

Abstract
Humanin (HN) inhibits neuronal death induced by various Alzheimer's disease (AD)-related insults via an unknown receptor on cell membranes. Our earlier study indicated that the activation of STAT3 was essential for HN-induced neuroprotection, suggesting that the HN receptor may belong to the cytokine receptor family. In this study, a series of loss-of-function tests indicated that gp130, the common subunit of receptors belonging to the IL-6 receptor family, was essential for HN-induced neuroprotection. Overexpression of ciliary neurotrophic factor receptor α (CNTFR) and/or the IL-27 receptor subunit, WSX-1, but not that of any other tested gp130-related receptor subunit, up-regulated HN binding to neuronal cells, whereas siRNA-mediated knockdown of endogenous CNTFR and/or WSX-1 reduced it. These results suggest that both CNTFR and WSX-1 may be also involved in HN binding to cells. Consistent with these results, loss-of-functions of CNTFR or WSX-1 in neuronal cells nullified their responsiveness to HN-mediated protection. In vitro–reconstituted binding assays showed that HN, but not the other control peptide, induced the hetero-oligomerization of CNTFR, WSX-1, and gp130. Together, these results indicate that HN protects neurons by binding to a complex or complexes involving CNTFR/WSX-1/gp130.
INTRODUCTION
Neuronal cell death is a prominent pathological feature of Alzheimer's disease (AD). Although the molecular mechanism underlying AD-related neuronal cell death remains elusive, it has been generally suggested that toxic amyloid-β peptides (Aβs) are closely linked to the AD-related neuronal cell death (Hardy and Selkoe, 2002). In support of this hypothesis, elevated levels of toxic Aβs have been shown to result in neuronal cell death in vitro (Yankner et al., 1989). In addition, several groups have shown that enforced expression of familial AD-linked mutant Aβ precursor protein (APP) and presenilin (PS) genes causes neuronal cell death in vitro (Wolozin et al., 1996; Yamatsuji et al., 1996; Zhao et al., 1997; Guo et al., 1999; Hashimoto et al., 2000; Nevé et al., 2000; McPhie et al., 2003; Laifenfeld et al., 2007).
Using a death-trap screening method, we identified a cDNA encoding the 24-amino acid peptide named Humanin (HN), from the remaining occipital lobe of an AD brain, that inhibits neuronal cell death induced by a London-type familial AD-related protein V642I-APP (Hashimoto et al., 2001a,b; Nishimoto et al., 2004; Matsuoka et al., 2006 for review). HN inhibits neuronal death induced in vitro by all AD-related neurotoxic insults examined to date, whereas it has limited neuroprotective activity against neurotoxic insults unrelated to AD (Hashimoto et al., 2001a,b; Nishimoto et al., 2004). Rattin, a rat homologue of HN, protects neurons from AD-related insults (Caricasole et al., 2002). Insulin-like growth factor-binding protein-3 enhances HN rescue activity from Aβ toxicity, whereas HN inhibited insulin-like growth factor-binding protein-3–induced apoptosis in human glioblastoma-A172 (Ikonen et al., 2003). In addition, multiple groups have demonstrated that HN derivatives inhibited memory impairment of AD model animals (Mamiya and Ukai, 2001; Krejcova et al., 2004; Chiba et al., 2005; Yamada et al., 2008), including transgenic mice expressing familial AD-causing genes (Chiba et al., 2009).
HN inhibits neuronal death from outside of cells via its putative receptor on the cell membrane (Hashimoto et al., 2001a,b; Yamagishi et al., 2003). A secretion-defective HN mutant L9R-HN does not show HN activity when expressed intracellularly by an expression vector, whereas a synthetic L9R-HN peptide suppresses AD-related neuronal death when added directly to the media (Hashimoto et al., 2001b). Independently, Guo et al. (2003) reported that HN inhibited Bax-mediated apoptosis by binding to Bax, suggesting that HN may suppress cell death through these two mechanisms. Ying et al. (2004) demonstrated that HN inhibited Aβ42-induced neurotoxicity by binding to G protein–coupled formylpeptide receptor-like-1 (FPRL-1) in PC12 pheochromocytoma cells and suggested that FPRL-1 is the receptor for HN in PC12 cells. Inconsistent with this report, our earlier study indicated that FPR2, the mouse orthologue of FPRL-1, was not essential for HN activity in F11 cells (Hashimoto et al., 2005b), suggesting the presence of a HN receptor other than FPRL-1 that mediates HN-induced neuroprotection. In addition, we found that the signal transducer and activator of transcription 3 (STAT3) is involved in HN activity (Hashimoto et al., 2005b).
Glycoprotein 130 (gp130) is a common component of cytokine receptors belonging to the interleukin-6 (IL-6) receptor family. Binding of IL-6 family cytokines to their receptor triggers intracellular signal cascades mediated by both Janus kinase (JAK)/STAT and RAS/mitogen-activated protein kinase (MAPK) signaling pathways (Taga and Kishimoto, 1997; Boulay et al., 2003). Another cytokine IL-27 that modifies Th-1 and Th-2 immunological responses (Yoshida et al., 2001; Pflanz et al., 2002; Villarino et al., 2004 for review) has been shown to transduce signals by binding to a WSX-1/gp130 receptor complex (Pflanz et al., 2004).
In this study, we demonstrate that HN transduces its signal by binding to an cytokine receptor complex or complexes involving CNTFR/WSX-1/gp130.
MATERIALS AND METHODS
Genes and Vectors
pcDNA3-V642I-APP was previously described (Hashimoto et al., 2001b). pCAG-human gp130, pCAG-human gp130 dn (dominant-negative) composed of the extracellular domain and the transmembrane domain of human gp130, and pCAG-mouse gp130 were as described (Fukada et al., 1996). C-terminally myc/6xHis-tagged human WSX-1 and mouse CREME9 cDNA were PCR-amplified from human and mouse embryo cDNAs (Biochain, Hayward, CA). A vector encoding C-terminally V5-tagged human CNTFR was purchased from Invitrogen (Carlsbad, CA). Rat IL-6Rα (pUCM18-rat IL-6R), purchased from American Type Culture Collection (Manassas, VA), was cloned into the pEF1/MycHis vector. A pcDNA3 vector encoding the extracellular domains (EDs) of human CNTFR (amino acids 1-390) fused in frame to the cDNA encoding the Fc region of human IgG and C-terminally tagged with 6xHis (CNTFR-ED-Fc-His) was as described (Fukada et al., 1996). A cDNA encoding the ED of mouse WSX-1 (amino acids 1-290) that contains the complete ligand-binding region was PCR-amplified with a sense primer (5′-CCGGAATTCACCATGAACCGGCTCCGGGTTGCACGC-3′) and an antisense primer (5′-CGGGGTACCGGAGACTAGGATAGGCTCTTCTACCTG-3′) and cloned into the mammalian expression vector pFLAG (WSX-1-ED-FLAG). A vector encoding p75NTR-FLAG was described previously (Tsukamoto et al., 2003; Hashimoto et al., 2004).
