

Abstract
The Rab7 GTPase promotes membrane fusion reactions between late endosomes and lysosomes. In previous studies, we demonstrated that Rab7 inactivation blocks growth factor withdrawal-induced cell death. These results led us to hypothesize that growth factor withdrawal activates Rab7. Here, we show that growth factor deprivation increased both the fraction of Rab7 that was associated with cellular membranes and the percentage of Rab7 bound to guanosine triphosphate (GTP). Moreover, expressing a constitutively GTP-bound mutant of Rab7, Rab7-Q67L, was sufficient to trigger cell death even in the presence of growth factors. This activated Rab7 mutant was also able to reverse the growth factor-independent cell survival conferred by protein kinase C (PKC) δ inhibition. PKCδ is one of the most highly induced proteins after growth factor withdrawal and contributes to the induction of apoptosis. To evaluate whether PKCδ regulates Rab7, we first examined lysosomal morphology in cells with reduced PKCδ activity. Consistent with a potential role as a Rab7 activator, blocking PKCδ function caused profound lysosomal fragmentation comparable to that observed when Rab7 was directly inhibited. Interestingly, PKCδ inhibition fragmented the lysosome without decreasing Rab7-GTP levels. Taken together, these results suggest that Rab7 activation by growth factor withdrawal contributes to the induction of apoptosis and that Rab7-dependent fusion reactions may be targeted by signaling pathways that limit growth factor-independent cell survival.
INTRODUCTION
In multicellular organisms, tissue homeostasis is enforced by the dependence of all cells on extrinsic growth factors for growth, proliferation, and survival (Raff, 1992). The molecular events that lead to apoptosis after growth factor deprivation are not completely understood. It is likely that growth factor withdrawal induces programmed cell death through multiple, parallel pathways. For example, maintaining Akt or mTOR activity, increasing Pim kinase signaling, or directly disabling apoptosis by overexpressing Bcl-XL rescues interleukin (IL)-3 dependent cell lines from death after growth factor withdrawal (Nunez et al., 1990; Vander Heiden et al., 1999; Plas et al., 2001; Edinger and Thompson, 2002, 2004; Fox et al., 2003). Interestingly, inactivating the small GTPase Rab7 also protects cells from growth factor withdrawal-induced death (Edinger et al., 2003). Rab7 promotes homotypic and heterotypic fusion reactions with lysosomes (Feng et al., 1995; Vitelli et al., 1997; Bucci et al., 2000). FL5.12 cells expressing either RNA interference (RNAi) targeting Rab7 or the dominant-negative (DN) Rab7 mutant Rab7-T22N exhibit significant IL-3–independent survival (Edinger et al., 2003). Moreover, when the tumor suppressor proteins p53 and Rb are also inactivated, inhibiting Rab7 function promotes colony formation by fibroblasts in soft agar. Although Rab7 facilitates growth factor withdrawal-induced apoptosis, whether Rab7 activity is regulated by growth factor signaling has not been evaluated.
Because GTPases only associate with their effector proteins when bound to guanosine triphosphate (GTP), the activity of Rab GTPases is regulated by their nucleotide binding state (Pfeffer and Aivazian, 2004; Grosshans et al., 2006). Specific guanine nucleotide exchange factor proteins activate Rabs by promoting the exchange of guanosine diphosphate (GDP) for GTP. GTPase-activating proteins (GAPs) fill the opposite role, accelerating the hydrolysis of the bound GTP to GDP. GTP binding status also influences the membrane insertion and extraction cycle of Rab proteins. GDP-bound Rabs are susceptible to extraction from membranes by GDP dissociation inhibitor (GDI), which masks the hydrophobic membrane anchor while Rab proteins are held inactive in the cytosol. Given this complex activation cycle, there are many points at which Rab7 activity might be controlled by signal transduction cascades.
Whether signal transduction cascades regulate Rab7 activity is of great interest given the role this protein is likely to play in multiple human diseases. In addition to its function in growth factor withdrawal-induced apoptosis (Edinger et al., 2003), Rab7 is also required for autophagy (Gutierrez et al., 2004; Colombo, 2007). Autophagy can suppress or accelerate tumor formation under different conditions (Mathew et al., 2007). Autophagy also helps to clear the protein aggregates that contribute to the pathogenesis of neurodegenerative diseases such as Huntington's and Alzheimer's disease (Kundu and Thompson, 2008; Mizushima et al., 2008). In keeping with the idea that Rab7 activity influences neuronal physiology, mutations in Rab7 have been linked to the ulcerating peripheral neuropathy Charcot-Marie-Tooth syndrome type 2B (Verhoeven et al., 2003; Houlden et al., 2004; Meggouh et al., 2006). Finally, due to its role in promoting lysosomal trafficking, Rab7 also helps to eliminate intracellular pathogens (Brumell and Scidmore, 2007; Philips et al., 2008). Given these potential links between Rab7 and multiple human diseases, we investigated whether Rab7 activity was regulated by growth factor receptor-dependent signal transduction.
MATERIALS AND METHODS
Antibodies, Plasmids, and Reagents
Antibodies were obtained from the following sources: Rab7, tubulin, and FLAG-M2 (Sigma-Aldrich, St. Louis, MO); green fluorescent protein (GFP) (Clontech, Mountain View, CA); calnexin, actin, and protein kinase C (PKC) δ (Santa Cruz Biotechnology, Santa Cruz, CA); hemagglutinin (Roche Diagnostics, Indianapolis, IN); and mouse immunoglobulin G (IgG) (Jackson ImmunoResearch Laboratories, West Grove, PA). A rabbit anti-GM130 antibody was generously provided by Christine Suetterlin (University of California–Irvine, Irvine, CA). AnnexinV-allophycocyanin conjugate was from eBioscience (San Diego, CA). GFP-Rab7, GFP-Rab7-Q67L, GFP-Rab7-T22N, and GFP-RILP were generously provided by Cecilia Bucci (Università del Salento, Lecce, Italy; canine Rab7 cDNAs, human RILP) and cloned into the EF6-V5-His vector (Invitrogen, Carlsbad, CA) or pBABEpuro for expression in FL5.12 cells. FLAG-Rab7 was generated by polymerase chain reaction (PCR) and cloned into EF6-V5-His. GFP-Rab7-Q67L was also cloned into pRevTRE (Clontech) and introduced into FL5.12 cells expressing rtTA from pUHD172-1 to allow doxycycline-inducible expression. HA-tagged dominant-negative PKCδ was kindly provided by Jae-Won Soh (Inha University, Man-gu Incheon, Korea) and cloned into the EF6-V5-His expression vector. Enhanced green fluorescent protein (EGFP) was expressed from pEGFP-C1 (Clontech). MicroRNA-adapted short hairpin RNA (shRNAmir) constructs targeting PKCδ were obtained from Open Biosystems (Huntsville, AL) in the pSM2 vector and cloned into the LMP vector (Open Biosystems). RNAi-1 is clone V2MM-63171 and RNAi-2 is clone V2MM-62352. Each hairpin produced similar knockdown when expressed from either the pSM2 or LMP vector. All chemicals were obtained from Sigma-Aldrich or EMD Biosciences (San Diego, CA). Bicinchoninic acid (BCA) reagents used for protein assays were from Pierce Chemical (Rockford, IL).
