van Berkel CG. Bosnoyan–Collins L. Alon N. Carreau M. Bender O. Demuth I. Schindler D. Pronk JC. Arwert F. Hoehn H. Digweed M. Buchwald M. Joenje H. The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nat Gene. 1998;20:281–283. doi: 10.1038/3093. [DOI] [PubMed] [Google Scholar]
- 16.de Winter JP. Leveille F. van Berkel CG. Rooimans MA. can Der WL. Steltenpool J. Demuth I. Morgan NV. Alon N. Bosnoyan–Collins L. Lightfoot J. Leegwater PA. Waisfisz Q. Komatsu K. Arwert F. Pronk JC. Mathew CG. Digweed M. Buchwald M. Joenje H. Isolation of a cDNA representing the Fanconi anemia complementation Group E gene. Am Hum Gene. 2000;67:1306–1308. doi: 10.1016/s0002-9297(07)62959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Winter JP. Rooimans MA. van Der WL. van Berkel CG. Alon N. Bosnoyan–Collins L. de Groot J. Zhi Y. Waisfisz Q. Pronk JC. Arwert F. Mathew CG. Scheper RJ. Hoatlin ME. Buchwald M. Joenje H. The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nat Gene. 2000;24:15–16. doi: 10.1038/71626. [DOI] [PubMed] [Google Scholar]
- 18.Doneshbod–Skibba G. Martin J. Shahidi N. Myeloid and erythroid colony growth in non-anemic patients with Fanconi's anemia. Br J Haematol. 1980;44:33–38. doi: 10.1111/j.1365-2141.1980.tb01181.x. [DOI] [PubMed] [Google Scholar]
- 19.Dufour C. Corcione A. Svahn J. Haupt R. Poggi V. Beka'ssy AN. Scime R. Pistorio A. Pistoia V. TNF-alpha and IFN-gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood. 2003;102:2053–2059. doi: 10.1182/blood-2003-01-0114. [DOI] [PubMed] [Google Scholar]
- 20.Ekbom A. Helmick C. Zack M. Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323:1228–1233. doi: 10.1056/NEJM199011013231802. [DOI] [PubMed] [Google Scholar]
- 21.Fagerlie SR. Diaz J. Christianson TA. McCartan K. Keeble W. Faulkner GR. Bagby GC. Functional correction of FA-C cells with FANCC suppresses the expression of interferon –inducible genes. Blood. 2001;97:3017–3024. doi: 10.1182/blood.v97.10.3017. [DOI] [PubMed] [Google Scholar]
- 22.Fagerlie S. Lensch MW. Pang Q. Bagby GC., Jr The Fanconi anemia group C gene product: signaling functions in hematopoietic cells. Exp Hematol. 2001;29:1371–1381. doi: 10.1016/s0301-472x(01)00755-x. [DOI] [PubMed] [Google Scholar]
- 23.Fiers W. Beyaert R. Declercq W. Vandenabeele P. More than one way to die: Apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999;18:7719–7730. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- 24.Freie B. Li X. Ciccone SL. Nawa K. Cooper S. Vogelweid C. Schantz L. Haneline LS. Orazi A. Broxmeyer HE. Lee SH. Clapp DW. Fanconi anemia type C and p53 cooperate in apoptosis and tumorigenesis. Blood. 2003;102:4146–4152. doi: 10.1182/blood-2003-03-0971. [DOI] [PubMed] [Google Scholar]
- 25.Freie BW. Ciccone SL. Li X. Plett PA. Orschell CM. Srour EF. Hanenberg H. Schindler D. Lee SH. Clapp DW. A role for the Fanconi anemia C protein in maintaining the DNA damage-induced G2 checkpoint. J Biol Chem. 2004;279:50986–50993. doi: 10.1074/jbc.M407160200. [DOI] [PubMed] [Google Scholar]
- 26.Fridman JS. Lowe SW. Control of apoptosis by p53. Oncogene. 22:9030–9040. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 27.Futaki M. Igarashi T. Watanabe S. Kajigaya S. Tatsuguchi A. Wang J. Liu JM. The FANCG Fanconi anemia protein interacts with CYP2E1: possible role in protection against oxidative DNA damage. Carcinogenesis. 2002;23:67–72. doi: 10.1093/carcin/23.1.67. [DOI] [PubMed] [Google Scholar]