Cells, Gene Expression, Induction of Cell Death, and Administration of HN
F11 neurohybrid cells are described in detail in earlier studies (Hashimoto et al., 2001b). The transient transfection to F11 cells was performed by two partially different procedures. F11 cells, seeded at 7 × 104/well in six-well plates in Ham's F-12 plus 18% fetal bovine serum (FBS; HF-18%) for 12–16 h, were transfected with indicated vectors for 3 h in the absence of serum and were then incubated with HF-18% for 2 h. At 5 h after the onset of transfection, culture media were replaced by HF-10% containing HN or its derivative in association with or without neutralizing antibodies. At 72 h after the onset of transfection, cell mortality and viability assays were performed (method I; Hashimoto et al., 2001b; Yamagishi et al., 2003). Alternatively, at 5 h after the onset of transfection, culture media were replaced by HF-10% not containing HN or its derivative. At 24 h after the onset of transfection, they were subsequently replaced by Ham's F12 plus N2 supplement (Invitrogen) containing HN or its derivative in association with or without neutralizing antibodies. At 72 h after the onset of transfection, cell mortality and viability assays were performed (method II). By the method II, HN treatment was started at 24 h after the onset of transfection. HN, administered through either method, shows a basically similar death-suppressive effect.
SH-SY5Y cells were grown in DMEM/Ham's F12 mixture (DMEM/F12) containing 10% FBS. SH-SY5Y cells were seeded at 2 × 105/well in six-well plates for 12–16 h, transfected with indicated vectors for 3 h in the absence of serum, and then replated onto 96-well plates at 2 × 104/well in DMEM/F12–10%. After a 16-h incubation, they were coincubated with or without 1 μM Aβ42 in the presence or absence of 10 μM HN. Indicated neutralizing antibodies were added if indicated. At 48 h after the onset of transfection, cells were harvested for cell mortality and viability assays.
Cell viability was measured by WST-8 assays and calcein-staining assays (Hashimoto et al., 2001a,b, 2005a,b). The WST-8 assay, performed using Cell Counting kit-8 (Dojindo, Osaka, Japan), was based on the ability of cells to convert a water-soluble 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2, 4-disulfophenyl)-2H-tetrazolium monosodium salt into a water-soluble formazan. Cells were treated with WST-8 reagent for 2 h at 37°C and at 450-nm absorbance was measured. Calcein staining was performed as described (Bozyczko-Coyne et al., 1993). In brief, 6 μM calcein AM (3′,6′-di-(O-acetyl)-2′,7′-bis [N,N-bis (carboxymethyl) aminomethyl] fluorescein, tetraacetoxymethyl ester; Dojindo) was added to cells, and more than 30 min after calcein AM treatment, calcein-specific fluorescence (excitation, 485 nm; emission, 535 nm) was observed by fluorescence microscopy or measured by a spectrofluorometer (Wallac 1420 ARVOsx Multilabel Counter, PerkinElmer, Wellesley, MA).
Cell mortality was measured by trypan blue exclusion assay (Hashimoto et al., 2000, 2001b). Cells were suspended by pipetting gently, and 50 μl of 0.4% trypan blue solution (Sigma) was mixed with 200 μl of cell suspension (final trypan blue concentration was 0.08%) at room temperature. The extent of cell viability shows a good negative correlation to that of cell death measured by cell death assays.
Peptides and Recombinant Cytokines
Synthetic HN, S14G-HN (HNG), C8A-HN (HNA), Aβ42, Aβ43, colivelin, and activity-dependent neurotrophic factor (ADNF) peptides were purchased from Peptide Institute (Minoh, Osaka, Japan). Because there are no differences in nature between HN and HNG activities except that HNG is ∼1000-fold more potent than HN (Hashimoto et al., 2001a,b), HNG was used in place of HN in some experiments. HNA is a HN mutant without HN activity. An experiment indicated that HNA was unable to bind to the HN receptor on cell membranes (Hashimoto et al., 2001b). Recombinant mouse gp130-ED-Fc-6xHis, rat IL-6, and recombinant C-terminally 6xHis-tagged human IL-27p28 (Phe29-Pro243) N-terminally fused to human EBI3 (recombinant human IL-27) were purchased from R&D Systems (Minneapolis, MN). Rat CNTF was from PeproTech EC (London, United Kingdom).
Antibodies
Two rabbit polyclonal antibodies to mouse WSX-1 and a rabbit polyclonal antibody to human WSX-1 were generated by immunization with synthetic peptides corresponding to the N-terminal 16-amino acid peptide of mouse WSX-1 (mWSX-1-N), the C-terminal 16-amino acid peptide of mouse WSX-1 (mWSX-1-C), and the N-terminal 16-amino acid peptide of human WSX-1 (hWSX-1), conjugated to keyhole limpet hemocyanin (Sigma-Aldrich, St. Louis, MO). Anti-phoshoSTAT3 (Tyr705), anti-myc monoclonal, and anti-V5 antibodies were purchased from Cell Signaling Technology (Beverly, MA), Biomol (Plymouth Meeting, PA), and Invitrogen, respectively. Antibodies to gp130 (C-20), CNTFRα (R-20), STAT3 (C-20), Bax (C-19), and anti-6xHis (H-15) were from Santa Cruz Biotechnology (Santa Cruz, CA). Neutralizing antibodies to mouse gp130, human gp130, and human CNTFR were purchased from R&D Systems. An antibody against APP (22C11) was from Chemicon (Temecula, CA).
HN-mediated Oligomerization of HN Receptor Subunits
To generate recombinant human CNTFR-ED-Fc-6xHis and mouse WSX-1-ED-FLAG, COS7 cells were transfected with a vector encoding each protein. At 48 h after transfection, conditioned media were harvested for precipitation with Ni NTA agarose (1:1 slurry; Invitrogen) or anti-FLAG antibody M2-conjugated agarose (1:1 slurry; Sigma-Aldrich). Bound CNTFR-ED-Fc-6xHis, washed with 10 mM imidazole solution, were eluted with 250 mM imidazole solution according to manufacturer's instructions. Eluted CNTFR-ED-Fc-6xHis was desalted by Zeba Desalting Column (Pierce, Rockford, IL) and then one-tenth volume of 10× PBS was added to the desalted protein solution. To induce the oligomerization, gp130-ED-Fc-6xHis (1 μg) and/or CNTFR-ED-Fc-6xHis (1 μg) were coincubated with WSX-1-ED-FLAG (estimated to be 5 μg) immobilized onto anti-FLAG mAb M2-conjugated agarose beads in 100 μl PBS containing 1% Brij 96 (Sigma-Aldrich) in the presence of a peptide at 37°C for 6 h. Oligomerized proteins were visualized by SDS-PAGE fractionation and immunoblot analysis after extensive washing with the same buffer.
For detection of the HN-induced dimerization between endogenous WSX-1 and gp130 in F11 cells, F11 cells were coincubated with HN or HNA for 60 min. A cross-linker, bis(sulfosuccinimidyl)suberate (1 mM; BS3; Pierce), was added during the last 30-min period of coincubation.