Cell Culture
FL5.12 cells were maintained at 250,000–750,000 cells/ml in RPMI 1640 medium (Mediatech, Herndon, VA) supplemented with 10% fetal calf serum (FCS) (Mediatech; HyClone Laboratories, Logan, UT; or Sigma-Aldrich), 500 pM recombinant mouse interleukin (IL)-3 (BD Biosciences Pharmingen, San Diego, CA; or eBioscience), 10 mM HEPES (Mediatech), 55 μM β-mercaptoethanol (Sigma-Aldrich), antibiotics, and l-glutamine (Mediatech). Bone marrow was isolated from the femurs of wild-type mice (C57BL6/Jx129 mixed background). Red blood cells were lysed with ACK buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM EDTA, pH 7.2–7.4) and bone marrow cells cultured in FL5.12 media supplemented with 2.5 nM recombinant IL-3. After 2 wk in culture, cells were infected with culture supernatant from Hox11 virus producer cells (Zinkel et al., 2005) kindly provided by Sandra Zinkel by way of Jeff Rathmell, Duke University, Durham, NC) to facilitate immortalization. The murine helper T-cell clone HT-2 (Watson, 1979) was generously supplied by Dr. Craig Walsh (University of California–Irvine) and cultured in RPMI 1640 medium supplemented with 10% FCS, 50 ng/ml IL-2 (kind gift of Dr. Craig Walsh), 10 mM HEPES, 55 μM β-mercaptoethanol, antibiotics, and l-glutamine.
Rab7 GTP Binding Assays
FL5.12 cells expressing low levels of FLAG-Rab7 were withdrawn from or maintained in IL-3 overnight (15 h). Cells were washed with phosphate-free RPMI 1640 medium (made from chemical components) and then labeled for 3 h in a humidified incubator at 37°C and 5% CO2 with 0.5 mCi/ml [32P]orthophosphate (MP Biomedicals, Irvine, CA) in phosphate-free RPMI 1640 medium supplemented with 10% dialyzed fetal bovine serum, 10 mM HEPES, 55 μM β-mercaptoethanol, antibiotics, and l-glutamine, with or without IL-3. Cells were washed with phosphate-free RPMI 1640 medium and resuspended in lysis buffer (50 mM HEPES, pH 7.4, 1% Triton X (TX)-100, 100 mM NaCl, 5 mM MgCl2, 1 mg/ml bovine serum albumin, 50 mM NaF, and 1 mM orthovanadate, supplemented with Complete protease inhibitors [Roche Diagnostics]), incubated on ice for 10 min., and spun at top speed in a 4°C microfuge. Then, the supernatant was transferred to a new tube. FLAG-M2–coated agarose beads (Sigma-Aldrich, prepared as recommended by the manufacturer) or protein A agarose beads (Invitrogen) and mouse IgG were added, and precipitations were incubated while rocking at 4°C for 2 h. Beads were washed twice with wash buffer (50 mM HEPES, pH 7.4, 500 mM NaCl, 5 mM MgCl2, 0.1% TX-100, 50 mM NaF, and 1 mM orthovanadate, supplemented with Complete protease inhibitors), and then guanine nucleotides were eluted at 68°C for 20 min in elution buffer (2 mM EDTA, 2 mM dithiothreitol (DTT), 0.2% SDS, 0.5 mM GDP, and 0.5 mM GTP). GTP and GDP were separated by thin layer chromatography (TLC) on polyethylenimine cellulose plates by using 0.5 M K2HPO4, pH 3.4. GDP and GTP standards were identified using UV light, and [32P]GTP and [32P]GDP were detected using a Storm PhosphorImager (GE Healthcare, Chalfont St. Giles, Buckinghamshire, United Kingdom) or Kodak XAR film (Eastman Kodak, Rochester, NY).
Glutathione Transferase-Rab Interacting Lysosomal Protein (GST-RILP) Pull-Downs
Nucleotides 658-897 (GenBank accession no. NM_001029938; amino acids 220-299) of the murine RILP protein constituting the Rab7 binding domain of RILP were fused to the C terminus of GST in the pGEX 4T-3 vector (Stratagene, La Jolla, CA). GST-RILP was transformed into Escherichia coli strain BL21. Then, 250 ml of Luria broth was inoculated with 1 ml of an overnight culture and grown at 37°C to an OD of 0.6–0.8. Isopropyl β-d-thiogalactoside was then added to a final concentration of 0.5 mM to induce protein production. The 250-ml culture was incubated for additional 3–4 h at 30°C, after which the bacteria were spun down, washed with cold (4°C) phosphate-buffered saline (PBS), resuspended in 5 ml of cold lysis buffer (25 mM Tris-HCl, pH 7.4, 1 M NaCl, 0.5 mM EDTA, 1 mM DTT, and 0.1% TX-100, with Complete protease inhibitors), and then sonicated. The bacterial lysates were cleared by centrifugation, and 5 ml of cold lysis buffer was added. Proteins were purified by adding 300 μl of a pre-equilibrated 50% slurry of glutathione-Sepharose 4B beads (GE Healthcare) to the lysate. Beads were incubated with lysates for 30 min at room temperature and then washed with lysis buffer and resuspended as a 50% slurry. Protein levels were quantified using the BCA assay. Mammalian cells to be analyzed in the pull-down were lysed in pull-down buffer (20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 1% TX-100, and protease inhibitors). Each pull-down was performed in 1 ml with 300 μg of cell lysate and 30 μg of beads pre-equilibrated in pull-down buffer. Beads were rocked overnight at 4°C, washed twice with cold pull-down buffer, and bound proteins were eluted by adding 2× Sample buffer with DTT and incubating at 72°C for 10 min.
Quantitative Reverse Transcription (RT)-PCR
Total RNA was isolated using the RNeasy Mini kit (QIAGEN, Valencia, CA). Approximately 0.5 μg of total RNA was analyzed in a total reaction volume of 30 μl, containing 150 nM gene-specific primers, 4 U of RNase Out (Invitrogen), 2.5 U of Superscript III RT (Invitrogen), and 1× quantitative PCR SYBR Green Mix (Abgene, Epsom, Surrey, United Kingdom). Reverse transcription was performed for 30 min at 48°C, and then PCR was performed using the following cycling parameters: 95°C for 10 min followed by 40 cycles of 15 s at 95°C, 30 s at 60°C, and 30 s at 72°C using an iCycler (Bio-Rad Laboratories, Hercules, CA). PKCδ mRNA was normalized to β-actin mRNA. The following primers were used for the reactions: PKCδ forward primer, CCTCCTGTACGAAATGCTCATC; PKCδ reverse primer, GTTTCCTGTTACTCCCAGCCT; β-actin forward primer, GGCTGTATTCCCCTCCATCG; and β-actin reverse primer, CCAGTTGGTAACAATGCCATGT. Primer sequences were taken from Primer Bank (http://pga.mgh.harvard.edu/primerbank/index.html).
Cellular Fractionation and Western Blotting
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer with Complete protease inhibitors. Equal amounts of protein were loaded onto NU-PAGE 10% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membranes. Western blots were either evaluated by chemiluminescence using horseradish peroxidase-coupled secondary antibodies (Cell Signaling Technology, Danvers, MA) and enhanced chemiluminescence (GE Healthcare) or by using the Odyssey infrared imaging system and IRDye680- or IRDye800CW-conjugated secondary antibodies (all from LI-COR, Lincoln, NE). Cellular fractionations were accomplished by resuspending cells in a small volume of buffer A (20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 250 mM sucrose, supplemented with Complete protease inhibitors) after washing with PBS. Cells were lysed by aspiration through a 26-gauge needle (20 times) and a 30-gauge needle (30 times). Nuclei and unlysed cells were cleared by serial spins at 1000 × g for 10 min. Cleared lysates were spun at 100,000 × g for 1 h at 4°C in a precooled TLS55 rotor (38,000 rpm; Beckman Coulter, Fullerton, CA). The supernatant collected after this spin was saved as the S100 fraction. The pellet was washed with 100 μl of fresh buffer A, resuspended in 500 μl, and spun for an additional 30 min at 100,000 × g. After removal of the wash, the pellet was lysed in RIPA buffer, incubated for 10 min on ice, and then spun for 10 min at top speed in a microfuge to remove insoluble material. The supernatant was collected as the P100 fraction. BCA assays were performed to allow loading of equal amounts of protein per lane. It should be noted that much less total protein was obtained from the P100 fraction (∼10% of total protein) than from the S100 fraction (∼90% of total protein). In all experiments, tubulin and calnexin localization was evaluated in parallel Western blots to confirm that the S100 and P100 fractions were not contaminated with membranes or cytosol, respectively.