- 28.Giaccia AJ. Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- 29.Gluckman E. Broxmeyer H. Auerbach A. Friedman H. Douglas G. Devergie A. Esperou H. Thierry D. Socie G. Lehn P. Cooper S. English D. Kurtzberg J. Bard J. Boyse E. Hematopoietic reconstitution in a patient with Fanconi's anemia by means of umbilical-cord blood from an HLA-identical sibling. N Engl J Med. 1989;321:1174–1178. doi: 10.1056/NEJM198910263211707. [DOI] [PubMed] [Google Scholar]
- 30.Hadjur S. Ung K. Wadsworth L. Dimmick J. Rajcan–Separovic E. Scott RW. Buchwald M. Jirik FR. Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding Fancc and Cu/Zn superoxide dismutase. Blood. 2001;98:1003–1011. doi: 10.1182/blood.v98.4.1003. [DOI] [PubMed] [Google Scholar]
- 31.Haneline LS. Broxmeyer HE. Cooper S. Hangoc G. Carreau M. Buchwald M. Clapp DW. Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from FAC−/− mice. Blood. 1998;91:4092–4098. [PubMed] [Google Scholar]
- 32.Haneline LS. Gobbett TA. Ramani R. Carreau M. Buchwald M. Yoder MC. Clapp DW Loss of Fancc function results in decreased hematopoietic stem cell repopulating ability. Blood. 1999;94:1–8. [PubMed] [Google Scholar]
- 33.Haneline LS. Li X. Ciccone SL. Hong P. Yang Y. Broxmeyer HE. Lee SH. Orazi A. Srour EF. Clapp DW. Retroviral-mediated expression of recombinant Fancc enhances the repopulating ability of Fancc−/− hematopoietic stem cells and decreases the risk of clonal evolution. Blood. 2003;101:1299–1307. doi: 10.1182/blood-2002-08-2404. [DOI] [PubMed] [Google Scholar]
- 34.Houghtaling S. Granville L. Akkari Y. Torimaru Y. Olson S. Finegold M. Grompe M. Heterozygosity for p53 (Trp53+/−) accelerates epithelial tumor formation in fanconi anemia complementation group D2 (Fancd2) knockout mice. Cancer Res. 2005;65:85–91. [PubMed] [Google Scholar]
- 35.Howlett NG. Taniguchi T. Olson S. Cox B. Waisfisz Q. De Die–Smulders C. Persky N. Grompe M. Joenje H. Pals G. Ikeda H. Fox EA. D'Andrea AD. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 36.Ichijo H. Nishida E. Irie K. ten Dijke P. Saitoh M. Moriguchi T. Takagi M. Matsumoto K. Miyazono K. Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 37.Jin S. Levine AJ. The p53 functional circuit. J Cell Sci. 2001;114:4139–4140. doi: 10.1242/jcs.114.23.4139. [DOI] [PubMed] [Google Scholar]
- 38.Joenje H. Arwert F. Eriksson AW. de Koning H. Oostra AB. Oxygen-dependence of chromosomal aberrations in Fanconi's anaemia. Nature. 1987;290:142–143. doi: 10.1038/290142a0. [DOI] [PubMed] [Google Scholar]
- 39.Joenje H. Levitus M. Waisfisz Q. D’ Andrea, Garcia–Higuera I, Pearson T, van Berkel CG, Rooimans MA, Morgan N, Mathew CG, and Arwert F. Complementation analysis in Fanconi anemia: Assignment of the reference FA-H patient to group A. Am J Hum Gene. 2000;67:759–762. doi: 10.1086/303067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jonkers J. Meuwissen R. van der Gulden H. Peterse H. van der Valk M. Bern A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 41.Kennedy RD. D'Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19:2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 42.Koh PS. Hughes GC. Faulkner GR. Keeble WW. Bagby GC. The Fanconi anemia group C gene product modulates apoptotic responses to tumor necrosis factor- and Fas ligand but does not suppress expression of receptors of the tumor necrosis factor receptor superfamily. Exp Hematol. 1999;27:1–8. doi: 10.1016/s0301-472x(98)00064-2. [DOI] [PubMed] [Google Scholar]