HN-binding Assay
Immunofluorescence-based binding assays without cell fixation were performed as described (Hashimoto et al., 2005b). In brief, cells were replated onto poly-l-lysine–coated 96-well plates (F11 cells, 7 × 103 cells/well) at 24 h after transfection. At 36 h after transfection, cells were incubated with indicated concentrations of biotin-labeled HN or biotin-labeled HNG in the presence or the absence of 100 μM of unlabeled HN or HNA. After 6 h of incubation at 37°C in 5% CO2, they were washed with PBS and stained with fluorescein-conjugated streptoavidin (Molecular Probes, Eugene, OR) in PBS for 2 h at 37°C. Immunofluorescence intensity was measured (excitation, 485 nm; emission, 535 nm) with a spectrofluorometer (1420 ARVOsx Multi Label Counter, Wallac).
Plasmid-based Small Interfering RNA and Real-Time PCR
Small interfering RNA (siRNA)-mediated gene knockdown and real-time PCR was performed according to the method as described (Sui et al., 2002, Kanekura et al., 2005). Plasmid vectors encoding siRNAs were described in detail in Supplementary Materials and Methods.
Detection of Tyr705-phosphorylated STAT3
F11 cells (2 × 105 cells in 6-cm dish) were transfected with a vector for 3 h in the absence of serum and then incubated with HF-18% for 2 h. Five hours after the onset of transfection, culture media were changed to HF-10%. At 24 h after the onset of transfection, culture media were replaced by Ham's F12 medium containing N2 supplement without FBS. At 72 h, cells were treated with indicated cytokines for 15 min at 37°C. Cells were harvested with a buffer containing 1% NP-40, 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM sodium orthovanadate, and protease inhibitor cocktail Complete (Roche Diagnostics, Alameda, CA) for immunoblot analysis with anti-phosphoSTAT3 (Tyr705) antibody (Cell Signaling) and total anti-STAT3 antibody (Santa Cruz Biotechnology).
Preparation of HN- and HNA-Sepharose and Pulldown Assays
Conjugation of a synthetic HN or HNA peptide with Sepharose 4B was performed according to the manufacturer's instruction (Amersham Pharmacia Biotech, Uppsala, Sweden). Briefly, 5 mg of a synthetic peptide was incubated with 3 ml of cyanogen bromide-activated Sepharose 4B in a coupling buffer (0.1 M NaHCO3 containing 0.5 M NaCl, pH 8.3) at 4°C overnight with constant rotation. Sepharose was then incubated in a blocking buffer (0.2 M glycine, pH 8.0) for 2 h at room temperature to eliminate nonspepcific binding. After blocking, Sepharose was washed with the coupling buffer, and a 0.1 M sodium acetate buffer (pH 4) containing 0.5 M NaCl. Sepharose 4B, conjugate with a peptide, was stored in the coupling buffer at 4°C.
F11 cells (7 × 104 cells/well in six-well plates), transfected with the V5-tagged human WSX-1-, human CNTFR-, or myc-tagged rat IL-6R–encoding plasmids, were harvested for pulldown assays using a lysis buffer (10 mM Tris-HCl, pH 7.5, containing 0.05% Tween 20, and protease inhibitor cocktail Complete; Roche, Manheim, Germany) at 48 h after transfection. For each pulldown assay, 100 μl lysate and 20 μl of 1:1 slurry of HN- or HNA-Sepharose was mixed.
Statistical Analyses
All cell-death experiments, cell viability experiments, binding experiments, and quantitative real-time PCR experiments were done (n = 3). All values in the figures of the in vitro studies are mean ± SD. Statistical analyses were carried out with Student's t test.
RESULTS
Involvement of gp130 and STAT3 in HN-mediated Neuroprotection
Activation of STAT3 is essential for HN activity (Hashimoto et al., 2005a). In agreement, the treatment of F11 neurohybrid cells with 10 μM HN or 100 nM S14G-HN (HNG), a 1000-fold potent derivative of HN (Hashimoto et al., 2001a,b) resulted in up-regulation of the Tyr705 phosphorylation level of STAT3 (representing activated STAT3; Figure 1A).
Figure 1.

gp130 is essential for HN-mediated protection against V642I-APP-induced death of F11 neurohybrid cells. (A) F11 cells, incubated in Ham's F12 medium containing N2 supplement without FBS for 48 h, were treated with 10 μM HN, 100 nM HNG, or 10 μM HNA at 37°C for 15 min and then harvested for immunoblot analysis with the phosphoSTAT3 (Tyr705) and total STAT3 antibodies. (B) F11 cells were transfected with 1.0 μg of the pcDNA3 vector (Vec) or pcDNA3-V642I-APP in association with 1.0 μg of the pCAG vector, pCAG-wild-type mouse gp130 (abbreviated wt), or pCAG-mouse gp130 extracellular domain plus transmembrane domain (dn). Death was induced by the method I in the presence or absence of HN (10 μM). Cell viability was determined with WST-8 assay at 72 h after transfection. Immunoblot analysis was done with an antibody to APP and gp130. Overexpression of wt-gp130 resulted in appearance of a degraded (or misfolded) gp130 product in addition to wt-gp130. (C) F11 cells were transfected with 1.0 μg of the pcDNA3 vector (Vec) or pcDNA3-V642I-APP in association with 1.0 μg of the pCAG vector (Vec) or pCAG-human wt-gp130, as indicated in B. HN (10 μM) and 10 μg of a neutralizing anti-mouse gp130 antibody (R&D Systems) or normal goat IgG were added at 5 h after the onset of transfection. Cell mortality was determined by trypan blue exclusion assay at 72 h after the onset of transfection. Immunoblot analysis was done with antibodies to APP and gp130 (Santa Cruz Biotechnology, C-20).
Several type I cytokines activate STAT3 by stimulating their specific receptors belonging to the IL-6 receptor family, which are composed of a common receptor subunit, gp130, and one or two unique subunits that are structurally similar to gp130 (Taga and Kishimoto 1997; Boulay et al., 2003; Villarino et al., 2004). Enforced expression of a dominant-negative gp130 mutant consisting of the extracellular domain and the transmembrane domain of human gp130 (dn) resulted in the complete inhibition of HN-mediated suppression of V642I-APP-induced death of F11 cells (Figure 1B). The addition of a neutralizing antibody to mouse gp130 inhibited the HN-mediated rescue of F11 cells from V642I-APP–induced death (Figure 1C). This inhibition was completely neutralized by simultaneous ectopic expression of human gp130 (Figure 1C) that was not recognized by the antibody (data not shown). All these results indicate that gp130 is essentially involved in HN-induced neuroprotection.