Flow Cytometry and Microscopy
Cells were analyzed on an LSR II flow cytometer (BD Biosciences, San Jose, CA). Viability was determined by vital dye exclusion (propidium iodide or 4,6-diamidino-2-phenylindole [DAPI]; Invitrogen). To evaluate lysosomal morphology, cells were stained with 500 nM LysoTracker Red (Invitrogen) for 30 min at 37°C and examined using an Eclipse TE2000 fluorescence microscope (Nikon, Tokyo, Japan) equipped with a CoolSNAP charge-coupled device camera.
RESULTS
Growth Factor Withdrawal Increases Rab7 Association with Membranes
We established previously that Rab7 inactivation by RNAi or through the expression of a dominant-negative mutant allows IL-3–dependent FL5.12 cells to survive in the absence of growth factor (Edinger et al., 2003). This result suggested that growth factor deprivation might lead to Rab7 activation. To test this idea, we evaluated Rab7 activity in growth factor withdrawn cells. In cell culture systems, growth factor withdrawal is sometimes simulated by depriving cells of serum. However, this is not an ideal approach. Serum is a complex mixture of growth factors, nutrients, vitamins, and other bioactive molecules. This means that, in addition to eliminating growth factors, serum withdrawal induces multiple, poorly defined forms of stress that complicate the interpretation of the results. To study the effects of growth factor deprivation in isolation, IL-3–dependent murine hematopoietic cell lines such as FL5.12 are often used. Removing IL-3 from the medium rapidly induces apoptosis in FL5.12 cells in the presence normal serum levels (McKearn et al., 1985; Nunez et al., 1990). We therefore chose to study the effects of growth factor signaling on Rab7 activity in FL5.12 cells.
Initially, we determined how growth factor withdrawal affected the subcellular localization of Rab7. Rab GTPases are recruited onto membranes as a part of their activation cycle; Rabs can only promote membrane fusion reactions when they are associated with membranes (Pfeffer and Aivazian, 2004; Grosshans et al., 2006). Thus, the relative distribution of Rab proteins between the cytosol and membranes is one indicator of their activation state. Because we have been unable to identify or develop antibodies to Rab7 that detect the endogenous protein by immunofluorescence microscopy, FL5.12 cells expressing a GFP-Rab7 fusion protein (Bucci et al., 2000) were generated. Cell lines expressing low levels of GFP-Rab7 were selected for these studies to minimize the chance that overexpression would cause aberrant localization of Rab7. GFP-Rab7 expressing FL5.12 cells were maintained in the presence or absence of IL-3, stained with LysoTracker Red (a fluorescent, acidotropic dye that selectively accumulates in cellular compartments of low pH), and the subcellular localization of Rab7 determined by fluorescence microscopy.
In the presence of IL-3, GFP-Rab7 was distributed both on membranes and diffusely in the cytosol (Figure 1A). In growth factor-deprived cells, in contrast, cytoplasmic staining was dramatically reduced and GFP-Rab7 only observed on lysosomal membranes. Importantly, over the time course of this experiment, the expression level of GFP-Rab7 was not altered by growth factor withdrawal (data not shown). These experiments also suggested that the membrane-associated Rab7 was active. In the presence of IL-3, many Rab7-positive, LysoTracker-negative vesicular structures were present (Figure 1A). These Rab7-positive vesicles that are not sufficiently acidified to retain LysoTracker Red are likely late endosomes. In IL-3–deprived cells, in contrast, all Rab7-positive structures were also LysoTracker-positive and thus are yellow in the merged image. The acidification of all Rab7-positive structures is consistent with an increased rate of late endosome–lysosome fusion. Together, these experiments suggest that growth factor withdrawal increases Rab7 association with membranes and activation state.
Figure 1.

Rab7 membrane association increases upon growth factor withdrawal. (A) Cells expressing low levels of GFP-Rab7 were incubated in the presence or absence of growth factor for 17 h, stained with LysoTracker Red, and examined by fluorescence microscopy. (B) Cells expressing low levels of GFP-RILP were examined as described in A. Bar, 10 μm. Representative images from three to four independent experiments are shown.
To confirm that endogenous Rab7 behaved similarly to GFP-Rab7, we evaluated the localization pattern of the Rab7 effector protein RILP. RILP associates with late endosome and lysosomal membranes by binding to the GTP-bound form of Rab7 (Cantalupo et al., 2001; Jordens et al., 2001). Because we were also unable to generate antibodies that detect RILP by immunofluorescence, we used cells expressing low levels of a GFP-RILP fusion protein (Cantalupo et al., 2001). The results with GFP-RILP were even more striking than what we observed with GFP-Rab7. IL-3 withdrawal virtually eliminated the diffuse, cytosolic staining of GFP-RILP seen in the presence of growth factor (Figure 1B). After growth factor deprivation, GFP-RILP was instead localized solely to the membranes of LysoTracker Red-positive structures. Importantly, the total cellular amount of GFP-RILP was not affected by growth factor withdrawal (Figure 2A). Because RILP is only recruited onto late endosome and lysosomal membranes by active Rab7-GTP, it was expected that RILP-positive structures would also stain with LysoTracker Red. The lack of LysoTracker-negative, RILP-positive structures also indicates that RILP was localized exclusively to lysosomes. RILP is also an effector protein for Rab34 in the Golgi (Wang and Hong, 2002), but we detected no colocalization with the Golgi marker GM130 (data not shown). In summary, our microscopic analyses demonstrate that Rab7 is recruited onto membranes and activated by growth factor deprivation.
Figure 2.