- 43.Kruyt FA. Hoshino T. Liu JM. Joseph P. Jaiswal AK. Youssoufian H. Abnormal microsomal detoxification implicated in Fanconi anemia group C by interaction of the FAC protein with NADPH cytochrome P450 reductase. Blood. 1998;92:3050–3056. [PubMed] [Google Scholar]
- 44.Kuper H. Adami HO. Trichopoulos D. Infections as a major preventable cause of human cancer. J Intern Med. 2000;248:171–183. doi: 10.1046/j.1365-2796.2000.00742.x. [DOI] [PubMed] [Google Scholar]
- 45.Kutler DI. Singh B. Satagopan J. Batish SD. Berwick M. Giampietro PF. Hanenberg H. Auerbach AD. A 20-year perspective on the International Fanconi Anemia Registry (IFAR) Blood. 2003;101:1249–1256. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- 46.Kutler DI. Wreesmann VB. Goberdhan A. Ben-Prat L. Satagopan J. Ngai I. Huvos AG. Giampietro P. Levran O. Pujara K. Diotti R. Carlson D. Huryn LA. Auerbach AD. Singh B. Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst. 2003;95:1718–1721. doi: 10.1093/jnci/djg091. [DOI] [PubMed] [Google Scholar]
- 47.Lensch MW. Rathbun RK. Olson SB. Jones GR. Bagby GC., Jr Selective pressure as an essential force in molecular evolution of myeloid leukemic clones: a view from the window of Fanconi anemia. Leukemia. 1999;13:1784–1789. doi: 10.1038/sj.leu.2401586. [DOI] [PubMed] [Google Scholar]
- 48.Levitus M. Rooimans MA. Steltenpool J. Cool NF. Oostra AB. Mathew CG. Hoatlin ME. Waisfisz Q. Arwert F. De Winter JP. Joenje H. Heterogeneity in Fanconi anemia: evidence for two new genetic subtypes. Blood. 2004;103(7):2498–2503. doi: 10.1182/blood-2003-08-2915. [DOI] [PubMed] [Google Scholar]
- 49.Levran O. Attwooll C. Henry RT. Milton KL. Neveling K. Rio P. Batish SD. Kalb R. Velleuer E. Barral S. Ott J. Petrini J. Schindler D. Hanenberg H. Auerbach AD. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 50.Li J. Sejas DP. Zhang X. Qiu Y. Nattamai KJ. Rani R. Rathbun KR. Geiger H. Williams DA. Bagby GC. Pang Q. TNF-α induces leukemic clonal evolution ex vivo in Fanconi anemia group C stem cells. J Clin Invest. 2007;117:3283–3295. doi: 10.1172/JCI31772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X. Yang Y. Yuan J. Hong P. Freie B. Orazi A. Haneline LS. Clapp DW. Continuous in vivo infusion of interferon-gamma (IFN-gamma) preferentially reduces myeloid progenitor numbers and enhances engraftment of syngeneic wild-type cells in Fancc−/− mice. Blood. 2004;104:1204–1209. doi: 10.1182/blood-2004-03-1094. [DOI] [PubMed] [Google Scholar]
- 52.Li Y. Youssoufian H. MxA overexpression reveals a common genetic link in four Fanconi anemia complementation groups. J Clin Invest. 1997;100:2873–2880. doi: 10.1172/JCI119836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu J. Fanconi's anemia. In: Young NS, editor. Bone Marrow Failure Syndromes. Philadelphia, PA; WB Saunders: 2000. pp. 47–68. [Google Scholar]
- 54.Liu TX. Howlett NG. Deng M. Langenau DM. Hsu K. Rhodes J. Kanki JP. D'Andrea AD. Look AT. Knockdown of zebrafish Fancd2 causes developmental abnormalities via p53-dependent apoptosis. Dev Cell. 2003;5:903–914. doi: 10.1016/s1534-5807(03)00339-3. [DOI] [PubMed] [Google Scholar]
- 55.Lohrum MA. Vousden KH. Regulation and activation of p53 and its family members. Cell Death Differ. 1999;6:1162–1168. doi: 10.1038/sj.cdd.4400625. [DOI] [PubMed] [Google Scholar]
- 56.Lo Ten Foe JR. Rooimans MA. Bosnoyan–Collins L. Alon N. Wijker M. Parker L. Lightfoot J. Carreau M. Callen DF. Savoia A. Cheng NC. van Berkel CG. Strunk MH. Gille JJ. Pals G. Kruyt FA. Pronk JC. Arwert F. Buchwald M. Joenje H. Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nature Gene. 1996;14:320–323. doi: 10.1038/ng1196-320. [DOI] [PubMed] [Google Scholar]
