

Abstract
Mutant connexins have been linked to hereditary congenital cataracts. One such mutant causes a proline-to-serine substitution at position 88 in human connexin 50 (CX50P88S). In transfected cells, CX50P88S does not form gap junctions, but localizes in cytoplasmic multilamellar structures. We studied the dynamics of formation and the stability of these structures in HeLa cells stably transfected with CX50P88S containing a tetracysteine motif appended to its C-terminus (HeLa-CX50P88S(Cys)4 cells). The tetracysteine motif binds the membrane-permeable biarsenical compounds, FlAsH and ReAsH, which become fluorescent upon binding allowing detection of CX50P88S(Cys)4 by fluorescence microscopy or by transmission electron microscopy after the ReAsH-driven fluorescent photoconversion of diaminobenzidine. CX50P88S structures were long-lived. Pulse labeling of HeLa-CX50P88S(Cys)4 cells with FlAsH followed by a chase and ReAsH labeling showed a differential distribution of the labels, with older CX50P88S surrounded by newly synthesized protein. Formation of CX50P88S accumulations was not affected by treatments that block ER-to-Golgi transport. Transmission electron microscopy and tomographic reconstruction revealed that CX50P88S accumulations corresponded to closely apposed circular or semicircular membrane stacks that were sometimes continuous with the rough endoplasmic reticulum. These results suggest that CX50P88S accumulations originate from the rough endoplasmic reticulum and that mutant protein is sequentially added resulting in long-lived cytoplasmic particles. The persistence of these particles in the lens may cause light scattering and the pulverulent cataracts observed in affected individuals.
Keywords: cataract, connexin, gap junction, tetracysteine, correlative microscopy
1. Introduction
The lens is an avascular organ that depends on gap junction mediated intercellular communication to maintain homeostasis and function (Goodenough, 1979). Gap junction channels are oligomeric assemblies of proteins called connexins (CX) and allow passage of ions and molecules of up to 1000 Da between adjacent cells. CX46 and CX50 are the most abundant connexins in fiber cells (Kistler et al., 1985; Paul et al., 1991; White et al., 1992). Similar to other integral membrane proteins, connexins are synthesized in the rough endoplasmic reticulum and follow the secretory pathway to the plasma membrane. Unlike many other integral membrane proteins, connexins have relatively short half-lives (reviewed in Berthoud et al., 2004), except in the lens in vivo where like other lens proteins they are expected to be long-lived. Both the ubiquitin/proteasomal and the lysosomal pathways have been implicated in connexin turnover (reviewed in Berthoud et al., 2004). Some disease-associated connexin mutants exhibit altered degradation (reviewed in Sáez et al., 2003).
The pathogenesis of cataracts (loss of lens transparency) has been associated with damage to various lens proteins and abnormalities of connexin-based intercellular communication. Inherited congenital cataracts have been mapped to several genes including those encoding CX46 and CX50, GJA3 and GJA8 (Reddy et al., 2004). One of these mutants results in a proline-to-serine substitution at position 88 in human connexin50 (CX50P88S) (Shiels et al., 1998). The proline at this position is conserved in all connexins suggesting its importance for connexin function, a hypothesis that has been substantiated for several connexins. In CX26, mutation of this proline to a leucine or a glycine leads to a marked decrease in conductance and reversal in the voltage-gating response when paired with wild type CX26 or CX32 (Suchyna et al., 1993). In CX32, mutagenesis experiments suggest that this proline acts as a flexible hinge that participates in voltage-dependent gating (Ri et al., 1999). In CX50, mutation of proline 88 to a serine (CX50P88S) or glutamine (CX50P88Q) apparently induces a conformational change that affects protein trafficking, because neither of these cataract-associated CX50 mutants traffics properly to the plasma membrane when expressed in connexin-deficient cells (Berthoud et al., 2003; Arora et al., 2006). Moreover, CX50P88S and to some extent CX50P88Q accumulate in the cytoplasm suggesting impaired degradation.
While cataract formation due to connexin mutations may occur due to impaired intercellular communication, formation of mutant protein aggregates may also contribute to the phenotype. The CX50P88S cytoplasmic accumulations are multilamellar structures (Berthoud et al., 2003). Such multilamellar structures may act as light scattering particles (Gilliland et al., 2001). Light scattering particles can be detrimental to lens function by interfering with the passage of light and its focusing onto the retina. Pathologically consequential particles should not only form, but also be stable.
To gain further insight into the formation and nature of these CX50P88S multilamellar structures, we implemented a strategy that allowed us to perform pulse-chase experiments to visualize the formation and stability of the multilamellar structures and to elucidate the ultrastructural relationship of these accumulations to different intracellular compartments.
2. Materials and methods
2.1. Chemicals
All chemicals were obtained from Sigma Chemical Co. (St. Louis, MO, USA) unless otherwise specified.
2.2. DNA constructs
A DNA construct coding for human CX50P88S with a tetracysteine motif (AEAAAREACCPGCCARA) appended to its carboxyl terminus (CX50P88S(Cys)4) was obtained by PCR using the LATAKARA kit (Invitrogen, Carlsbad, CA, USA). The PCR product was fully sequenced at the Cancer Research Center Sequencing Facility of the University of Chicago to insure that the PCR reaction did not introduce unwanted mutations. Then, the DNA construct was subcloned into pSFFV-neo (Rup et al., 1993).
2.3. Cell culture
HeLa cells were stably transfected with CX50P88S(Cys)4 (HeLa-CX50P88S(Cys)4) and grown in MEM supplemented with nonessential amino acids, 10% fetal bovine serum (FBS), 2 mM glutamine, 10 units/ml penicillin G and 10 μg/ml streptomycin sulfate supplemented with 1 mg/ml Geneticin (Invitrogen).