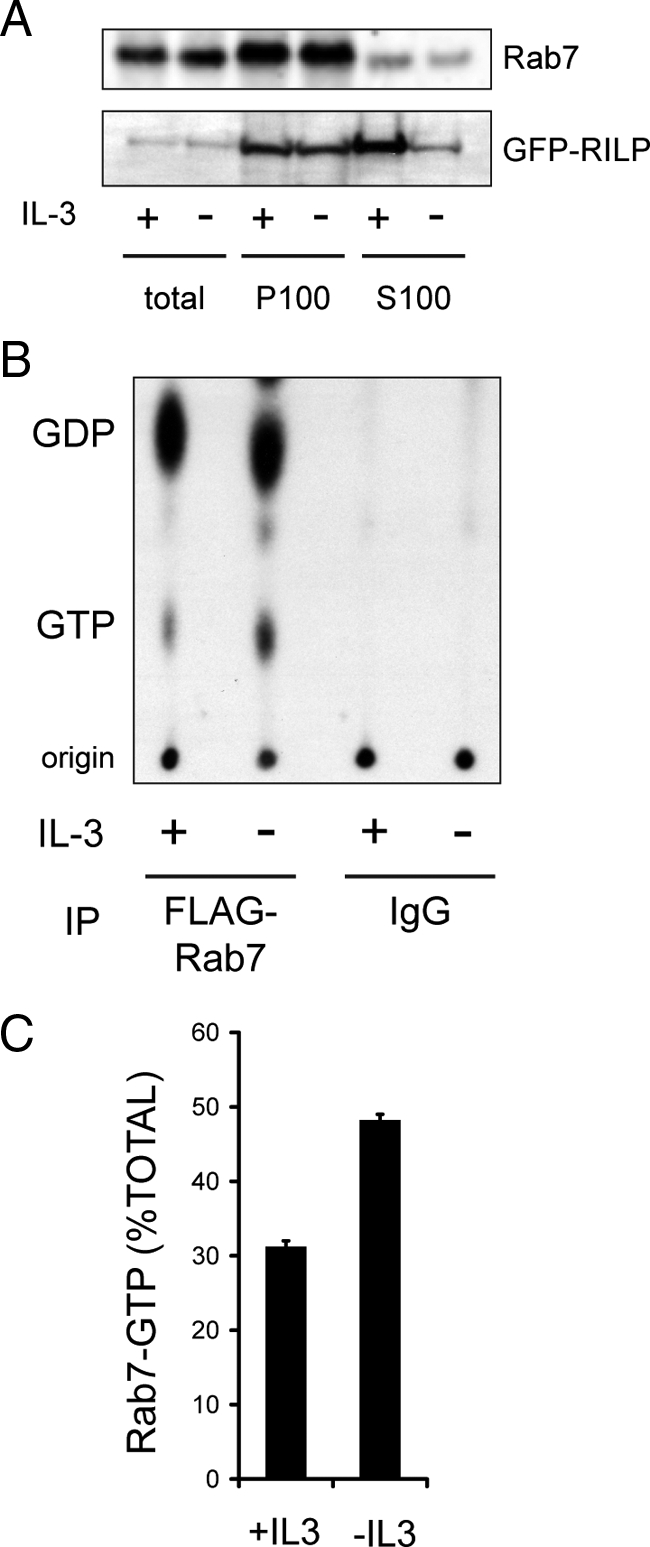
Rab7 is activated by growth factor withdrawal. (A) The localization of endogenous Rab7 or GFP-RILP was evaluated in FL5.12 cells after 18 h in the presence or absence of IL-3. An equal amount of protein (25 μg) was loaded in each lane; after ultracentrifugation, ∼90% of the total protein was in the S100 and ∼10% was present in the P100 fraction. The purity of cytosolic (S100) and membrane (P100) fractions was confirmed by evaluating the localization of tubulin (S100 only) and calnexin (P100 only). Cells expressed Bcl-XL to eliminate the confounding effects of apoptosis following growth factor deprivation. The results shown are representative of four independent experiments. (B) Cells expressing low levels of FLAG-Rab7 were maintained in or withdrawn from IL-3 for 15 h, labeled with [32P]orthophosphate, and Rab7 immunoprecipitates were evaluated for levels of [32P]GTP and [32P]GDP by TLC and autoradiography. Parallel samples were precipitated with nonspecific mouse IgG as a negative control. (C) The results in B were quantified using a PhosphorImager. The results shown are representative of two separate orthophosphate labeling experiments where GDP and GTP levels in immunoprecipitations were evaluated in triplicate TLC lanes. Error bars, SD.
Because endogenous Rab7 can be detected by Western blot, we also evaluated how growth factor availability affected the distribution of the endogenous protein by subcellular fractionation. Cells were separated into cytosolic (S100) and membrane (P100) fractions. In the presence of IL-3, Rab7 was present in both the S100 and P100 fractions (Figure 2A). This distribution was consistent with the results obtained by fluorescence microscopy (Figure 1). Following IL-3 withdrawal, a slight but reproducible decrease in the amount of Rab7 in the cytosolic S100 fraction was detected (Figure 2A). The amount of Rab7 in the total cell lysate and P100 fractions increased mildly or remained the same following IL-3 withdrawal in 4 independent experiments. As our RILP anti-serum is not sensitive enough to detect the endogenous RILP protein by Western blot, we examined the effect of growth factor withdrawal on the localization of GFP-RILP. Although no change in the levels of GFP-RILP in the P100 fraction was detected, the level of GFP-RILP in the S100 fraction declined after growth factor deprivation, suggesting that RILP was lost from the cytosol as it bound to GTP-Rab7 on lysosomal membranes. Thus, both fluorescence microscopy and subcellular fractionation studies suggest an increased association of Rab7 and its effector protein, RILP, with membranes after growth factor deprivation.
Growth Factor Withdrawal Increases the Fraction of Rab7 That Is Bound to GTP
The acidification of all Rab7-positive structures and the movement of RILP onto lysosomal membranes (Figure 1) suggested that Rab7-GTP levels were increased by growth factor withdrawal. To test this prediction, we evaluated Rab7-GTP levels directly in the presence and absence of IL-3. FL5.12 cells stably expressing low levels of FLAG-Rab7 were used to facilitate the immunoprecipitation of Rab7. Briefly, cells were maintained in the presence or absence of IL-3 for 15 h, labeled with [32P]orthophosphate for 3 h, FLAG-Rab7 was immunoprecipitated, and the associated GTP/GDP was eluted and separated by TLC using unlabeled GDP and GTP as standards. Parallel, nonspecific precipitations using mouse immunoglobulin confirmed that the [32P]GTP and [32P]GDP present in immunoprecipitates was bound to Rab7. Autoradiography (Figure 2B) and quantification using a PhosphorImager (Figure 2C) indicated that the fraction of Rab7 associated with GTP increased from 31% in IL-3 replete cells to 48% in IL-3–deprived cells. These results are consistent with our subcellular localization studies and in good accord with recent observations in HeLa cells (23% of Rab7 and 70% of the constitutively active Rab7 mutant Q67L was GTP bound) (Spinosa et al., 2008). Similar results have been obtained with Rab5 (21% of wild-type Rab5 and 63% of the activated mutant Rab5-Q79L was GTP-bound) (Stenmark et al., 1994). The yeast Rab Ypt1 is also primarily in the inactive form during log phase growth (28% GTP-bound) (Richardson et al., 1998). Therefore, our results are consistent with those published for other Rabs and suggest that IL-3 withdrawal increases the amount of Rab7 in the active, GTP-bound state.
Evaluating the nucleotide binding status of Rab7 in orthophosphate-labeled cells is technically challenging and requires large amounts of radioactivity. To facilitate additional studies of Rab7 nucleotide binding state, we developed an effector protein pull-down assay for Rab7 similar to those commonly used to measure the activity of other GTPases such as Ras (Taylor et al., 2001). Using the crystal structure of Rab7 bound to RILP as a guide (Wu et al., 2005), we fused GST to the Rab7 binding domain of RILP. To validate that this construct precipitates only GTP-bound Rab7, we determined whether it isolated constitutively GDP-bound (Rab7-T22N) and GTP-bound (Rab7-Q67L) Rab7 mutants from cell extracts. GST-RILP–coated beads precipitated Rab7-Q67L and wild-type Rab7 but not Rab7-T22N from cell lysates, although all three proteins were expressed at similar levels (Figure 3A). Thus, GST-RILP specifically binds to GTP-Rab7.
Figure 3.