- 57.Lowy DR. Gillison ML. A new link between Fanconi anemia and human papillomavirus-associated malignancies. J Natl Cancer Inst. 2003;95:1648–1650. doi: 10.1093/jnci/djg125. [DOI] [PubMed] [Google Scholar]
- 58.Luna–Fineman S. Shannon KM. Lange BJ. Childhood monosomy 7: epidemiology, biology, and mechanistic implications. Blood. 1995;85:1985–1989. [PubMed] [Google Scholar]
- 59.Macciò A. Lai P. Santona MC. Pagliara L. Melis GB. Mantovani G. High serum levels of soluble IL-2 receptor, cytokines, and C-reactive protein correlate with impairment of T cell response in patients with advanced epithelial ovarian cancer. Gynecol Oncol. 1998;69:248–252. doi: 10.1006/gyno.1998.4974. [DOI] [PubMed] [Google Scholar]
- 60.Macdougall IC. Cooper AC. Erythropoitin resistence: the role of inflammation and pro-inflammatory cytokines. Nephrol Dial Transplant. 2002;17:39–43. doi: 10.1093/ndt/17.suppl_11.39. [DOI] [PubMed] [Google Scholar]
- 61.Maciejewski JP. Selleri C. Sato T. Anderson S. Young NS. Increased expression of Fas antigen on bone marrow CD34+ cells of patients with aplastic anaemia. Br J Haematol. 1995;91:245–252. doi: 10.1111/j.1365-2141.1995.tb05277.x. [DOI] [PubMed] [Google Scholar]
- 62.Mackay IR. Rose NR. Autoimmunity and lymphoma: tribulations of B cells. Nat Immunol. 2001;2:793–795. doi: 10.1038/ni0901-793. [DOI] [PubMed] [Google Scholar]
- 63.Mantovani G. Macciò A. Pisano M. Versace R. Lai P. Esu S. Massa E. Ghiani M. Dessi D. Melis GB. Del Giacco DS. Tumor-associated lympho-monocytes from neoplastic effusions are immunologically defective in comparison with patient autologous PBMCs but are capable of releasing high amounts of various cytokines. Int J Cancer. 1997;71:724–731. doi: 10.1002/(sici)1097-0215(19970529)71:5<724::aid-ijc6>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 64.Mantovani G. Macciò A. Madeddu C. Mura L. Gramigano G. Lusso MR. Mulas C. Mudu MC. Murgia V. Camboni P. Massa E. Ferreli L. Contu P. Rinaldi A. Sanjust E. Atzei D. Elsener B. Quantitative evaluation of oxidative stress, chronic inflammatory indices and leptin in cancer patients: correlation with stage and performance status. Int J Cancer. 2002;98:84–91. doi: 10.1002/ijc.10143. [DOI] [PubMed] [Google Scholar]
- 65.Marx J. Cancer research. Inflammation and cancer: the link grows stronger. Science. 2004;306:966–968. doi: 10.1126/science.306.5698.966. [DOI] [PubMed] [Google Scholar]
- 66.Meetei AR. de Winter JP. Medhurst AL. Wallisch M. Waisfisz Q. van de Vrugt HJ. Oostra AB. Yan Z. Ling C. Bishop CE. Hoatlin ME. Joenje H. Wang W. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003;35:165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 67.Meetei AR. Levitus M. Xue Y. Medhurst AL. Zwaan M. Ling C. Rooimans MA. Bier P. Hoatlin M. Pals G. de Winter JP. Wang W. Joenje H. X-linked inheritance of Fanconi anemia complementation group B. Nat Gene. 2004;36:1219–1224. doi: 10.1038/ng1458. [DOI] [PubMed] [Google Scholar]