2.4. Immunofluorescence
Cells were grown on coverslips and allowed to reach 80-90% confluence. Cells were rinsed with phosphate buffered saline pH 7.4 (PBS), fixed in 4% paraformaldehyde in PBS for 15 min, and permeabilized in 1% Triton X-100 in PBS for 15 min. Cells were subsequently incubated in 4% normal goat serum,1% Triton X-100 in PBS for 10 min at room temperature and then in rabbit polyclonal anti-CX50 antibodies (Berthoud et al., 2003)overnightat4 °C. Cells were rinsed six times with PBS and then incubated in Cy3- or Cy2-conjugated anti-rabbit IgG antibodies (Jackson ImmunoResearch, West Grove, PA, USA) at room temperature. After 45 min, cells were rinsed 6 times with PBS, and coverslips were mounted with 2% n-propylgallate in PBS:glycerol (1:1).
Confocal images were obtained using a Leica SP2 AOBS (Leica Microsystems Inc., Bannockburn, IL, USA) laser scanning confocal system and a 63 X (n.a.: 1.4) oil objective using settings corresponding to the excitation and emission wavelengths of the fluorescent labels. Images were collected by sequential scanning using single laser-line excitation to eliminate the possibility of fluorescence bleeding from one channel into the other. Images illustrating the overlap of the fluorescent signals were generated using Image J software (http://rsb.info.nih.gov/ij/). Composite figures were assembled using Adobe Photoshop software (Adobe Systems, San Jose, CA).
2.5. Cell labeling
Labeling of cells with fluorescein-based arsenical hairpin binder (FlAsH) or resorufin-based arsenical hairpin binder (ReAsH) was performed as previously described (Gaietta et al., 2002). HeLa cells expressing CX50P88S(Cys)4 grown on coverslips were rinsed three times with Hank's balanced salt solution (HBSS; Invitrogen) and then incubated at 37 °C for 1 h with 0.5 μM FlAsH or 2 h with 0.25 μM ReAsH in HBSS containing 12.5 μM 1,2-ethanedithiol (EDT2). Then, cells were rinsed with HBSS containing 250 or 750 μM EDT2 (for ReAsH- or FlAsH-labeled cells, respectively), followed by rinses with HBSS alone. Cells labeled with FlAsH or ReAsH were then fixed with 4% paraformaldehyde, rinsed in PBS and coverslips mounted using 2% n-propyl gallate in PBS:glycerol (1:1). Specimens were analyzed by confocal microscopy.
For the pulse-chase experiments analyzing the stability of the CX50P88S accumulations, HeLa-CX50P88S(Cys)4 cells were pulse-labeled with FlAsH, returned to normal growth media and fixed at different times of chase. Cells were then permeabilized and stained with DAPI to visualize the nuclei. Specimens were studied with a Zeiss Plan Apochromat 40X objective (n.a., 1.0) in an Axioplan 2 microscope (Carl Zeiss, München, Germany) equipped with a mercury lamp and images were captured with an Axiocam digital camera (Carl Zeiss) using Zeiss Axiovision software. The number of FlAsH-labeled structures and the number of cells per visual field were quantified by counting FlAsH-labeled accumulations and DAPI-stained nuclei in each field using Image J software for analysis of particles. A minimum of ten fields were counted for each time point.
For the pulse-chase experiments studying the formation of CX50P88S accumulations, cells were first labeled with FlAsH (or ReAsH), returned to normal culture medium for different time intervals and then labeled with ReAsH (or FlAsH). As a control, de novo protein synthesis was blocked by cycloheximide treatment; HeLa-CX50P88S(Cys)4 cells were pulse-labeled with FlAsH (or ReAsH) and treated with 10 μg/ml cycloheximide throughout the time of chase. After 2-4h, cells were labeled with ReAsH (or FlAsH) in HBSS containing 12.5 μM EDT2 and 10 μg/ml cycloheximide. After labeling with the second biarsenical compound, cells were fixed and analyzed by confocal microscopy. Three-dimensional reconstruction after deconvolution of Z-series was computed using the non-blind deconvolution routine within the Leica LAS-AS software package or a measured point spread function with the Image J software. Composite figures were assembled using Adobe Photoshop software (Adobe Systems).
2.6. Inhibition of endoplasmic reticulum-to-Golgi transport
2.6.1. Reduced temperature
After pulse labeling HeLa-CX50P88S(Cys)4 cells with FlAsH, cells were incubated for 24 h either at 15 °C or 37 °C followed by labeling with ReAsH. After ReAsH labeling, cells were fixed and analyzed by confocal microscopy as described above.
2.6.2. Treatment with N-[2-((p-Bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide (H89)
HeLa-CX50P88S(Cys)4 cells were labeled with FlAsH, incubated in the presence or absence of N-[2-((p-Bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide (H89; Calbiochem, San Diego, CA) for 4 h and then labeled with ReAsH. After ReAsH labeling, cells were fixed and analyzed by confocal microscopy as described above.
2.7. ReAsH-driven photoconversion of diaminobenzidine and transmission electron microscopy
ReAsH-labeled cells were fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate, pH 7.4 at 4 °C. After 20 min, the cells were rinsed in 0.1 M sodium cacodylate pH 7.4 and incubated for 30-45 min with blocking buffer (10 mM KCN, 10 mM aminotriazole, 0.01% hydrogen peroxide, 50 mM glycine in 0.1 M sodium cacodylate pH 7.4). This buffer was then replaced with 1 mg/ml diaminobenzidine (DAB) in blocking buffer, and photoconversion was performed using intense illumination (75 W xenon arc lamp without neutral density filters) focused through the microscope objective. Rinsing and further preparation of the sample for transmission electron microscopy were performed as described in Gaietta et al. (2002). Several 80-nm thin sections were obtained and examined on a JEOL 1200 electron microscope (JEOL USA, Peabody, MA) operated at 80 kV.