GST-RILP is a specific probe for Rab7-GTP. (A) human embryonic kidney 293T cells were transfected with plasmids encoding GFP alone or GFP-tagged Rab7, Rab7-Q67L, or Rab7-T22N. The Rab7 binding domain of RILP coupled to GST (GST-RILP) or GST alone were added to cell lysates and used to precipitate the GTP-bound form of Rab7. A representative blot is shown from four independent experiments. (B) Endogenous Rab7-GTP was isolated as described in A from FL5.12 cells maintained in the presence or absence of IL-3 for 18 h. Cells expressed Bcl-XL to eliminate the confounding effects of cell death. Western blots were analyzed using the Odyssey Infrared Imaging System. The gel shown is representative of more than five independent experiments. (C) Quantification of the results shown in B. The amount of Rab7 detected in the GST-RILP pull-down was expressed as a fraction of the input and normalized to the +IL-3 sample. Error bars are SD of triplicate gels. (D) IL-3 was withdrawn from IL-3–dependent bone marrow cultures for 8 h and Rab7-GTP was isolated using GST-RILP–coated beads. Quantification of the results was performed as described in C. Results shown are representative of two independent experiments. (E) IL-2 was withdrawn from HT-2 cells for 8 h and Rab7-GTP was isolated using GST-RILP–coated beads. Quantification of the results in was performed as described in C. Results shown are representative of three independent experiments. All pull-downs were performed when cultures were 85% viable or greater.
We next used GST-RILP to evaluate the GTP binding state of endogenous Rab7 in growth factor-replete and -deprived FL5.12 cells. Consistent with results obtained by immunofluorescence microscopy (Figure 1), subcellular fractionation (Figure 2A), and direct GTP binding assays (Figure 2, B and C), growth factor withdrawal increased Rab7-GTP binding by approximately twofold (Figure 3, B and C). This increase occurred independently of growth factor withdrawal-induced apoptosis as Rab7 activation was observed in cells overexpressing the antiapoptotic protein Bcl-XL. We also tested whether IL-3 regulates Rab7 nucleotide binding state in primary murine bone marrow cells. IL-3 withdrawal from IL-3 dependent bone marrow cultures markedly increased Rab7-GTP binding levels before the induction of apoptosis (Figure 3D). To test whether growth factors other than IL-3 modulate Rab7-GTP binding status, we used the HT-2 murine helper T-cell line that is dependent on IL-2 for survival (Watson, 1979). IL-2 withdrawal from HT-2 cells also increased Rab7-GTP levels before cell death (Figure 3E). Together, the data presented in Figures 1–3 demonstrate that Rab7 is activated by growth factor withdrawal.
Rab7 Activation Can Induce Cell Death
We have previously shown that inactivating Rab7 protects cells from growth factor withdrawal-induced apoptosis (Edinger et al., 2003). In the context of our present findings, these results suggested to us that Rab7 activation by growth factor withdrawal might contribute to the induction of apoptosis. To test this idea, we generated cell lines stably expressing the GTP-bound mutant GFP-Rab7-Q67L. These cell lines did not display decreased viability relative to control lines expressing GFP alone (data not shown). We reasoned that cells might be able to adapt to increased Rab7 activity when GFP-Rab7-Q67L was constitutively expressed. Thus, we generated cell lines where GFP-Rab7-Q67L expression was regulated by a doxycycline-inducible promoter (Figure 4A). When GFP-Rab7-Q67L expression was induced by doxycycline addition, viability declined in a dose-dependent manner despite the presence of IL-3 in the medium (Figure 4B). As expected, doxycycline itself was not toxic to control cells. Rab7-Q67L induction killed cells apoptotically as determined by AnnexinV staining and by evaluating nuclear morphology (Figure 4, C and D). These results suggest that acute Rab7 activation by growth factor withdrawal contributes to the induction of apoptosis.
Figure 4.

Acute Rab7 activation can induce cell death. (A) FL5.12 cell lines expressing GFP-Rab7-Q67L in response to doxycycline addition were evaluated for GFP-Rab7-Q67L expression level by flow cytometry. The mean GFP-fluorescence from three independent experiments is graphed. Error bars, SD. (B) The viability of the cell lines shown in A was evaluated 48 h after doxycycline addition. Error bars, SD. Equivalent results were obtained in more than five independent experiments. (C) AnnexinV and DAPI staining was evaluated in inducible GFP-Rab7-Q67L cells maintained in the presence or absence of doxycycline for 48 h. Data shown are representative of three independent experiments. (D) Inducible GFP-Rab7-Q67L–expressing cells were treated with doxycycline or maintained without drug, stained with Hoechst33342 (H342) and propidium iodide (PI) at 48 h, and evaluated by fluorescence microscopy. Hoechst33342 stains the nuclei of all cells, whereas propidium iodide selectively stains dead cells.
Activating Rab7 Reverses the Growth Factor-independent Survival Seen in Cells with Reduced PKCδ Activity
If Rab7 activation after growth factor withdrawal helps to trigger apoptosis, we hypothesized that activating Rab7 might induce death in cells protected from growth factor withdrawal by alterations in growth factor receptor-dependent signal transduction. The tumor suppressor PKCδ is one of the most highly induced genes after growth factor withdrawal from FL5.12 cells (Gschwendt, 1999; Brachat et al., 2000; Jackson and Foster, 2004). We confirmed that PKCδ induction contributed to growth factor withdrawal-induced apoptosis under our experimental conditions. Using quantitative RT-PCR, we detected a dramatic induction of PKCδ mRNA after IL-3 deprivation (Figure 5A). PKCδ protein levels were also increased by growth factor withdrawal, although not to the same degree (Figure 5B). To determine whether PKCδ up-regulation contributed to apoptosis, we inhibited PKCδ by expressing a dominant-negative mutant or by targeting the protein for knockdown via RNAi. FL5.12 cells expressing DN-PKCδ (Figure 5C) were protected from death after IL-3 withdrawal (Figure 5D). Cell lines expressing shRNAmir targeting PKCδ were similarly protected (Figure 5, E and F). Thus, PKCδ induction contributes to growth factor withdrawal-induced apoptosis in FL5.12 cells.
Figure 5.

PKCδ induction contributes to growth factor withdrawal-induced cell death. (A) FL5.12 cells were maintained in the presence or absence of IL-3 for the indicated times. Total RNA was isolated and PKCδ mRNA levels determined by quantitative RT-PCR. Results are normalized to actin mRNA (levels of this transcript were minimally affected by growth factor withdrawal). Cells expressed Bcl-XL to prevent cell death. (B) FL5.12 cells expressing Bcl-XL were evaluated for PKCδ protein induction over time. (C) FL5.12 cells stably expressing HA-tagged DN-PKCδ were evaluated by Western blot. Cells expressing empty vector (VEC) were used as a control. (D) IL-3 was withdrawn from the cells in C for the indicated period, and cell viability determined by vital dye exclusion and flow cytometry. (E) FL5.12 cells stably expressing shRNAmir targeting PKCδ were evaluated for PKCδ expression by Western blot. Clones expressing two different hairpins targeting PKCδ are shown as well as two clones expressing VEC. (F) IL-3 was withdrawn from the cells in E, and cell viability was determined by vital dye exclusion and flow cytometry. Error bars, SD. In all cases, a representative experiment is shown where similar results were obtained in at least three independent experiments.
To determine whether Rab7-Q67L could reverse the growth factor-independent cell survival of cells with reduced PKCδ activity, we introduced GFP-Rab7-Q67L into the cell lines expressing dominant-negative PKCδ and PKCδ RNAi. Two clones expressing different levels of GFP-Rab7-Q67L (Figure 6A) but identical amounts of DN-PKCδ (Figure 6B) were evaluated. Although substantial growth factor-independent cell survival was observed in cells expressing DN-PKCδ, expression of GFP-Rab7-Q67L reversed this effect in a dose-dependent manner (Figure 6C). Similarly, GFP-Rab7-Q67L introduction into PKCδ RNAi-expressing cells (Figure 6, D and E) reversed growth factor-independent cell survival (Figure 6F). In PKCδ RNAi-expressing cells, higher levels of GFP-Rab7-Q67L expression were attained (Figure 6, A vs. D) consistent with the more complete reversal of growth factor-independent cell survival in PKCδ RNAi cell lines (Figure 6, C vs. F). From these experiments, we conclude that activating Rab7 reverses the growth factor-independent cell survival conferred by PKCδ inactivation.