Previously, we established an Aβ42-induced neuronal death system using F11 cells ectopically expressing p75NTR. Using this system, we showed that p75NTR behaved as a receptor for Aβ42-mediated neuronal death and that HN inhibited Aβ42-induced neuronal death (Tsukamoto et al., 2003; Hashimoto et al., 2004). In addition, we found that the addition of Aβ42 similarly induced death of SH-SY5Y cells (human neuroblastoma cells) expressing p75NTR and that HN inhibited this Aβ42-induced death of SH-SY5Y cells expressing p75NTR (unpublished data). Using the latter system, we examined whether HN inhibited Aβ42 neurotoxicity via the same gp130-meditaed pathway. As shown in Figure 2, A and B, Aβ42 treatment induced marked reduction in the cell viability of SH-SY5Y cells expressing p75NTR (Figure 2C), but not in SH-SY5Y cells lacking p75NTR, indicating that Aβ42 treatment induced death of SH-SY5Y cells via p75NTR. HN protected SH-SY5Y cells from this Aβ42-induced death. We found that the addition of neutralizing antibody to human gp130, but not control IgG, inhibited HN-induced rescue from Aβ42 toxicity (Figure 2, A and B). In addition, we found that the addition of the recombinant extracellular domain of the human gp130, which was thought to be a dominant-negative gp130, nullified HN rescue activity in SH-SY5Y cells (data not shown). Together, these results strongly supports the idea that gp130 is essential for HN activity in these cells. Further, calcein fluorescence analysis indicated that HN significantly increased calcein fluorescence levels of SH-SY5Y cells only in the absence of Aβ42 (Figure 2B). This result also suggests that HN may have a slight but statistically significant potentiating effect on growth of SH-SY5Y cells.
Figure 2.

gp130 is essential for HN-mediated protection against Aβ42-induced death of SH-SY5Y cells expressing p75NTR. Human SH-SY5Y cells, transfected with 1.0 μg of the pFLAG vector (Vec) or pFLAG-human p75NTR, were replated onto 96-well dished at 3 h after the onset of transfection. At 24 h after the start of transfection, they were coincubated with 1 μM Aβ42 together with or without 10 μM HN in the presence of 1 μL of 1 mg/ml neutralizing mouse mAb to human gp130 or control mouse IgG. At 48 h after the start of transfection, representative microscopic pictures of the calcein-stained cells were taken (A) and cell viability was determined with calcein-staining assays (B). Immunoblot analysis was done with an antibody to FLAG for the detection of p75NTR-FLAG (C). DDW: distilled water. * p < 0.05; ** p < 0.01; *** p < 0.001; n.s., not significant.
HN Binds to CNTFR and the IL-27 Receptor WSX-1
We examined whether treatment with known IL-6 family cytokines was able to mimic HN-mediated neuroprotection. Treatment with mouse cardiotropin-1, rat IL-6, mouse IL-11, human oncostatin M, mouse leukemia-inhibitory factor (LIF), or human CNTF, at effective concentrations (100 ng/ml), did not inhibit V642I-APP–induced death (data not shown).
Receptors belonging to the IL-6 receptor family have one or two subunits other than gp130. To further identify HN receptor subunits other than gp130, we performed a series of immunofluorescence-based binding assays (Hashimoto et al., 2005) using biotin-labeled HN or HNG after a series of known gp130-coupling proteins, and WSXWS-motif–containing putative gp130-coupling proteins were separately overexpressed together with human gp130 in F11 cells. As shown in Figure 3A, overexpression of the human IL-27 receptor, WSX-1, which appears to require gp130 as a coreceptor (Pflanz et al., 2004), or overexpression of human CNTFR in association with gp130 increased the binding of HN or HNG to F11 cells. In contrast, overexpression of CREME9, an uncharacterized putative gp130-coupled receptor (CRL4), with gp130 did not increase the binding of HN or HNG to F11 cells. We further observed that overexpression of both CNTFR and WSX-1 in association with gp130 resulted in a synergistic increase in the binding of both HN and HNG to F11 cells (Figure 3A).
Figure 3.

Enforced expression of CNTFR or WSX-1 in F11 cells increases the association between HN and F11 cells. (A) F11 cells were cotransfected with pEF1/MycHis-mouse CREME9 (0.5 μg) plus the pcDNA3 vector (0.5 μg) plus pCAG-human gp130 (1.0 μg), pEF1/MycHis-human WSX-1 (0.5 μg) plus the pcDNA3 vector (0.5 μg) plus pCAG-human gp130 (1.0 μg), the pEF1/MycHis vector (0.5 μg) plus pcDNA3.1/GS-human CNTFR (0.5 μg) plus pCAG-human gp130 (1.0 μg), or pEF1/MycHis-human WSX-1 (0.5 μg) plus pcDNA3.1/GS-human CNTFR (0.5 μg) plus pCAG-human gp130 (1.0 μg), for 3 h in the absence of serum. The cells were incubated in HF-18% thereafter. At 24 h after the onset of transfection, cells were replated onto poly-l-lysine–coated 96-well plates (7 × 103 cells/well). At 36 h after the start of transfection, they were used for immunofluorescence-based binding assay with 10 μM biotin-HN or 10 nM biotin-HNG. Immunoblot analysis was done with a mixture of an anti-human gp130 antibody and an anti-myc mAb (CREME9 and WSX-1), or an anti-V5 mAb (CNTFR). (B) F11 cells, cotransfected with 0.25 μg of the pcDNA3.1/GS vector, 0.25 μg of the pEF1/MycHis vector, and 0.5 μg of the pCAG vector (left panel) or F11 cells, cotransfected with 0.25 μg of pcDNA3.1/GS-human CNTFR, 0.25 μg of pEF1/MycHis-human WSX-1, and 0.5 μg of pCAG-human gp130 (right panel), were replated onto poly-l-lysine–coated 96-well plates at 24 h after transfection. After 12 h of incubation, they were used for immunofluorescence-based binding assay with stepwise increasing concentrations of biotin-HN in the presence or absence of 100 μM of unlabeled HN or HNA as a competitor.
Binding assays with stepwise increasing concentrations of biotin-HN (Figure 3B) indicated that the fluorescence intensity representing the association between HN and the endogenous HN receptors increased in a manner dependent on the HN concentration, and this increase was largely inhibited by an excess amount of unlabeled HN, but not by that of C8A-HN (HNA), an HN derivative without HN activity (Hashimoto et al., 2001 a and b). The dissociation constant (Kd; EC50) of the association between HN and the HN receptor was estimated to be 1–10 μM. This finding is consistent with accumulated biochemical data indicating that HN shows its full neuroprotective activity at a concentration of 10 μM (Hashimoto et al., 2001b). We observed that the Kd of the association between HNG and the HN receptor was estimated to be 1–10 nM (data not shown), confirming that HNG is a 1000-fold more potent HN derivative. In addition, we examined the association between HN and F11 cells overexpressing CNTFR/WSX-1/gp130 (Figure 3B, right panel). Immunofluorescence intensity numbers were sevenfold up-regulated by overexpression of CNTFR, WSX-1, and gp130. We found that HN bound to the F11 cells overexpressing CNTFR/WSX-1/gp130 with an approximate Kd of 1–10 μM that was similar to that for the association between HN and the endogenous HN receptor. This finding indirectly supports the idea that CNTFR and WSX-1 in association with gp130 constitute the HN receptor.
Using in vitro pulldown assays with HN (or HNA) immobilized onto Sepharose 4B beads, we also found that HN coprecipitated with CNTFR and WSX-1, but did not coprecipitate with IL-6Rα, ectopically expressed in COS7 cells (Figure 4). In contrast, HNA did not coprecipitate with CNTFR, WSX-1, or IL-6Rα (Figure 4). These results confirm that HN binds to CNTFR and WSX-1 in a specific manner in vitro.
Figure 4.