2.8. Electron tomographic data acquisition, reconstruction and analysis
Fiducial markers of 20 nm colloidal gold for post acquisition alignment were applied to both sides of the thick sections prior to EM imaging. Two tilt series (1 single-axis tilt, 1 double-axis tilt) of ReAsH photoconverted CX50P88S accumulations were recorded with a JEOL 4000EX intermediate-voltage electron microscope (JEOL USA) operated at 400 kV. Following standard procedures for plastic embedded specimens as detailed recently in Perkins et al. (2008), the sections were irradiated before initiating a tilt series in order to limit anisotropic specimen thinning during image collection. Individual images were recorded on a Spectral Instruments 1100 series 16 megapixel CCD camera (Spectral Instruments, Tucson, AZ) at a nominal microscope magnification of 15,000X using angular increments of 2° from -60° to +60° about an axis perpendicular to the axis of the microscope. For the double-tilt series, the grid was subsequently rotated 90° after the first tilt series was acquired and a second tilt series was collected with the same angular increment and range. The IMOD package (Kremer et al., 1996) was used for rough alignment with the fine alignment and reconstruction performed using the TxBR package (Lawrence et al., 2006). In both cases, the sampling size was 0.89 nm/pixel. The calculated section thicknesses for these two volumes were ~0.12 μM (single tilt tomogram) and ~0.25 μM (double tilt tomogram). Segmentation was performed using the IMOD package and surface and volume renderings for visualization and animations were performed with the Amira package (Mercury/TGS, San Diego, CA). Original tilt images, tomographic reconstructions, animations and segmentations have been deposited in the Cell Centered Database (Martone et al., 2008; CCDB; http://ccdb.ucsd.edu/CCDBWebSite/index.html) and can be found under Project Number P2020. Individual data sets, volumes and segmented models can be accessed by microscopy product codes MP6347 (single tilt data set) and MP6348 (double tilt data set).
3. Results
3.1. Validation of biarsenical labeling for the study of CX50P88S
The biarsenical compounds FlAsH and ReAsH bind to tetracysteine-tagged proteins. The validity of this approach to detect tetracysteine-tagged CX50P88S was tested by determining the relationship between structures recognized by the anti-CX50 antibodies and those recognized by FlAsH. HeLa cells stably transfected with CX50P88S(Cys)4 were labeled with FlAsH and then subjected to immunofluorescence using anti-CX50 antibodies (Fig. 1). FlAsH labeled one to several cytoplasmic structures per cell that had a diameter of 0.4-2.9 μM. Most, if not all, accumulations analyzed by confocal microscopy contained a lumen that was readily apparent depending on magnification and the “z” level (height) and orientation of confocal sectioning relative to the accumulation. Confocal microscopy analysis of these cells revealed that the same intracellular structures were recognized by the anti-CX50 antibodies (Fig. 1B) and FlAsH (Fig. 1A). In the merged images, near perfect overlap between the two signals was observed (Fig. 1C). Dual labeling experiments performed with ReAsH and anti-CX50 antibodies yielded similar results (data not shown). These results demonstrated the specificity of FlAsH/ReAsH to label CX50P88S(Cys)4.
Fig. 1.
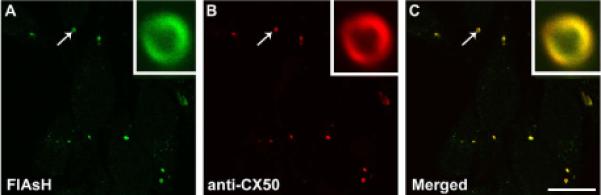
CX50P88S(Cys)4 accumulations are detected by biarsenical reagents and anti-CX50 antibodies. Confocal images of HeLa-CX50P88S(Cys)4 cells that were labeled with FlAsH and then subjected to immunofluorescence using rabbit polyclonal anti-CX50 antibodies and Cy3-conjugated goat anti-rabbit IgG antibodies. The same structures labeled by FlAsH (A) were also recognized by anti-CX50 antibodies (B) as shown by the overlap of the two labels in the merged image (C). The insets represent a magnified view of the structure indicated by the arrow. Scale bar, 22 μm.
The biarsenical-labeled accumulations were similar in size and shape to those formed by untagged CX50P88S when expressed by itself or in combination with wild type CX50 in HeLa cells (Berthoud et al., 2003) implying that the tetracysteine tag did not alter the trafficking or aggregation behavior of the mutant protein. This was further confirmed after transient transfection of CX50P88S(Cys)4 into HeLa cells stably expressing untagged CX50P88S. In these cells, the FlAsH-label co-localized with some of the pre-existing anti-CX50 immunoreactive accumulations (data not shown). Taken together, these results validate the use of FlAsH/ReAsH to study the stability and mechanism of formation of the CX50P88S accumulations.
3.2. CX50P88S accumulations are very stable
To determine the stability of the intracellular structures, HeLa-CX50P88S(Cys)4 cells were pulse-labeled with FlAsH for 1 h, followed by rinsing and incubation in label-free medium, and the persistence of FlAsH-labeled structures was examined by fluorescence microscopy after different times of chase (Fig. 2). Many FlAsH-labeled accumulations were observed immediately after pulse labeling (zero time of chase; Fig. 2A). The number of accumulations per visual field gradually decreased with increasing times of chase (Fig. 2B-D). Quantification of these data is shown in Fig. 2E. Analysis of the data revealed that by 120 h of chase (the longest time of chase studied), the number of accumulations decreased to about 25% of the initial value. Assuming that the fluorescence intensity of the FlAsH-labeled structures did not decrease over time, it was possible to calculate an apparent half-life of 67 h for CX50P88S(Cys)4 bound to FlAsH.