Figure 6.

Rab7 activation reverses the growth factor-independent survival of PKCδ-deficient cells. (A) Cell lines stably expressing both DN-PKCδ and Rab7-Q67L were generated. GFP-Rab7-Q67L levels were determined by flow cytometry. The mean of three independent experiments is shown. Error bars, SD. (B) Equivalent DN-PKCδ expression was confirmed by Western blotting with anti-HA antibodies. (C) Growth factor withdrawal was performed on the clones evaluated in A and B. Cell viability was determined by vital dye exclusion and flow cytometry. A representative experiment of three is shown. Error bars, SD. (D) GFP-Rab7-Q67L was stably expressed the PKCδ RNAi-1 cell line (see Figure 5E). Mean GFP-Rab7-Q67L expression level from three experiments is expressed on the same scale as in Figure 7A. (E) PKCδ knockdown was confirmed in the clones evaluated in D by Western blotting for PKCδ. (F) The clones examined D and E were withdrawn from growth factor, and cell viability was determined by vital dye exclusion and flow cytometry. A representative experiment of three independent experiments is shown. Error bars, SD.
PKCδ Inhibition Fragments the Lysosome but Does Not Decrease Rab7-GTP Levels
The ability of Rab7-Q67L to kill PKCδ-deficient cells in the absence of growth factors suggested that reducing PKCδ activity might alter Rab7 activity. When Rab7 is inhibited, lysosomal fragmentation occurs (Figure 7, A and B; Edinger et al., 2003). This change in lysosomal morphology is readily detected in FL5.12 cells because, unlike many adherent cell lines, they have only three to five large lysosomes per cell. We stained cells with reduced PKCδ activity with LysoTracker Red and quantified the number of lysosomes per cell. Strikingly, cells expressing either dominant-negative PKCδ or shRNAmir targeting PKCδ exhibited lysosomal fragmentation (Figure 7, A and B). The degree of fragmentation was equivalent to that observed when Rab7 function was directly inhibited by expressing the dominant-negative mutant Rab7-T22N. This result is consistent with the proposal that PKCδ positively regulates Rab7-dependent lysosomal fusion reactions. We therefore tested whether PKCδ induction was required for the increase in Rab7-GTP levels associated with growth factor deprivation (Figures 2, B and C, and 3, B–E). Surprisingly, PKCδ knockdown produced a mild elevation in Rab7-GTP levels in the presence of IL-3 (Figure 7, C and D). The increase in Rab7-GTP that follows growth factor withdrawal was, however, reduced. This result indicates that the lysosomal fragmentation observed in cells with reduced PKCδ activity (Figure 7, A and B) is not the result of alterations in Rab7 nucleotide binding state.
Figure 7.

PKCδ inhibition fragments the lysosome without decreasing Rab7-GTP levels. (A) Lysosomal morphology was evaluated in FL5.12 cells expressing the indicated constructs using LysoTracker Red. RNAi-1 and -2 are expressing independent shRNAmir targeting PKCδ. (B) Quantification of the lysosomal fragmentation shown in A. Error bars, SEM. (C) Control cells and PKCδ shRNAmir-expressing cells were evaluated for Rab7-GTP levels by using GST-RILP in the presence of IL-3 or after 9 h of growth factor withdrawal. Control cells expressed Bcl-XL to prevent cell death; RNAi-expressing cells were fully viable at 9 h. Western blots were performed using the Odyssey gel imaging system. The results shown are representative of three independent experiments. (D) Quantification of results shown in C. Error bars, SD.
DISCUSSION
The data presented here establish that Rab7 activity is regulated by growth factor availability. Rab7 and its effector protein RILP shift from the cytosol onto lysosomal membranes after growth factor withdrawal (Figures 1 and 2A). The increased colocalization of GFP-Rab7 and LysoTracker Red in the absence of IL-3 (Figure 1A) suggests that this membrane-associated Rab7 is active. Increased Rab7-GTP levels in growth factor-deprived cells (Figures 2, B and C, and 3, B–E) further support this conclusion. Although this is the first demonstration that Rab7 activity is regulated by extrinsic signals, the activity of other Rab proteins is controlled by signal transduction cascades. For example, Rab5 association with GDI and the activity of the Rab5 GAP are regulated by mitogen-activated protein kinase and epidermal growth factor receptor signaling, respectively (Lanzetti et al., 2000; Cavalli et al., 2001). In contrast, Rab4 association with membranes but not GTP-binding is negatively regulated by the mitotic kinase, CDC2 (Bailly et al., 1991). Thus, Rab7 can now be added to the list of endocytic Rabs that are regulated by signal transduction cascades.
These studies also extend our previous work showing that inhibiting Rab7 blocks growth factor withdrawal-induced cell death by demonstrating for the first time that Rab7 activation can induce apoptosis. The finding that Rab7 activation can kill cells may have implications for human disease. Activating mutations in Rab7 have been associated with the sensory neuropathy Charcot-Marie-Tooth syndrome type 2B (Spinosa et al., 2008). Our present findings suggest that increased neuronal cell death might contribute to the pathogenesis of this form of the disease. Decreased Rab7 activity might contribute to the ability of tumor cells to survive in the absence of growth factors. Because the growth factor-independent survival seen in PKCδ-deficient cells can be reversed by Rab7 activation, it is also possible that leukemias or brain tumors with reduced PKCδ expression (Oncomine database; Rhodes et al., 2004) might be susceptible to Rab7 activation. PKCδ promotes apoptosis not only in response to growth factor withdrawal but also in cells exposed to gamma irradiation, UV-C irradiation, and chemotherapeutics (Matassa et al., 2001; Humphries et al., 2006). Thus, drugs that directly activate Rab7 might be useful in conjunction with these modalities. Whether activating Rab7 increases autophagy either directly or indirectly would also be interesting to determine. Autophagosome degradation depends on Rab7-mediated fusion with the lysosome. Disruptions in autophagy are involved in cancer, neurodegenerative diseases, and pathogen clearance (Levine and Kroemer, 2008; Mizushima et al., 2008) suggesting that alterations in Rab7 activation state could be important in these conditions as well. In summary, our present results demonstrate that Rab7 activation state is regulated, a finding with potential implications for multiple human diseases.
Why does a constitutively GTP-bound Rab7 mutant reverse the growth factor-independent cell survival of PKCδ-deficient cells if PKCδ inhibition does not decrease Rab7-GTP binding? The simplest explanation may be that parallel cell death pathways are activated by PKCδ and Rab7-Q67L. However, increased Rab7-GTP levels in PKCδ-deficient cells also do not rule out the possibility that PKCδ inactivation inhibits Rab7-dependent fusion reactions. For example, inactivating the Rab7 effector RILP dramatically increases Rab7-GTP levels while blocking Rab7-dependent lysosomal fusion (Cantalupo et al., 2001; Jordens et al., 2001; Peralta and Edinger, unpublished data). Because it contains an evolutionarily conserved PKCδ consensus phosphorylation site, we favor the hypothesis that PKCδ promotes lysosomal fusion through effects on Vps39. Although its precise function remains ambiguous, Vps39 promotes lysosomal fusion reactions in both yeast and mammalian cells (Raymond et al., 1992; Wada et al., 1992; Wurmser et al., 2000; Caplan et al., 2001; Poupon et al., 2003; Rink et al., 2005). Thus, PKCδ might facilitate lysosomal fusion reactions by phosphorylating and activating Vps39. Additional studies will be required to determine whether PKCδ promotes lysosomal fusion by activating Vps39 or by altering the activity of other substrates.