- 68.Meetei AR. Medhurst AL. Ling C. Xue Y. Singh TR. Bier P. Steltenpool J. Stone S. Dokal I. Mathew CG. Hoatlin M. Joenje H. de Winter JP. Wang W. A human ortholog of archael DNA repair protein HEF is defective in. Fanconi anemia complementation group M Nat Genet. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mukhopadhyay SS. Leung KS. Hicks MJ. Hastings PJ. Youssoufian H. Plon SE. Defective mitochondrial per-oxiredoxin-3 results in sensitivity to oxidative stress in. Fanconi anemia J Cell Biol. 2006;175:225–235. doi: 10.1083/jcb.200607061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakamura H. Nakamura K. Yodoi J. Redox regulation of cellular activation. Annu Rev Immunol. 1997;15:351–369. doi: 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- 71.Nakata S. Matsumura I. Tanaka H. Ezoe S. Satoh Y. Ishikawa J. Era T. Kanakura Y. NF-kappa B family proteins participate in multiple steps of hematopoiesis through elimination of reactive oxygen species. J Biol Chem. 2004;279:55578–55586. doi: 10.1074/jbc.M408238200. [DOI] [PubMed] [Google Scholar]
- 72.Otsuki T. Nagakura S. Wang J. Bloom M. Grompe M. Liu JM. Tumor necrosis factor- and CD95 ligation suppress erythropoiesis in Fanconi anemia C gene knockout mice. J Cell Physiol. 1999;179:79–86. doi: 10.1002/(SICI)1097-4652(199904)179:1<79::AID-JCP10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 73.Pagano G. Degan P. d'Ischia M. Kelly F J. Nobili B. Pallardó F V. Youssoufian H. Zatterale A. Oxidative stress as a multiple effector in Fanconi anaemia clinical phenotype. Eur J Haematol. 2005;75:93–100. doi: 10.1111/j.1600-0609.2005.00507.x. [DOI] [PubMed] [Google Scholar]
- 74.Pang Q. Keeble W. Christianson TA. Faulkner GR. Bagby GC. FANCC interacts with Hsp70 to protect hematopoietic cells from IFN-γ/TNF-α-mediated cytotoxicity. EMBO J. 2001;20:4478–4489. doi: 10.1093/emboj/20.16.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pang Q. Keeble W. Diaz J. Christianson TA. Fagerlie S. Rathbun RK. Faulkner G R. O'Dwyer M. Bagby GC. The role of double-stranded RNA dependent protein kinase (PKR) in mediating hypersensitivity of Fanconi anemia complementation group C cells to interferon-γ, tumor necrosis factor-α, and double stranded RNA. Blood. 2001;97:1644–1652. doi: 10.1182/blood.v97.6.1644. [DOI] [PubMed] [Google Scholar]
- 76.Pang Q. Christianson TA. Keeble W. Koretsky T. Bagby GC. The anti-apoptotic function of Hsp70 in the interferon-inducible double-stranded RNA-dependent protein kinase-mediated death signaling pathway requires the Fanconi anemia protein, FANCC. J Biol Chem. 2002;277:49638–49643. doi: 10.1074/jbc.M209386200. [DOI] [PubMed] [Google Scholar]
- 77.Park SJ. Ciccone SL. Beck BD. Hwang B. Freie B. Clapp DW. Lee SH. Oxidative stress/damage induces multimerization and interaction of Fanconi anemia proteins. J Biol Chem. 2004;279:30053–30059. doi: 10.1074/jbc.M403527200. [DOI] [PubMed] [Google Scholar]
- 78.Rathbun R K. Faulkner G R. Ostroski M H. Christianson TA. Hughes G. Jones G. Cahn R. Maziarz R. Royle G. Keeble W. Heinrich MC. Grompe M. Tower PA. Bagby GC. Inactivation of the Fanconi anemia group C gene augments interferon-gamma-induced apoptotic responses in hematopoietic cells. Blood. 1997;90:974–985. [PubMed] [Google Scholar]
- 79.Rathbun RK. Christianson TA. Faulkner GR. Jone G. Keeble W. O'Dwyer M. Bagby GC. Interferon—induced apoptotic responses of Fanconi anemia group C hematopoietic progenitor cells involve caspase 8-dependent activation of caspase 3 family members. Blood. 2000;96:4204–4211. [PubMed] [Google Scholar]
- 80.Reid S. Schindler D. Hanenberg H. Barker K. Hanks S. Kalb R. Neveling K. Kelly P. Seal S. Freund M. Wurm M. Batish SD. Lach FP. Yetgin S. Neitzel H. Ariffin H. Tischkowitz M. Mathew CG. Auerbach AD. Rahman N. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2006;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 81.Rosselli F. Sanceau J. Wietzerbin J. Moustacchi E. Abnormal lymphokine production: a novel feature of the genetic disease Fanconi anemia. I. Involvement of interleukin-6. Hum Genet. 1992;89:42–48. doi: 10.1007/BF00207040. [DOI] [PubMed] [Google Scholar]