Fig. 2.
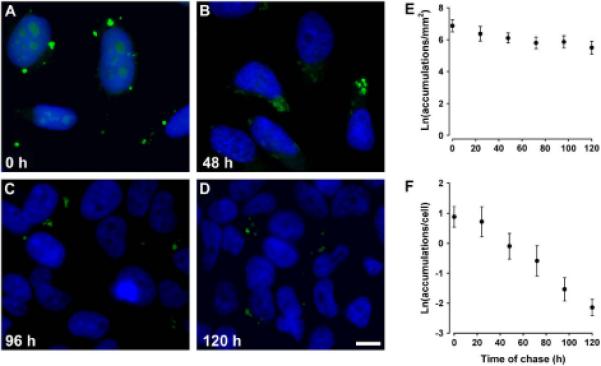
CX50P88S accumulations are long-lived. (A-D) Photomicrographs of HeLa-CX50P88S(Cys)4 cells that were pulse-labeled with FlAsH, chased in normal growth medium for 0 (A), 48 (B), 96 (C) or 120 (D) h, fixed, and stained with DAPI to visualize cell nuclei. (E, F) Semi-logarithmic graphs show the quantification of the number of FlAsH-labeled CX50P88S accumulations per mm2 (E) or per number of cells (F) as a function of the time of chase in hours. Scale bar, 20 μm.
We also quantified the number of accumulations per cell. On average, 3.6 FlAsH-labeled structures per cell were detected after the pulse (zero time of chase). After 24h, this number had decreased to 3.1, and continued decreasing to reach an average value of 2.2 by 120 h (plotted as natural logarithm in Fig. 2F). This decrease is likely due to both the increase in cell number and the decrease in the number of FlAsH-labeled accumulations at longer chase times. These results demonstrated that the accumulations were long-lived and that long chase periods could be used to study their formation.
3.3. Mechanism of formation of CX50P88S accumulations
To study the mechanism of formation of CX50P88S accumulations, HeLa-CX50P88S(Cys)4 cells were sequentially labeled first with FlAsH and then with ReAsH to identify pre-existing and newly synthesized proteins, respectively. At all time points studied (from 4 to 96 h), many of the FlAsH-labeled accumulations were also labeled with ReAsH and appeared yellow in the merged images (shown for 96 h in Fig. 3B-D, arrow). However, after deconvolution of single accumulations, limited intermingling between the FlAsH-and ReAsH-labeled materials was observed. Each structure showed an unlabeled center encircled by an internal layer labeled with FlAsH and then an external layer labeled with ReAsH (Fig. 3E-H). When the sequence of labeling with the biarsenical reagents was reversed, the internal layer was demarcated by ReAsH while the external layer was labeled by FlAsH (data not shown).
Fig. 3.
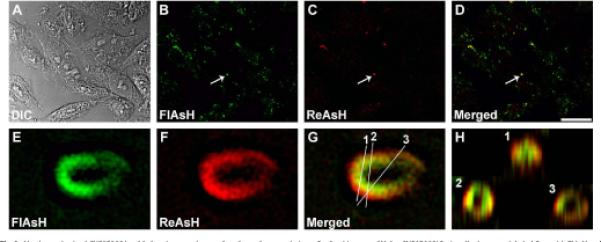
Newly synthesized CX50P88S is added to the outer layers of preformed accumulations. Confocal images of HeLa-CX50P88S(Cys)4 cells that were labeled first with FlAsH and 96 h later with ReAsH. (A) DIC, (B) FlAsH labeling, (C) ReAsH labeling, (D) FlAsH and ReAsH merged image. (E-H) Three-dimensional reconstruction of a z-series after deconvolution of the CX50P88S accumulation indicated by the arrow in panels B-D acquired at higher magnification. The insets in panel H represent the three-dimensional volume projection of the CX50P88S accumulation at the levels indicated by the white lines numbered 1, 2 and 3 in panel G. Scale bar, 24 mm for A-D, and 1 μm for E-H.
To prove that the ReAsH-labeled material corresponded to newly synthesized protein, HeLa-CX50P88S(Cys)4 cells were treated with cycloheximide (an inhibitor of protein synthesis) starting immediately after labeling with the first biarsenical compound (FlAsH). Under these conditions, no material labeled with the second biarsenical compound was observed (data not shown).
Incubation of mammalian cells at temperatures lower than 16 °C causes a block in ER-to-Golgi transport of newly synthesized proteins leading to their accumulation in the ER to Golgi intermediate compartment, ERGIC (Saraste and Kuismanen, 1984; Appenzeller-Herzog and Hauri, 2006). To test whether ER-to-Golgi transport was required for formation of CX50P88S accumulations or addition of newly synthesized protein to already formed accumulations, we performed a pulse-chase experiment at 15 °C. HeLa-CX50P88S(Cys)4 cells were labeled first with FlAsH, and then they were incubated in labeling-free medium either at 15 °Cor 37 °C followed by labeling with ReAsH. Similar to the results obtained at 37 °C(Fig. 4A-C), when cells were incubated at 15 °C, most of the FlAsH-labeled accumulations were also labeled with ReAsH (Fig. 4D-F, arrows), and some accumulations were labeled with ReAsH only (Fig. 4D-F, arrowheads).
Fig. 4.