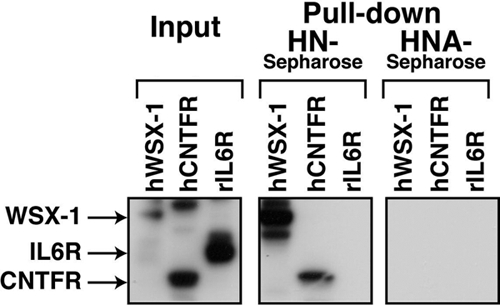
HN binds to WSX-1 and CNTFR. F11 cells were transfected with 0.5 μg of pEF1/MycHis-rat IL-6R, pcDNA3.1/GS-human CNTFR, or pEF1/MycHis-human WSX-1. At 48 h after transfection, cells were harvested for pulldown assays with HN or HNA-conjugated Sepharose4B. Precipitants were subjected to immunoblot analysis with a mixture of an anti-myc mAb to detect myc-tagged rat IL-6Rα and human WSX-1, and an anti-V5 mAb to detect V5-tagged human CNTFR.
Both HN Rescue Activity and HN Binding to F11 Cells Are Inhibited by the Loss of Function of Endogenous CNTFR or WSX-1
To examine the involvement of CNTFR or WSX-1 in HN-induced neuroprotection, we generated plasmid-based siRNAs that knocked down endogenous expression of both proteins (Sui et al., 2002). WSX-1 is expressed in neuronal cells including F11 cells (Supplementary Figure S1A). The efficacy of the siRNAs was tested by the measurement of mRNA levels using quantitative real-time PCR and of protein levels with immunoblot analysis (Supplementary Figure S1B). It was also confirmed by the finding that enforced expression of CNTFR or WSX-1 siRNA inhibited the recombinant CNTF-induced or the IL-27-induced Tyr705 phosphorylation of STAT3, respectively (Figure 5A). Consistent with the hypothesis that CNTFR and WSX-1 are involved in HN binding to F11 cells, the siRNA-mediated reduction of endogenous WSX-1 or CNTFR expression attenuated HNG-induced Tyr705 phosphorylation of STAT3 (Figure 5A). This result suggests that CNTFR and WSX-1 are essential for HN activity. In agreement, we further found that the siRNA-mediated knockdown of endogenous WSX-1 was effective in suppressing the HN- or HNG-induced protection of F11 cells from death induced by V642I-APP (Figure 5B). Similarly, the siRNA-mediated reduction of endogenous CNTFR attenuated HN or HNG activity to ∼20% of the control levels (Figure 5B), suggesting that the siRNA-mediated reduction of endogenous CNTFR may not be sufficient to suppress the HN or HNG activity perfectly. On the other hand, the knockdown of FPR2, the mouse homologue of FPRL-1, did not reduce HN activity (Figure 5B), consistent with the results in our earlier study (Hashimoto et al., 2005a).
Figure 5.

WSX-1 and CNTFR are essential for HN activity in F11 cells. (A) F11 cells were transfected with 0.5 μg of the pRNA-U6.1/Shuttle vector (indicated Vec) or the vector encoding mCNTFR siRNA, mWSX-1 siRNA, or mFPR2 siRNA. Cells were thereafter incubated in HF-18%. In some experiments, F11 cells were cotransfected with 0.5 μg of mCNTFR siRNA- and 0.5 μg of mWSX-1 siRNA-encoding vectors. Total amounts of transfected vectors were kept at 1.0 μg by the addition of appropriate amounts of the backbone vector. At 24 h after the onset of transfection, culture media were replaced by Ham's F12 media containing N2 supplement without FBS. At 72 h after the onset of transfection, cells were incubated with 100 nM HNG, 100 ng/ml human CNTF, or 1 μM human IL-27 at 37°C for 15 min and then harvested for immunoblot analysis with the anti-phosphoSTAT3 (Tyr705) and anti-total STAT3 antibodies. (B) F11 cells were cotransfected with 0.5 μg of the pcDNA3 vector (Vector) or pcDNA3-V642I APP together with 0.5 μg of the pRNA-U6.1/Shuttle vector (indicated empty) or a vector encoding mWSX-1 siRNA, mouse CNTFR siRNA, or mFPR2 siRNA. Cell death was induced by the method II with or without 10 μM HN or 10 nM HNG. WST-8 assays were performed at 72 h after the onset of transfection. Immunoblot analysis was done with the antibody to APP (bottom panel).
In addition, we found that a neutralizing antibody to human CNTFR or a polyclonal antibody to the N-terminal 16-amino acid peptide of human WSX-1 blocked HN-induced inhibition of Aβ42 toxicity in SH-SY5Y cells (Figure 6, A and B), which expresses WSX-1 and CNTFR (data not shown). These results indicate that CNTFR or WSX-1 are essential for HN-induced rescue of human neuroblastoma cells from Aβ42 toxicity.
Figure 6.

WSX-1 and CNTFR are essential for HN-mediated protection against Aβ42-induced death of SH-SY5Y cells expressing p75NTR. SH-SY5Y cells, transfected with 1.0 μg of the pFLAG vector (Vec) or pFLAG-human p75NTR, were replated onto 96-well dishes at 3 h after transfection. At 24 h after the start of transfection, they were coincubated with 1 μM Aβ42 together with or without 10 μM HN. One microliter of 1 mg/ml neutralizing goat polyclonal antibody mAb to human CNTFR or control goat IgG was added in A, and 1 μL of rabbit polyclonal antibody to the N-terminal 16 peptide of human WSX-1 or preimmune serum was added in B. At 48 h after the start of transfection, the cell viability was determined with calcein-staining assays. Microscopic pictures of the cells were also taken. DDW, distilled water. * p < 0.05; ** p < 0.01; *** p < 0.001; n.s., not significant.
HN-binding assays with F11 cells showed that the knockdown of endogenous CNTFR or WSX-1 significantly reduced the association between HN and F11 cells, whereas the knockdown of FPR2 did not reduce it significantly (Figure 7). The knockdown of both CNTFR and WSX-1 down-regulated the level of the binding of HNG to F11 cells to ∼20% of the control.
Figure 7.

Knockdown of WSX-1 and/or CNTFR reduces the association between HNG and F11 cells. F11 cells were transfected with 0.5 μg of the pRNA-U6.1/Shuttle vector or a vector encoding mWSX-1 siRNA, mCNTFR siRNA, mFPR2 siRNA, or mLIFR siRNA 3 h in the absence of serum and were thereafter incubated with HF-18%. In an experiment, 0.5 μg of mWSX-1 siRNA and mCNTFR siRNA-encoding vectors were cotransfected. Total amounts of transfected vectors were kept at 1.0 μg by the addition of appropriate amounts of the vector. At 24 h after transfection, cells were replated to poly-l-lysine–coated 96-well plates. At 36 h after the start of transfection, they were used for immunofluorescence-based HN-binding assay with indicated concentrations of biotin-HNG together with or without 100 nM of unlabeled HNG.
Using siRNAs that knock down mouse IL-6R and leukemia-inhibitory factor receptor (LIFR) specifically, we further found that the IL-6– or CNTF-induced phosphorylation of STAT3, but not the HNG-induced phosphorylation of STAT3, was inhibited by siRNA-mediated knockdown of endogenous expression of IL-6R or LIFR (Supplementary Figure S2). We also found that the knockdown of LIFR did not significantly reduce the binding of HNG to F11 cells (Figure 7). These results do not support a model in which IL-6R or LIFR is a component of the HN receptor.