- 82.Rosselli F. Sanceau J. Gluckman E. Wietzerbin J. Moustacchi E. Abnormal lymphokine production: a novel feature of the genetic disease Fanconi anemia. II. In vitro and in vivo spontaneous overproduction of tumor necrosis factor alpha. Blood. 1994;83:1216–1225. [PubMed] [Google Scholar]
- 83.Rubin CM. Arthur DC. Woods WG. Lange BJ. Nowell PC. Rowley JD. Nachman J. Bostrom B. Baum ES. Suarez CR. Shah NR. Morgan E. Mauer HS. McKenzie SE. Larson RA. Le Beau MM. Therapy-related myelodysplastic syndrome and acute myeloid leukemia in children: correlation between chromosomal abnormalities and prior therapy. Blood. 1991;78:2982–2988. [PubMed] [Google Scholar]
- 84.Saadatzadeh MR. Bijangi–Vishehsaraei K. Hong P. Bergmann H. Haneline LS. Oxidant hypersensitivity of Fanconi anemia type C-deficient cells is dependent on a redox-regulated apoptotic pathway. J Biol Chem. 2004;279:16805–16812. doi: 10.1074/jbc.M313721200. [DOI] [PubMed] [Google Scholar]
- 85.Schindler D. Hoehn H. Fanconi anemia mutation causes cellular susceptibility to ambient oxygen. Am J Hum Genet. 1988;43(4):429–435. [PMC free article] [PubMed] [Google Scholar]
- 86.Schultz JC. Shahidi NT. Tumor necrosis factor-alpha overproduction in Fanconi's anemia. Am J Hematol. 1993;42:196–201. doi: 10.1002/ajh.2830420211. [DOI] [PubMed] [Google Scholar]
- 87.Sejas DP. Rani R. Qiu Y. Zhang X. Fagerlie SR. Nakano H. Williams DA. Pang Q. Inflammatory reactive oxygen species-mediated hematopoietic suppression in Fancc-deficient mice. J Immunol. 2007;178:5277–5287. doi: 10.4049/jimmunol.178.8.5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Si Y. Ciccone S. Yang FC. Yuan J. Zeng D. Chen S. van de Vrugt H. Critser J. Arwert F. Haneline LS. Clapp DW. Continuous in vivo infusion of interferon-gamma (IFN-gamma) enhances engraftment of syngeneic wild-type cells in Fanca−/− and Fancg−/− mice. Blood. 2006;108:4283–4287. doi: 10.1182/blood-2006-03-007997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smogorzewska A. Matsuoka S. Vinciguerra P. McDonald ER., 3rd Hurov KE. Luo J. Ballif BA. Gygi SP. Hofmann K. D'Andrea AD. Elledge SJ. Identification of the FANCI Protein, a Monoubiquitinated FANCD2 Paralog Required for DNA Repair. Cell. 2007;129:1–13. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stark R. Andre C. Thierry D. Cherel M. Galibert F. Gluckman E. The expression of cytokine and cytokine receptor genes in long-term bone marrow culture in congenital and acquired bone marrow hypoplasias. Br J Haematol. 1993;83:560–566. doi: 10.1111/j.1365-2141.1993.tb04691.x. [DOI] [PubMed] [Google Scholar]
- 91.Strathdee CA. Gavish H. Shannon WR. Buchwald M. Cloning of cDNAs for Fanconi's anaemia by functional complementation. Nature. 1992;356:763–767. doi: 10.1038/356763a0. [DOI] [PubMed] [Google Scholar]
- 92.Suematsu N. Tsutsui H. Wen J. Kang D. Ikeuchi M. Ide T. Hayashidani S. Shiomi T. Kubota T. Hamasaki N. Takeshita A. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 18; 2003;107:1418–1423. doi: 10.1161/01.cir.0000055318.09997.1f. [DOI] [PubMed] [Google Scholar]
- 93.Tak PP. Zvaifler NJ. Rheumatoid arthritis, p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today. 2000;21:78–82. doi: 10.1016/s0167-5699(99)01552-2. [DOI] [PubMed] [Google Scholar]