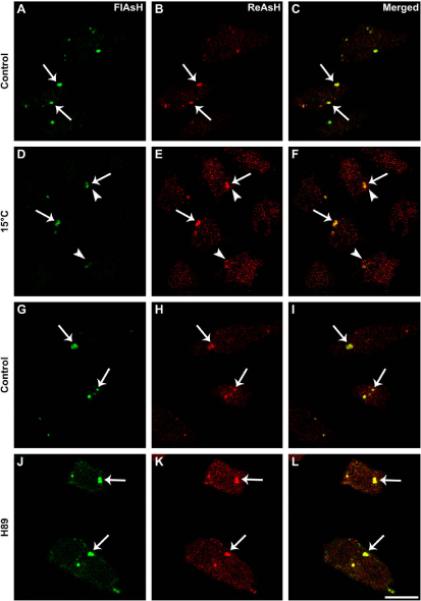
Blockade of ER-to-Golgi transport does not affect formation of CX50P88S accumulations. (A-F) Confocal images of HeLa-CX50P88S(Cys)4cells that were labeled with FlAsH, incubated for 24 h at either 37 °C (A-C) or 15 °C (D-F) and then labeled with ReAsH. (G-L) Confocal images of HeLa-CX50P88S(Cys)4 cells that were labeled with FlAsH, incubated for 4 h in the absence (G-I) or presence (J-L) of 50 μM H89 and subsequently labeled with ReAsH. Accumulations labeled with both FlAsH and ReAsH are indicated by arrows. The arrowheads in panels D-F indicate accumulations that labeled with ReAsH only. Scale bar, 20 mm.
Next, we tested whether exit from the ER was required for formation of CX50P88S(Cys)4 accumulations. We used H89, a compound that inhibits ER export by preventing recruitment of Sec13 to ER export sites (Jamora et al., 1999; Lee and Lindstedt, 2000). HeLa-CX50P88S(Cys)4 cells were incubated first with FlAsH to label pre-existing proteins, treated with 50 μM H89 for 4 h, and then labeled with ReAsH to identify newly synthesized proteins (Fig. 4G-L). Similar to the results obtained in untreated cultures, most of the FlAsH-labeled accumulations (Fig. 4G and J, arrows) were labeled also with ReAsH (Fig. 4, compare panels G and H with panels J and K, arrows) and appeared yellow on the merged images (Fig. 4I and L, arrows).
3.4. Ultrastructural analysis of CX50P88S(Cys)4 aggregates
In order to clarify the origin of CX50P88S accumulations, ReAsH-labeled structures in HeLa-CX50P88S(Cys)4 cells were used to drive the photoconversion of diaminobenzidine at labeled structures which were then studied by transmission electron microscopy. In these samples, labeled circular or semicircular membrane stacks were seen within the cytoplasm (Fig. 5A, B). Most of these membrane stacks had a lumen; in the ones in which the lumen was not apparent, the thin section seemed to graze the top of the accumulation. These structures had a similar size and shape to the CX50P88S accumulations previously observed (Berthoud et al., 2003). In most instances, they contained individual membrane sheets closely apposed to each other forming structures that could be described as stacks of gap junctions. The average center-tocenter spacing between these double-layered membranes as measured in these thin section electron micrographs was 30.8 nm (standard deviation = 1.9 nm, n = 12). In some specimens, the plane of sectioning allowed the observation of continuity between the rough endoplasmic reticulum and membranes containing photoconverted material (Fig. 5A, arrow). These structures contained only two membranes (Fig. 5A).
Fig. 5.
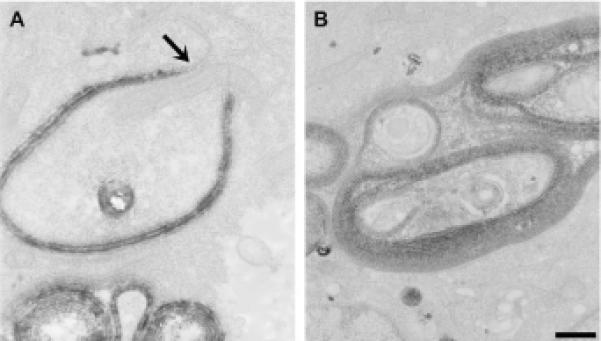
Single and double lamellar membrane stacks are the first step in formation of CX50P88S multilamellar accumulations. High magnification views of CX50P88S accumulations show labeled membrane stacks. (A, B) Thin section electron micrographs of ReAsH-driven photoconverted DAB at labeled CX50P88S(Cys)4. Specific staining is highest in the most densely packed CX50P88S(Cys)4 structures and is not evident in other organelles such as mitochondria and surrounding vesicles. The arrow in (A) indicates rough ER giving rise to these cytosolic accumulations. (B) These multilamellar accumulations can have complex geometries. Scale bar, 376 nm for A, and 500 nm for B.
To examine further the continuity of the membranes within these aggregates, we used electron tomographic reconstruction methods to obtain three-dimensional volumes of several larger structures (Fig. 6). For these tomograms, sections were post-stained with uranyl acetate to provide extra contrast for the other cytoplasmic components. Fig. 6 shows three representative slices (A, B, C corresponding to top, middle and bottom of the volume, respectively) going through the tomographic double-tilt volume of one typical, large aggregate. The concentric double membrane layers of the aggregate appear to form “onion skin” sheets that do not form a closed structure along the long axis. The differential staining between the outside and inside is most likely due to impeded diffusion of DAB into the interior of these accumulations. The center-to-center spacing at the outermost layers is w30 nm. Another prominent feature is the apposition of ER and ER-derived vesicles with these accumulations. Typically, cytoplasm containing ribosomes and vesicles was observed at the interior of the aggregate; the electron dense material within these vesicles resembled that of degraded proteins. The vesicles immediately outside the aggregate may be part of the formation and addition mechanism (and/or components that will be recycled into the ER). In these slices, flat sheets of ER appeared to collapse together to form a double layer gap junction-like assembly. Fig. 6D-F shows three slices from another tomogram containing two multilaminar accumulations that have become joined structures through the addition of CX50P88S(Cys)4 containing membranes from the same piece of rough ER (arrow).