Complex Formation of HN Receptor Subunits
Taking the above results together, we hypothesized that the HN receptor might be a hetero-oligomer (or hetero-oligomers) composed of CNTFR/WSX-1/gp130. To examine whether this hypothesis is true, we tried to show the HN-dependent association of recombinant EDs of these three components that contain regions essential for ligand binding in vitro. After coincubating three recombinant C-terminally tagged ED proteins in the presence of HN, HNG, or ADNF (Chiba et al., 2005) as a negative control at various concentrations, we performed pulldown analysis. The results showed that only in the presence of 10 μM HN, 100 nM HNG, or 10 μM HNG, human gp130-ED-Fc-6xHis as well as human CNTFR-ED-Fc-6xHis were coimmunoprecipitated with mouse WSX-1-ED-FLAG that were immobilized onto anti-FLAG mAb M2-conjugated agarose beads (Figure 8A). Using another coimmunoprecipitation assay, in which CNTFR-ED/Fc/6xHis or gp130-ED/Fc/6xHis was mixed with mouse WSX-1-ED-FLAG immobilized on the beads in the presence or the absence of 10 μM HN, we observed that the dimerization between WSX-1 and CNTFR, but not the dimerization between WSX-1 and gp130, was induced by HN treatment in vitro (Figure 8B). These results suggest that HN induced the dimerization between WSX-1 and CNTFR initially and, then, induced the oligomerization of the three subunits. We confirmed this finding by performing similar pulldown experiments using CNTFR-ED-FLAG instead of WSX-1-ED-FLAG and WSX-1-ED-Fc-6xHis instead of CNTFR-ED-Fc-6xHis (data not shown).
Figure 8.

Complex formation of the HN receptor subunits. (A and B) Human CNTFR-ED-Fc-6xHis (CNTFR- ED; 1 μg) and/or mouse gp130-ED-Fc-6xHis (gp130-ED; 1 μg) were coincubated with mouse WSX-1-ED-FLAG (WSX-1-ED; estimated to be 5 μg) beforehand immobilized onto M2 anti-FLAG antibody-conjugated agarose in 100 μl PBS containing 1% Brij 96 for 6 h at 37°C in the presence or absence of indicated concentrations of HN, HNG, or ADNF (A) or in the presence or absence of 10 μM HN or ADNF (B). Washed precipitates were then subjected to immunoblot analysis with anti-6xHis antibody (for CNTFR-ED and gp130-ED) or anti-FLAG (M2) antibody (for WSX-1-ED-FLAG). CNTFR-ED (0.5 μg), gp130-ED (0.5 μg), and WSX-1-ED-FLAG (2.5 μg) immobilized onto M2 anti-FLAG antibody-conjugated agarose were separately subjected to immunoblot analysis as inputs.
DISCUSSION
In the present study, our results show that gp130 and two gp130-related receptor subunits, CNTFR and WSX-1, are essential for cellular responsiveness to HN-mediated neuroprotection (Figures 1, 2, 5, and 6) and that CNTFR and WSX-1 mediate HN binding to neuronal cells (Figures 3 and 7). In vitro pulldown analysis also indicates that HN bind to CNTFR or WSX-1 (Figure 4). Finally, we demonstrate that HN treatment induced the dimerization between CNTFR and WSX-1 (Figure 8, A and B) as well as the dimerization between WSX-1 and gp130 (Figure 8B). Based on the finding that HN induced the dimerization between WSX-1 and gp130 only in the presence of CNTFR (Figure 8, A with B), it is likely that HN initially induces the dimerization between WSX-1 and CNTFR and then induces the hetero-trimerization of CNTFR/WSX-1/gp130, at least transiently. A receptor belonging to the IL-6 receptor family with three subunits has been already known. The receptor for CNTF consists of three subunits CNTFR/LIFR/gp130. Accordingly, it is not extraordinary that the HN receptor consists of three subunits, CNTFR/WSX-1/gp130. However, it is also possible that HN inhibits neuronal cell death by simultaneously interacting with two (or three) cytokine receptor dimers consisting of gp130/WSX-1, gp130/CNTFR, or CNTFR/WSX-1. Therefore, it remains to be experimentally proved whether HN induces the hetero-trimerization of CNTFR/WSX-1/gp130.
Using coimmunoprecipitation assays with the gp130 antibody, we further demonstrated that the addition of 10 μM HN induced the coprecipitation of endogenous WSX-1 with endogenous gp130 in F11 cells (Supplementary Figure S3). However, we were unable to show that HN induced the coprecipitation of endogenous CNTFR with gp130 (negative data not shown) possibly because the molecular size of CNTFR is nearly equal to the IgG heavy chain and CNTFR coimmunoprecipitated with gp130 is not clearly separated from the IgG heavy chain by immunoblot analysis. In addition, we were unable to confirm the HN-induced association between endogenous CNTFR and WSX-1 in F11 cells or other living cells (negative data not shown). This result may be due to the lack of appropriate antibodies that recognize WSX-1 or CNTFR in the CNTFR/WSX-1/gp130 complexes.
Taken together, these findings suggest that the HN receptor involved in the HN-mediated rescue of neuronal death is a novel receptor or receptor complexes composed of CNTFR/WSX-1/gp130. Currently, however, we cannot completely exclude two other possibilities regarding the nature of the HN receptor on the cell membrane. First, the HN receptor complex(es) may involve another unknown gp130-related coreceptor subunit in addition to CNTFR, WSX-1, and gp130. Second, in addition to the complex or complexes composed of CNTFR/WSX-1/gp130, HN binds to different types of receptor(s) and transduces distinct neuroprotective signals that are simultaneously essential for HN activity. These possibilities require future investigation.
Gp130 and CNTFR have been shown to be expressed ubiquitously in neuronal cells. WSX-1 is expressed in all neuronal cells examined including F11 cells (Supplementary Figure S1), SH-SY5Y cells (data not shown), and primary cortical neurons (data not shown). These observations are consistent with our hypothesis that a hetero-oligomer or hetero-oligomers composed of WSX-1/CNTFR/gp130 is involved in the HN signaling in neuronal cells. CNTFR, but not CNTF, knockout mice suffer from motor neuronal death (DeChiara et al., 1995). Combined with the fact that HN inhibits some types of motor neuronal death (unpublished observation), this finding suggests that CNTFR is a component of the HN receptor complex.
FPRL-1 has been reported to be a HN receptor that mediates the toxicity by Aβ42 in PC12 cells (Ying et al., 2004). This report suggests that different mechanisms may underlie the HN neuroprotective activity in different cells. Although the results of our preliminary experiment has indicated that overexpression of dominant-negative STAT3 almost completely nullifies HN-mediated rescue of death of PC12 cells induced by Aβ42 (unpublished observation), it remains possible that HN binding to FPR2 or a related molecule activates the gp130/STAT3 signaling axis indirectly (cross-talk).