- 94.Timmers C. Taniguchi T. Hejna J. Reifsteck C. Locas L. Bruun D. Thayer M. Cox B. Olson S. D'Andrea AD. Moses R. Grompe M. Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol Cell. 2001;7:241–248. doi: 10.1016/s1097-2765(01)00172-1. [DOI] [PubMed] [Google Scholar]
- 95.Tischkowitz MD. Hodgson SV. Fanconi anaemia. J Med Genet. 2003;40:1–10. doi: 10.1136/jmg.40.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tischkowitz M. Dokal I. Fanconi anaemia and leukaemia—clinical and molecular aspects. Br J Haematol. 2004;126(2):176–191. doi: 10.1111/j.1365-2141.2004.05023.x. [DOI] [PubMed] [Google Scholar]
- 97.Umeda T. Hino O. Molecular aspects of human hepatocarcinogenesis mediated by inflammation: from hyper-carcinogenic state to normo- or hypocarcinogenic state. Oncology. 2002;62:38–42. doi: 10.1159/000048274. [DOI] [PubMed] [Google Scholar]
- 98.Ventura JJ. Cogswell P. Flavell RA. Baldwin AS Jr. Davis RJ. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004;18:2905–2915. doi: 10.1101/gad.1223004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wajant H. Pfizenmaier K. Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 100.Walsh CE. Nienhuis AW. Samulski RJ. Brown MG. Miller JL. Young NS. Liu JM. Phenotypic correction of Fanconi anemia in human hematopoietic cells with a recombinant adeno-associated virus vector. J Clin Invest. 1994;94:1440–1448. doi: 10.1172/JCI117481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang J. Otsuki T. Youssoufian H. Foe JL. Kim S. Devetten M. Yu J. Li Y. Dunn D. Liu JM. Overexpression of the Fanconi anemia group C gene (FAC) protects hematopoietic progenitors from death induced by Fas-mediated apoptosis. Cancer Res. 1998;58:3538–3541. [PubMed] [Google Scholar]
- 102.West RR. Stafford DA, White AD, Bowen DT, and Padua RA. Cytogenetic abnormalities in the myelodysplastic syndromes and occupational or environmental exposure. Blood. 2000;95:2093–2097. [PubMed] [Google Scholar]
- 103.Whitney MA. Royle G. Low MJ. Kelly MA. Axthelm MK. Reifsteck C. Olsen S. Braun RE. Heinrich MC. Rathbun RK. Bagby GC. Grompe M. Germ cell defects and hematopoietic hypersensitivity to -interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996;88:49–58. [PubMed] [Google Scholar]
- 104.Xia B. Dorsman JC. Ameziane N. de Vries Y. Rooimans MA. Sheng Q. Pals G. rrami A. Gluckman E. Llera J. Wang W. Livingston DM. Joenje H. de Winter JP. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2006;39:159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 105.Zhang QS. Eaton L. Snyder ER. Houghtaling S. Mitchell JB. Finegold M. Van Waes C. Grompe M. Tempol protects against oxidative damage and delays epithelial tumor onset in Fanconi anemia mice. Cancer Res. 2008;68:1601–1608. doi: 10.1158/0008-5472.CAN-07-5186. [DOI] [PubMed] [Google Scholar]
- 106.Zhang X. Li J. Sejas DP. Pang Q. Hypoxia-reoxygenation induces premature senescence in FA bone marrow hematopoietic cells. Blood. 2005;106:75–85. doi: 10.1182/blood-2004-08-3033. [DOI] [PubMed] [Google Scholar]
- 107.Zhang X. Li J. Sejas DP. Pang Q. The ATM/p53/p21 pathway influences cell fate decision between apoptosis and senescence in reoxygenated hematopoietic progenitor cells. J Biol Chem. 2050;280:19635–19640. doi: 10.1074/jbc.M502262200. [DOI] [PubMed] [Google Scholar]
- 108.Zhang X. Sejas DP. Qiu Y. Williams DA. Pang Q. Inflammatory ROS promote and cooperate with Fanconi anemia mutation for hematopoietic senescence. J Cell Science. 2007;120:1572–1583. doi: 10.1242/jcs.003152. [DOI] [PMC free article] [PubMed] [Google Scholar]