Fig. 6.
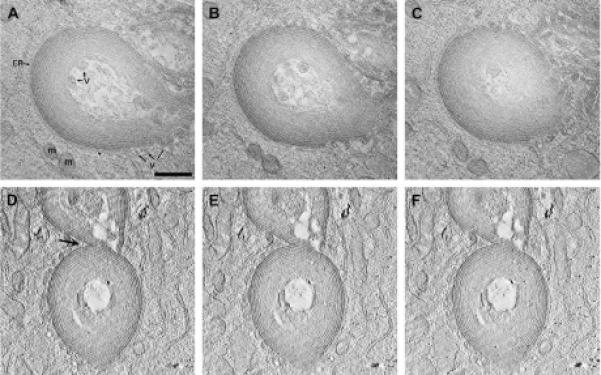
Three-dimensional reconstructions of multilamellar accumulations. Thick sections of photoconverted CX50P88S(Cys)4 accumulations were prepared for electron tomography. Two tomograms were reconstructed and analyzed. (A-C) Slices from the top, middle and bottom parts of the double tilt reconstruction are shown to illustrate the “onion skin” appearance of these structures. Also indicated in the slice shown in (A) are the close apposition of ER membranes (ER), mitochondria (m) and vesicles (v). (D-F) On occasion, two accumulations were joined together by a piece of ER attached to both (see arrow), as seen in three slices from this single tilt tomogram. Scale bar, 0.5 μm.
Segmenting the volume into several components helps elucidate the complexity of the structure. Fig. 7 shows surface shaded segmentations derived from manual tracing of the membranes in the double tilt tomogram presented in Fig. 6A-C. Models were scaled in the z direction to account for irradiation-induced specimen shrinkage during data acquisition. Double-layered membranes that were precisely traced (gap junction-like assemblies), single membranes (rough ER) and vesicles are color-coded dark blue, green and red, respectively. The set of interior double-layered membranes that were not stained strongly enough to permit precise segmentation are indicated in light blue; for these double-layers, the tracings were estimated based on measurements and shape from the outer, more accurately traced double-layers (dark blue). The impression from these three-dimensional reconstructions is that these accumulations may be more cylindrical than spherical (see Fig. 6C). The layering of the “gap junction-like” structures is easily seen in the cross-sectional view shown in Fig. 7B. These graphic representations show the high density packing of CX50P88S-containing membranes that would contribute to a strong light scattering object, especially when accumulations cluster as shown in Fig. 6D-F. The vesicles seen in thin section and slice views (Fig. 6) appear tubular when viewed in three dimensions (Fig. 7A,C). When the adhering ER and vesicles are displayed (Fig. 7C), there is clear separation between these two classes of cytoplasmic components. The image shown in Fig. 7D is a representation of the outermost double membrane with two pieces of ER (green and yellow) at either end that fuse to make this “gap junction-like” structure. Thus, these multilamellar accumulations have a distinct, compact and organized structure derived from the ER. Animations of the series of slices through these two volumes as well as visualization of the segmented model in 3D can be accessed in the Supplementary Information (S1 and S2).
Fig. 7.

Analysis of the three-dimensional organization of the CX50P88S multilamellar accumulations. The membranes in the volume shown in Fig. 6A-C were traced in order to visualize the continuity of the various membrane compartments. Computer graphic representations are shown as surface shaded models. (A) Overview of the entire structure. Blue membranes represent double bilayers. Membranes that could be accurately traced on the volume slices are represented in dark blue; membranes in which the staining was not strong enough to permit precise segmentation were approximated based on the spacings of the dark blue double bilayers and are represented in light blue. Single membranes, most likely ER or of ER origin, are represented in green. Tubular vesicles found at the periphery and the interior of the accumulations are represented in red. In individual volume slices and thin sections, these tubules appeared circular (see Fig. 6). Scale bar, 0.5 μm. (B) Cross-sectional view of the accumulation presented in (A) at the plane indicated by the arrow shown at 1.5x that of (A). (C) Models of single membranes (ER) and tubules show segregation of these two structures. (D) Two ER membranes (green and yellow) fuse at the outermost edge to form a double layered structure (dark blue).
4. Discussion
In this study, we examined the process by which the cataract-associated mutant, CX50P88S, forms cytoplasmic accumulations. We found that these structures are very stable and are formed by sequential addition of membrane embedded protein derived from the endoplasmic reticulum.
Most wild type connexins have relatively short half-lives (1.5-5 h) (reviewed in Berthoud et al., 2003) implying that within 24 h essentially all of the connexin within the cell has been renewed. The half-lives of lens connexins show more variation (from hours to days); short- and long-lived pools have been determined in lens cells in culture after differentiation into lentoids (Berthoud et al., 1999) and a long-lived pool has been reported in lens in organ culture (Jiang and Goodenough, 1998). In stably transfected HeLa cells, treatment with cycloheximide leads to a dramatic decrease in levels of wild type human CX50 by 21 h. In contrast, the decrease in CX50P88S levels is much less pronounced; they remain close to the initial values after 21 h of cycloheximide treatment (Berthoud et al., 2003). In agreement with these results, in our current studies, mutant CX50P88S pulse-labeled with FlAsH decreased with time of chase, but it was still visible even 120 h after labeling. These results demonstrate that CX50P88S is very long-lived, and it is much longer lived than the wild type protein, implying impaired degradation of this mutant protein. The calculated apparent half-life of the FlAsH-labeled structures was almost 70 h. This value may even be an underestimation, because it assumes that fluorescence intensity and the number of protein-bound FlAsH molecules (for a highly and locally concentrated protein) are directly related and that the fluorescence of FlAsH does not decrease with time. It also assumes that we can detect all of the CX50P88S within the cell.