Bax has been reported to be another HN receptor mediating HN anti–cell death activity (Guo et al., 2003). This finding suggests that HN may exhibit its neuroprotective activity both by binding to the cell-membrane HN receptor complex and by intracellularly binding to Bax. We found that siRNA-mediated knockdown of endogenous Bax did not reduce the binding of HN to F11 cells (Supplementary Figure S4). Although this result indicates that a major portion of extracellularly added HN binds to the membrane HN receptor, a minor portion of HN may enter the cytoplasm and contributed to the inhibition of neuronal cell death. Unfortunately, we were unable to examine whether Bax knockdown attenuated HN rescue activity because the knockdown of endogenous Bax directly attenuated AD-related neuronal death independently of HN rescue activity (unpublished observation).
Crystallographic analysis revealed that the stable complex of IL-6/IL-6R/gp130, formed by the binding of IL-6 to its receptor, is a hexamer containing two copies of each component (Boulanger and Garcia, 2004). The intracellular domains of “homodimerized” gp130 mediate IL-6 signals by interacting with JAKs. In contrast, CNTF, LIF, or cardiotropin-1 binding to its specific receptor results in heterodimerization between LIFR and gp130. Heterodimerized LIFR/gp130 mediates their signals. Similarly, HN-induced hetero-oligomerization of HN/CNTFR/WSX-1/gp130 may trigger signals via the intracytoplasmic domains of heterodimerized WSX-1 and gp130. Consistent with this possibility, increased doses of IL-27, which is believed to exert its activity by inducing heterodimerized WSX-1 and gp130 (Pflanz et al., 2004), mimicked HN-induced neuroprotection (unpublished observation). In addition, as mentioned above, HN induces the dimerization of CNTFR and WSX in the absence of gp130 (Figure 8B). This result suggests that putative “homodimerized WSX-1” may function as a signal transducer. In addition, considering that highly up-regulated homodimerization of gp130, induced by the addition of large amounts of IL-6 and soluble IL-6Rα, completely mimics HN activity (Niikura et al., 2004), we suggest that homodimerized gp130 may be another signaling transducer.
Type I cytokines, whose receptors belong to the IL-6 receptor family, contain four α helices consisting of 10–20 amino acids (Boulay et al., 2003), which are involved in interaction with their receptors. A structural study using NMR and circular dichroism spectroscopy has indicated that, in the less polar environment of 30% 2,2,2-trifluoroethanol solutions, HN, a 24-amino acid peptide, readily adopts a helical structure with a long-range order spanning residues Gly5 to Leu18, corresponding to the core domain of HN (Hashimoto et al., 2001a,b; Benaki et al., 2005). HN has been shown to be multimerized to be active (Terashita et al., 2003). Accordingly, it is hypothesized that near the cell membrane, multimerized HNs may acquire a four-helix structure, resembling type I cytokines, and bind to receptors belonging to the IL-6 receptor family on the cell membrane.
The activation of the gp130-STAT3 axis is essential for HN activity. However, because an IL-6 family cytokine simultaneously activates multiple signaling cascades including multiple JAK/STAT cascades as well as MAPK and other signal pathways by binding to a gp130-containing receptor (Fukada et al., 1996; Taga and Kishimoto, 1997), HN may activates multiple signaling pathways including negative and positive feedback cascades simultaneously. Treatment with HN, as well as IL-27, induces the up-regulation of p-STAT1 (Tyr701) levels (unpublished observation). To explain the specific neuroprotective activity of HN not fully mimicked by other cytokines (data not shown), we could currently hypothesize that HN-mediated orchestrated activation of multiple pathways including the JAK/STAT3 axis ultimately results in HN activity in neurons as a whole.
In conclusion, we provide evidence that HN or an HN-like molecule protects neurons from AD-related neuronal death by binding to the receptor complex(es) involving CNTFR/WSX-1/gp130.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to Drs. Etsuro Ogata, and Yoshiomi Tamai, and Yumi Tamai for indispensable support. We especially thank Takako Hiraki and Tomo Yoshida-Nishimoto for essential assistance. This work was supported in part by grants from the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-02-0168) on April 22, 2009.
REFERENCES
- Benaki D., Zikos C., Evangelou A., Livaniou E., Vlassi M., Mikros E., Pelecanou M. Solution structure of humanin, a peptide against Alzheimer's disease-related neurotoxicity. Biochem. Biophys. Res. Commun. 2005;329:152–160. doi: 10.1016/j.bbrc.2005.01.100. [DOI] [PubMed] [Google Scholar]
- Bozyczko-Coyne D., McKenna B. W., Connors T. J., Neff N. T. A rapid fluorometric assay to measure neuronal survival in vitro. J. Neurosci. Methods. 1993;50:205–216. doi: 10.1016/0165-0270(93)90009-g. [DOI] [PubMed] [Google Scholar]
- Boulanger M. J., Garcia K. C. Shared cytokine signaling receptors: structural insights from the gp130 system. Adv. Protein Chem. 2004;68:107–146. doi: 10.1016/S0065-3233(04)68004-1. [DOI] [PubMed] [Google Scholar]
- Boulay J-L, O'Shea J. J., Paul W. E. Molecular phylogeny within type I cytokines and their cognate receptors. Immunity. 2003;19:159–163. doi: 10.1016/s1074-7613(03)00211-5. [DOI] [PubMed] [Google Scholar]
- Caricasole A., Bruno V., Cappuccio I., Melchiorri D., Copani A., Nicoletti F. A novel rat gene encoding a Humanin-like peptide endowed with broad neuroprotective activity. FASEB J. 2002;16:1331–1333. doi: 10.1096/fj.02-0018fje. [DOI] [PubMed] [Google Scholar]
- Chiba T., Yamada M., Hashimoto Y., Sato M., Sasabe J., Kita Terashita K., Aiso S., Nishimoto I., Matsuoka M. Development of a femtomolar-acting Humanin derivative named Colivelin by attaching ADNF to its N terminus: characterization of Colivelin-mediated neuroprotection against Alzheimer's disease-relevant insults in vitro and in vivo. J. Neurosci. 2005;25:10252–10261. doi: 10.1523/JNEUROSCI.3348-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T., Yamada M., Sasabe J., Terashita K., Shimoda M., Matsuoka M., Aiso S. Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol. Psychiatr. 2009;14:206–222. doi: 10.1038/mp.2008.105. [DOI] [PubMed] [Google Scholar]
- DeChiara T. M., et al. Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell. 1995;83:313–322. doi: 10.1016/0092-8674(95)90172-8. [DOI] [PubMed] [Google Scholar]
- Fukada T., Hibi M., Yamanaka M., Takahashi-Tezuka M., Fujitani Y., Yamaguchi T., Nakajima K., Hirano T. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130 involvement of STAT3 in anti-apoptosis. Immunity. 1996;5:449–460. doi: 10.1016/s1074-7613(00)80501-4. [DOI] [PubMed] [Google Scholar]