Regardless of the molecular mechanism involved, lysosomal fragmentation upon PKCδ inhibition might help to explain several phenotypes that have been documented in PKCδ−/− cells. PKCδ−/− cells exhibit defects in major histocompatibility complex class II and CD1d-mediated antigen presentation (Chen et al., 2004; Brutkiewicz et al., 2007), both processes that depend on normal lysosomal function (Bertram et al., 2002). The resistance of B cells from PKCδ knockout mice to growth factor withdrawal (Mecklenbrauker et al., 2002; Miyamoto et al., 2002) may also be due in part to disruptions in lysosomal trafficking (Edinger et al., 2003). Finally, lysosomal fragmentation might help to explain the susceptibility of PKCδ−/− macrophages to Listeria monocytogenes. PKCδ−/− macrophages produce increased levels of bacteriocidal nitric oxide, proinflammatory cytokines, and chemokines, yet fail to confine Listeria to phagosomes (Schwegmann et al., 2007). Our results suggest that, in the absence of PKCδ, Listeria-containing phagosomes may not fuse with lysosomes, allowing the organism to persist intracellularly (Brumell and Scidmore, 2007). The Rab7-dependent process of autophagy is an important mechanism for clearing intracellular pathogens (Gutierrez et al., 2004; Colombo, 2007). Thus, PKCδ might also facilitate clearance of Listeria and other intracellular pathogens by promoting the Rab7-dependent fusion of autophagosomes and lysosomes. Our results raise the interesting possibility that activating Rab7 might reverse these phenotypes associated with PKCδ deficiency or facilitate pathogen clearance from wild-type cells.
ACKNOWLEDGMENTS
We thank David Fruman for comments on the manuscript, Craig Walsh for advice and the HT-2 cells, and Brent Martin for excellent technical assistance. This work was supported by Public Health Service grants from the National Cancer Institute (K08 CA100526 to A.L.E. and F31 CA126494 to K.R.R.), by an American Cancer Society Institutional Research Grant to University of California–Irvine (ACS-IRG 98-279-04), and by a University of California Cancer Research Coordinating Committee Grant (to A.L.E.).
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-09-0911) on April 22, 2009.
REFERENCES
- Bailly E., McCaffrey M., Touchot N., Zahraoui A., Goud B., Bornens M. Phosphorylation of two small GTP-binding proteins of the Rab family by p34cdc2. Nature. 1991;350:715–718. doi: 10.1038/350715a0. [DOI] [PubMed] [Google Scholar]
- Bertram E. M., Hawley R. G., Watts T. H. Overexpression of rab7 enhances the kinetics of antigen processing and presentation with MHC class II molecules in B cells. Int. Immunol. 2002;14:309–318. doi: 10.1093/intimm/14.3.309. [DOI] [PubMed] [Google Scholar]
- Brachat A., Pierrat B., Brungger A., Heim J. Comparative microarray analysis of gene expression during apoptosis-induction by growth factor deprivation or protein kinase C inhibition. Oncogene. 2000;19:5073–5082. doi: 10.1038/sj.onc.1203882. [DOI] [PubMed] [Google Scholar]
- Brumell J. H., Scidmore M. A. Manipulation of rab GTPase function by intracellular bacterial pathogens. Microbiol. Mol. Biol. Rev. 2007;71:636–652. doi: 10.1128/MMBR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brutkiewicz R. R., Willard C. A., Gillett-Heacock K. K., Pawlak M. R., Bailey J. C., Khan M. A., Nagala M., Du W., Gervay-Hague J., Renukaradhya G. J. Protein kinase C delta is a critical regulator of CD1d-mediated antigen presentation. Eur. J. Immunol. 2007;37:2390–2395. doi: 10.1002/eji.200737124. [DOI] [PubMed] [Google Scholar]
- Bucci C., Thomsen P., Nicoziani P., McCarthy J., van Deurs B. Rab 7, a key to lysosome biogenesis. Mol. Biol. Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantalupo G., Alifano P., Roberti V., Bruni C. B., Bucci C. Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J. 2001;20:683–693. doi: 10.1093/emboj/20.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan S., Hartnell L. M., Aguilar R. C., Naslavsky N., Bonifacino J. S. Human Vam6p promotes lysosome clustering and fusion in vivo. J. Cell Biol. 2001;154:109–122. doi: 10.1083/jcb.200102142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli V., Vilbois F., Corti M., Marcote M. J., Tamura K., Karin M., Arkinstall S., Gruenberg J. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol. Cell. 2001;7:421–432. doi: 10.1016/s1097-2765(01)00189-7. [DOI] [PubMed] [Google Scholar]
- Chen Y. W., Lang M. L., Wade W. F. Protein kinase C-alpha and -delta are required for FcalphaR (CD89) trafficking to MHC class II compartments and FcalphaR-mediated antigen presentation. Traffic. 2004;5:577–594. doi: 10.1111/j.1600-0854.2004.00202.x. [DOI] [PubMed] [Google Scholar]
- Colombo M. I. Autophagy: a pathogen driven process. IUBMB Life. 2007;59:238–242. doi: 10.1080/15216540701230503. [DOI] [PubMed] [Google Scholar]
- Edinger A. L., Cinalli R. M., Thompson C. B. Rab7 prevents growth factor-independent survival by inhibiting cell-autonomous nutrient transporter expression. Dev. Cell. 2003;5:571–582. doi: 10.1016/s1534-5807(03)00291-0. [DOI] [PubMed] [Google Scholar]
- Edinger A. L., Thompson C. B. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell. 2002;13:2276–2288. doi: 10.1091/mbc.01-12-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edinger A. L., Thompson C. B. An activated mTOR mutant supports growth factor-independent, nutrient-dependent cell survival. Oncogene. 2004;23:5654–5663. doi: 10.1038/sj.onc.1207738. [DOI] [PubMed] [Google Scholar]
- Feng Y., Press B., Wandinger-Ness A. Rab 7, an important regulator of late endocytic membrane traffic. J. Cell Biol. 1995;131:1435–1452. doi: 10.1083/jcb.131.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox C. J., Hammerman P. S., Cinalli R. M., Master S. R., Chodosh L. A., Thompson C. B. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 2003;17:1841–1854. doi: 10.1101/gad.1105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans B. L., Ortiz D., Novick P. Rabs and their effectors: achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. USA. 2006;103:11821–11827. doi: 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwendt M. Protein kinase C delta. Eur. J. Biochem. 1999;259:555–564. doi: 10.1046/j.1432-1327.1999.00120.x. [DOI] [PubMed] [Google Scholar]
- Gutierrez M. G., Munafo D. B., Beron W., Colombo M. I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- Houlden H., King R. H., Muddle J. R., Warner T. T., Reilly M. M., Orrell R. W., Ginsberg L. A novel RAB7 mutation associated with ulcero-mutilating neuropathy. Ann. Neurol. 2004;56:586–590. doi: 10.1002/ana.20281. [DOI] [PubMed] [Google Scholar]
- Humphries M. J., Limesand K. H., Schneider J. C., Nakayama K. I., Anderson S. M., Reyland M. E. Suppression of apoptosis in the protein kinase Cδ null mouse in vivo. J. Biol. Chem. 2006;281:9728–9737. doi: 10.1074/jbc.M507851200. [DOI] [PubMed] [Google Scholar]
- Jackson D. N., Foster D. A. The enigmatic protein kinase Cdelta: complex roles in cell proliferation and survival. FASEB J. 2004;18:627–636. doi: 10.1096/fj.03-0979rev. [DOI] [PubMed] [Google Scholar]