Our current ultrastructural studies confirm our previous observation (Berthoud et al., 2003) that the CX50P88S accumulations contain multiple layers of membranes. The thickness of these double layers is consistent with that measured for gap junctions. Tomographic analysis showed that these multilayered structures corresponded to several concentric, regularly spaced membrane layers. The differential distribution of newly synthesized and older CX50P88S molecules within individual double-labeled accumulations indicated that new protein (labeled with ReAsH) was added to the periphery of pre-existing accumulations (labeled with FlAsH). These results obtained after sequential labeling with FlAsH and ReAsH imply that CX50P88S does not freely diffuse (or it has very limited diffusion) within the membrane in these structures.
We have previously shown that CX50P88S accumulations can form in the presence of Brefeldin A, a drug that disrupts the Golgi compartment and protein trafficking to the plasma membrane (Berthoud et al., 2003). Blockade of transport of newly synthesized proteins between compartments of the secretory pathway can be achieved by lowering the temperature at which cells are incubated. Decreasing the temperature to 15 °C allows proteins to leave the ER, but they cannot enter the Golgi cisternae leading to their accumulation in the ERGIC (Saraste and Kuismanen, 1984; Appenzeller-Herzog and Hauri, 2006). Such inhibition of ER to Golgi transport did not prevent addition of newly synthesized CX50P88S protein to pre-existing accumulations (or formation of new ones), suggesting that CX50P88S accumulations originated in a pre-Golgi compartment (which could be the ER or the ERGIC). The presence of ReAsH-labeled accumulations after treatment with H89 to inhibit ER export suggests that the newly synthesized CX50P88S does not need to exit the ER to become part of accumulations. Thus, these results support the hypothesis that CX50P88S accumulations originate from the rough endoplasmic reticulum. They also suggest that this protein is resistant to ER-associated degradation.
Our ultrastructural studies show continuity between the rough endoplasmic reticulum and a two-layered structure containing photoconverted ReAsH material (Fig. 5). This observation further suggests that the accumulations of CX50P88S originate from the rough endoplasmic reticulum. Moreover, double label immunostaining does not show the presence of ER-resident proteins in CX50P88S accumulations (Berthoud et al., 2003). Tomographic analysis of these accumulations indicate that they have an organized and compact layering of “gap junction-like” double membranes with ER membranes at the outermost edges and tubular membrane structures at the ER periphery and the interior of the accumulations.
Taking together the results from the ultrastructural studies and the pulse-chase experiments, we have developed a model illustrating the steps involved in the generation and persistence of CX50P88S accumulations (Fig. 8). We propose that a CX50P88S accumulation originates from rough endoplasmic reticulum membrane containing CX50P88S (which fails to reach the plasma membrane). Initially, ER membrane containing CX50P88S forms an out-pocketing. As additional protein is synthesized, membrane containing CX50P88S is added to the initial out-pocketing increasing its size and the abundance of CX50P88S. This process continues and the out-pocketing collapses on itself and forms a sheet-like (gap junction-like) outgrowth. When this reaches a critical size, it folds upon itself forming a tube-like structure (or nascent accumulation). During generation of the nascent accumulation, ER-resident membrane proteins are excluded and likely recycled back to the rough endoplasmic reticulum; this would explain the lack of co-localization of CX50P88S immunoreactivity with that of endoplasmic reticulum markers observed previously (Berthoud et al., 2003). The nascent accumulation may act as a “nucleation particle” for the subsequent addition of CX50P88S-containing membranes producing the multilayered structures that we have observed. An alternative model in which CX50P88S-containing membranes are added randomly to pre-existing circular structures is not supported by our studies.
Fig. 8.

Proposed mechanism of formation of CX50P88S cytosolic accumulations. CX50P88S is synthesized in the rough endoplasmic reticulum (rER). This mutant protein does not leave the rER compartment or follow the normal pathway of wild type CX50 to the plasma membrane (1). Continuing synthesis of CX50P88S leads to the formation of an out-pocketing from the rER membrane in which the mutant protein becomes concentrated (2). The size of the out-pocketing increases as more CX50P88S protein is synthesized (3). This leads to a highly localized concentration of CX50P88S which provokes the collapse of the ER membrane from opposite sides, displacement of the ER luminal contents, and formation of a sheet-like (gap junction-like) double membrane structure (4). This double membrane structure grows by continued addition of more CX50P88S-containing vesicles (not shown in the diagram) until it reaches a critical size where it folds upon itself to form a tubular structure or nascent accumulation (5).
These studies imply that the CX50P88S mutation likely causes lens pathology through mechanisms beyond loss of intercellular communication. The loss of lens transparency characteristic of cataracts can also occur by the presence of multilamellar structures or large protein aggregates. Cytoplasmic CX50P88S accumulations are long-lived multilamellar structures that can cause light scattering, may act as nucleation particles for accumulation/aggregation of other proteins and cause cellular damage, all factors that can contribute to cataract formation.