- Jordens I., Fernandez-Borja M., Marsman M., Dusseljee S., Janssen L., Calafat J., Janssen H., Wubbolts R., Neefjes J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001;11:1680–1685. doi: 10.1016/s0960-9822(01)00531-0. [DOI] [PubMed] [Google Scholar]
- Kundu M., Thompson C. B. Autophagy: basic principles and relevance to disease. Annu. Rev. Pathol. 2008;3:427–455. doi: 10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- Lanzetti L., Rybin V., Malabarba M. G., Christoforidis S., Scita G., Zerial M., Di Fiore P. P. The Eps8 protein coordinates EGF receptor signalling through Rac and trafficking through Rab5. Nature. 2000;408:374–377. doi: 10.1038/35042605. [DOI] [PubMed] [Google Scholar]
- Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matassa A. A., Carpenter L., Biden T. J., Humphries M. J., Reyland M. E. PKCδ is required for mitochondrial-dependent apoptosis in salivary epithelial cells. J. Biol. Chem. 2001;276:29719–29728. doi: 10.1074/jbc.M100273200. [DOI] [PubMed] [Google Scholar]
- Mathew R., Karantza-Wadsworth V., White E. Role of autophagy in cancer. Nat. Rev. Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKearn J. P., McCubrey J., Fagg B. Enrichment of hematopoietic precursor cells and cloning of multipotential B-lymphocyte precursors. Proc. Natl. Acad. Sci. USA. 1985;82:7414–7418. doi: 10.1073/pnas.82.21.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecklenbrauker I., Saijo K., Zheng N. Y., Leitges M., Tarakhovsky A. Protein kinase Cdelta controls self-antigen-induced B-cell tolerance. Nature. 2002;416:860–865. doi: 10.1038/416860a. [DOI] [PubMed] [Google Scholar]
- Meggouh F., Bienfait H. M., Weterman M. A., de Visser M., Baas F. Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology. 2006;67:1476–1478. doi: 10.1212/01.wnl.0000240068.21499.f5. [DOI] [PubMed] [Google Scholar]
- Miyamoto A., Nakayama K., Imaki H., Hirose S., Jiang Y., Abe M., Tsukiyama T., Nagahama H., Ohno S., Hatakeyama S., Nakayama K. I. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature. 2002;416:865–869. doi: 10.1038/416865a. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez G., London L., Hockenbery D., Alexander M., McKearn J. P., Korsmeyer S. J. Deregulated Bcl-2 gene expression selectively prolongs survival of growth factor-deprived hemopoietic cell lines. J. Immunol. 1990;144:3602–3610. [PubMed] [Google Scholar]
- Pfeffer S., Aivazian D. Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 2004;5:886–896. doi: 10.1038/nrm1500. [DOI] [PubMed] [Google Scholar]
- Philips J. A., Porto M. C., Wang H., Rubin E. J., Perrimon N. ESCRT factors restrict mycobacterial growth. Proc. Natl. Acad. Sci. USA. 2008;105:3070–3075. doi: 10.1073/pnas.0707206105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plas D. R., Talapatra S., Edinger A. L., Rathmell J. C., Thompson C. B. Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J. Biol. Chem. 2001;276:12041–12048. doi: 10.1074/jbc.M010551200. [DOI] [PubMed] [Google Scholar]
- Poupon V., Stewart A., Gray S. R., Piper R. C., Luzio J. P. The role of mVps18p in clustering, fusion, and intracellular localization of late endocytic organelles. Mol. Biol. Cell. 2003;14:4015–4027. doi: 10.1091/mbc.E03-01-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff M. Social controls on cell survival and death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Raymond C. K., Howald-Stevenson I., Vater C. A., Stevens T. H. Morphological classification of the yeast vacuolar protein sorting mutants: evidence for a prevacuolar compartment in class E vps mutants. Mol. Biol. Cell. 1992;3:1389–1402. doi: 10.1091/mbc.3.12.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D. R., Yu J., Shanker K., Deshpande N., Varambally R., Ghosh D., Barrette T., Pandey A., Chinnaiyan A. M. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C. J., Jones S., Litt R. J., Segev N. GTP hydrolysis is not important for Ypt1 GTPase function in vesicular transport. Mol. Cell. Biol. 1998;18:827–838. doi: 10.1128/mcb.18.2.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink J., Ghigo E., Kalaidzidis Y., Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Schwegmann A., Guler R., Cutler A. J., Arendse B., Horsnell W. G., Flemming A., Kottmann A. H., Ryan G., Hide W., Leitges M., Seoighe C., Brombacher F. Protein kinase C delta is essential for optimal macrophage-mediated phagosomal containment of Listeria monocytogenes. Proc. Natl. Acad. Sci. USA. 2007;104:16251–16256. doi: 10.1073/pnas.0703496104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinosa M. R., Progida C., De Luca A., Colucci A. M., Alifano P., Bucci C. Functional characterization of Rab7 mutant proteins associated with Charcot-Marie-Tooth type 2B disease. J. Neurosci. 2008;28:1640–1648. doi: 10.1523/JNEUROSCI.3677-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H., Parton R. G., Steele-Mortimer O., Lutcke A., Gruenberg J., Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S. J., Resnick R. J., Shalloway D. Nonradioactive determination of Ras-GTP levels using activated ras interaction assay. Methods Enzymol. 2001;333:333–342. doi: 10.1016/s0076-6879(01)33067-7. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M. G., Chandel N. S., Schumacker P. T., Thompson C. B. Bcl-XL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol. Cell. 1999;3:159–167. doi: 10.1016/s1097-2765(00)80307-x. [DOI] [PubMed] [Google Scholar]
- Verhoeven K., De Jonghe P., Coen K., Verpoorten N., Auer-Grumbach M., Kwon J. M., FitzPatrick D., Schmedding E., De Vriendt E., Jacobs A., Van Gerwen V., Wagner K., Hartung H. P., Timmerman V. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003;72:722–727. doi: 10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitelli R., Santillo M., Lattero D., Chiariello M., Bifulco M., Bruni C. B., Bucci C. Role of the small GTPase Rab7 in the late endocytic pathway. J. Biol. Chem. 1997;272:4391–4397. doi: 10.1074/jbc.272.7.4391. [DOI] [PubMed] [Google Scholar]
- Wada Y., Ohsumi Y., Anraku Y. Genes for directing vacuolar morphogenesis in Saccharomyces cerevisiae. I. Isolation and characterization of two classes of vam mutants. J. Biol. Chem. 1992;267:18665–18670. [PubMed] [Google Scholar]
- Wang T., Hong W. Interorganellar regulation of lysosome positioning by the Golgi apparatus through Rab34 interaction with Rab-interacting lysosomal protein. Mol. Biol. Cell. 2002;13:4317–4332. doi: 10.1091/mbc.E02-05-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J. Continuous proliferation of murine antigen-specific helper T lymphocytes in culture. J. Exp. Med. 1979;150:1510–1519. doi: 10.1084/jem.150.6.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M., Wang T., Loh E., Hong W., Song H. Structural basis for recruitment of RILP by small GTPase Rab7. EMBO J. 2005;24:1491–1501. doi: 10.1038/sj.emboj.7600643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurmser A. E., Sato T. K., Emr S. D. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J. Cell Biol. 2000;151:551–562. doi: 10.1083/jcb.151.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkel S. S., Hurov K. E., Ong C., Abtahi F. M., Gross A., Korsmeyer S. J. A role for proapoptotic BID in the DNA-damage response. Cell. 2005;122:579–591. doi: 10.1016/j.cell.2005.06.022. [DOI] [PubMed] [Google Scholar]