Acknowledgements
We are grateful to Masako Terada and James Obiyashi for their help with the tomographic processing and visualization process. We thank Benjamin Heilbrunn for generating the HeLa-CX50P88S(Cys)4 cells and Peter Minogue for performing the transient transfection experiments. This work was supported by NIH grants RO1-EY08368 (ECB), GM072881 (GES) and GM065937 (GES), and a grant from the Children's Research Foundation (AL). Some of the work included here was conducted at the National Center for Microscopy and Imaging Research at San Diego, which is supported by National Institutes of Health Grant RR04050 awarded to Dr. Mark Ellisman.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version, at doi:10.1016/j.exer.2008.11.024
References
- Appenzeller-Herzog, Hauri H-P. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J. Cell Sci. 2006;119:2173–2183. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- Arora A, Minogue PJ, Liu X, Reddy MA, Ainsworth JR, Bhattacharya SS, Webster AR, Hunt DM, Ebihara L, Moore AT, Beyer EC, Berthoud VM. A novel GJA8 mutation is associated with autosomal dominant lamellar pulverulent cataract: further evidence for gap junction dysfunction in human cataract. J. Med. Genet. 2006;43:e2. doi: 10.1136/jmg.2005.034108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud VM, Bassnett S, Beyer EC. Cultured chicken embryo lens cells resemble differentiating fiber cells in vivo and contain two kinetic pools of connexin56. Exp. Eye Res. 1999;68:475–484. doi: 10.1006/exer.1998.0635. [DOI] [PubMed] [Google Scholar]
- Berthoud VM, Minogue PJ, Guo J, Williamson EK, Xu X, Ebihara L, Beyer EC. Loss of function and impaired degradation of a cataract-associated mutant connexin50. Eur. J. Cell Biol. 2003;82:209–221. doi: 10.1078/0171-9335-00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud VM, Minogue PJ, Laing JG, Beyer EC. Pathways for degradation of connexins and gap junctions. Cardiovasc. Res. 2004;62:256–267. doi: 10.1016/j.cardiores.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- Gilliland KO, Freel CD, Lane CW, Fowler WC, Costello MJ. Multilamellar bodies as potential scattering particles in human age-related nuclear cataracts. Mol. Vis. 2001;7:120–130. [PubMed] [Google Scholar]
- Goodenough DA. Lens gap junctions: a structural hypothesis for nonregulated low-resistance intercellular pathways. Invest. Ophthalmol. Vis. Sci. 1979;18:1104–1122. [PubMed] [Google Scholar]
- Jamora C, Yamanouye N, Van Lint J, Laudenslager J, Vandenheede J, Faulkner D, Malhotra V. Gbg-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell. 1999;98:59–68. doi: 10.1016/S0092-8674(00)80606-6. [DOI] [PubMed] [Google Scholar]
- Jiang JX, Goodenough DA. Phosphorylation of lens-fiber connexins in lens organ cultures. Eur. J. Biochem. 1998;255:37–44. doi: 10.1046/j.1432-1327.1998.2550037.x. [DOI] [PubMed] [Google Scholar]
- Kistler J, Kirkland B, Bullivant S. Identification of a 70,000-D protein in lens membrane junctional domains. J. Cell Biol. 1985;101:28–35. doi: 10.1083/jcb.101.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Lawrence A, Bouwer JC, Perkins G, Ellisman MH. Transform-based backprojection for volume reconstruction of large format electron microscope tilt series. J. Struct. Biol. 2006;154:144–167. doi: 10.1016/j.jsb.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Lee TH, Lindstedt AD. Potential role for protein kinases in regulation of bidirectional endoplasmic reticulum-to-Golgi transport revealed by protein kinase inhibitor H89. Mol. Biol. Cell. 2000;11:2577–2590. doi: 10.1091/mbc.11.8.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martone ME, Tran J, Wong WW, Sargis J, Fong L, Larson S, Lamont SP, Gupta A, Ellisman MH. The cell centered database project: an update on building community resources for managing and sharing 3D imaging data. J. Struct. Biol. 2008;161:220–231. doi: 10.1016/j.jsb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul DL, Ebihara L, Takemoto LJ, Swenson KI, Goodenough DA. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J. Cell Biol. 1991;115:1077–1089. doi: 10.1083/jcb.115.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins GA, Sosinsky GE, Ghassemzadeh S, Perez A, Jones Y, Ellisman MH. Electron tomographic analysis of cytoskeletal cross-bridges in the paranodal region of the node of Ranvier in peripheral nerves. J. Struct. Biol. 2008;161:469–480. doi: 10.1016/j.jsb.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy MA, Francis PJ, Berry V, Bhattacharya SS, Moore AT. Molecular genetic basis of inherited cataract and associated phenotypes. Surv. Ophthalmol. 2004;49:300–315. doi: 10.1016/j.survophthal.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Ri Y, Ballesteros JA, Abrams CK, Oh S, Verselis V, Weinstein H, Bargiello TA. The role of a conserved proline residue in mediating conformational changes with voltage gating of Cx32 gap junctions. Biophys. J. 1999;76:2887–2898. doi: 10.1016/S0006-3495(99)77444-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rup DM, Veenstra RD, Wang H-Z, Brink PR, Beyer EC. Chick connexin-56, a novel lens gap junction protein. Molecular cloning and functional expression. J. Biol. Chem. 1993;268:706–712. [PubMed] [Google Scholar]
- Sáez JC, Berthoud VM, Brañes MC, Martínez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Saraste J, Kuismanen E. Pre- and post-Golgi vacuoles operate in the transport of Semliki Forest virus membrane glycoproteins to the cell surface. Cell. 1984;38:535–549. doi: 10.1016/0092-8674(84)90508-7. [DOI] [PubMed] [Google Scholar]
- Shiels A, Mackay D, Ionides A, Berry V, Moore A, Bhattacharya S. A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q. Am. J. Hum. Genet. 1998;62:526–532. doi: 10.1086/301762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchyna TM, Xu LX, Gao F, Fourtner CR, Nicholson BJ. Identification of a proline residue as a transduction element involved in voltage gating of gap junctions. Nature. 1993;365:847–849. doi: 10.1038/365847a0. [DOI] [PubMed] [Google Scholar]
- White TW, Bruzzone R, Goodenough DA, Paul DL. Mouse Cx50, a functional member of the connexin family of gap junction proteins, is the lens fiber protein MP70. Mol. Biol. Cell. 1992;3:711–720. doi: 10.1091/mbc.3.7.711. [DOI] [PMC free article] [PubMed] [Google Scholar]