- Guo B., Zhai D., Cabezas E., Welsh K., Nouraini S., Satterthwait A. C., Reed J. C. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423:456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- Guo Q., Fu W., Sopher B. L., Miller M. W., Ware C. B., Martin G. M., Mattson M. P. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat. Med. 1999;5:101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- Hardy J., Selkoe D. J. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Laifenfeld D., Patzek L. J., McPhie D. L., Chen Y., Levites Y., Cataldo A. M., Neve R. L. Rab5 mediates an amyloid precursor protein signaling pathway that leads to apoptosis. J. Neurosci. 2007;27:7141–7153. doi: 10.1523/JNEUROSCI.4599-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y., Niikura T., Ito Y., Nishimoto I. Multiple mechanisms underlie neurotoxicity by different types of Alzheimer's disease mutations of amyloid precursor protein. J. Biol. Chem. 2000;275:34541–34551. doi: 10.1074/jbc.M005332200. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y., Niikura T., Ito Y., Sudo H., Hata M., Arakawa E., Abe Y., Kita Y., Nishimoto I. Detailed characterization of neuroprotection by a rescue factor Humanin against various Alzheimer's disease-relevant insults. J. Neurosci. 2001a;21:9235–9245. doi: 10.1523/JNEUROSCI.21-23-09235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y., et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Aβ. Proc. Natl. Acad. Sci. USA. 2001b;98:6336–6341. doi: 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y., et al. Molecular characterization of neurohybrid cell death induced by Alzheimer's amyloid-beta peptides via p75NTR/PLAIDD. J Neurochem. 2004;90:549–558. doi: 10.1111/j.1471-4159.2004.02513.x. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y., Chiba T., Yamada M., Nawa M., Kanekura K., Suzuki H., Terashita K., Aiso S., Nishimoto I., Matsuoka M. Transforming growth factor β2 is a neuronal cell death-inducing ligand for amyloid-β precursor protein. Mol. Cell. Biol. 2005a;25:9304–9317. doi: 10.1128/MCB.25.21.9304-9317.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y., Suzuki H., Aiso S., Niikura T., Nishimoto I., Matsuoka M. Involvement of tyrosine kinases and STAT3 in Humanin-mediated neuroprotection. Life Sci. 2005b;77:3092–3104. doi: 10.1016/j.lfs.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Ikonen M., Liu B., Hashimoto Y., Ma L., Lee K. W., Niikura T., Nishimoto I., Cohen P. Interaction between the Alzheimer's survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc. Natl. Acad. Sci. USA. 2003;100:13042–13047. doi: 10.1073/pnas.2135111100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekura K., Hashimoto Y., Kita Y., Sasabe J., Aiso S., Nishimoto I., Matsuoka M. A Rac1/phosphatidylinositol 3-kinase/Akt3 anti-apoptotic pathway, triggered by AlsinLF, the product of the ALS2 gene, antagonizes Cu/Zn-superoxide dismutase (SOD1) mutant-induced motoneuronal cell death. J. Biol. Chem. 2005;280:4532–4543. doi: 10.1074/jbc.M410508200. [DOI] [PubMed] [Google Scholar]
- Krejcova G., Patocka J., Slaninova I. Effect of humanin analogues on experimentally induced impairment of spatial memory in rats. J. Pept. Sci. 2004;10:636–639. doi: 10.1002/psc.569. [DOI] [PubMed] [Google Scholar]
- Mamiya T., Ukai M. [Gly(14)]-Humanin improved the learning and memory impairment induced by scopolamine in vivo. Br. J. Pharmacol. 2001;134:1597–1599. doi: 10.1038/sj.bjp.0704429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M., Hashimoto Y., Aiso S., Nishimoto I. Humanin and colivelin: neuronal-death-suppressing peptides for Alzheimer's disease and amyotrophic lateral sclerosis. CNS Drug Rev. 2006;12:113–122. doi: 10.1111/j.1527-3458.2006.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhie D. L., Coopersmith R., Hines-Peralta A., Chen Y., Ivins K. J., Manly S. P., Kozlowski M. R., Neve K. A., Neve R. L. DNA synthesis and neuronal apoptosis caused by familial Alzheimer's disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J. Neurosci. 2003;23:6914–6927. doi: 10.1523/JNEUROSCI.23-17-06914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevé RL, McPhie D. L., Chen Y. Alzheimer's disease: a dysfunction of the amyloid precursor protein. Brain Res. 2000;886:54–66. doi: 10.1016/s0006-8993(00)02869-9. [DOI] [PubMed] [Google Scholar]
- Niikura T., Yamada M., Chiba T., Aiso S., Matsuoka M., Nishimoto I. Characterization of V642I-AβPP-induced cytotoxicity in primary neurons. J. Neurosci. Res. 2004;77:54–62. doi: 10.1002/jnr.20139. [DOI] [PubMed] [Google Scholar]
- Nishimoto I., Matsuoka M., Niikura T. Unravelling the role of Humanin. Trends Mol. Med. 2004;10:102–105. doi: 10.1016/j.molmed.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Pflanz S., et al. IL-27, a heterodimeric cytokine composed of EB13 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity. 2002;16:779–790. doi: 10.1016/s1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- Pflanz S., Hibbert L., Mattson J., Rosales R., Vaisberg E., Bazan J. F., Phillips J. H., McClanahan T. K., de Waal Malefyt R., Kastelein R. A. WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J Immunol. 2004;172:2225–2231. doi: 10.4049/jimmunol.172.4.2225. [DOI] [PubMed] [Google Scholar]
- Sui G., Soohoo C., Affar E. B., Gay F., Shi Y., Forrester W. C., Shi Y. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl. Acad. Sci. USA. 2002;99:5515–5520. doi: 10.1073/pnas.082117599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taga T., Kishimoto T. gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- Terashita K., et al. Two Ser residues distinctly regulate the rescue function of Humanin, an inhibiting factor of Alzheimer's disease-related neurotoxicity: functional potentiation by isomerization and dimerization. J. Neurochem. 2003;85:1521–1538. doi: 10.1046/j.1471-4159.2003.01797.x. [DOI] [PubMed] [Google Scholar]
- Tsukamoto E., Hashimoto Y., Kanekura K., Niikura T., Aiso S., Nishimoto I. Characterization of the toxic mechanism triggered by Alzheimer's amyloid-beta peptides via p75 neurotrophin receptor in neuronal hybrid cells. J. Neurosci. Res. 2003;73:627–636. doi: 10.1002/jnr.10703. [DOI] [PubMed] [Google Scholar]
- Villarino A. V., Huang E., Hunter C. A. Understanding the pro- and anti-inflammatory properties of IL-27. J. Immunol. 2004;173:715–720. doi: 10.4049/jimmunol.173.2.715. [DOI] [PubMed] [Google Scholar]
- Wolozin B., Iwasaki K., Vito P., Ganjei J. K., Lacana E., Sunderland T., Zhao B., Kusiak J. W., Wasco W., D'Adamio L. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- Yamada M., Chiba T., Sasabe J., Terashita K., Aiso S., Matsuoka M. Nasal colivelin treatment ameliorates memory impairment related to Alzheimer's disease. Neuropsychopharmacology. 2008;33:2020–2033. doi: 10.1038/sj.npp.1301591. [DOI] [PubMed] [Google Scholar]
- Yamagishi Y., Hashimoto Y., Niikura T., Nishimoto I. Identification of essential amino acids in Humanin, a neuroprotective factor against Alzheimer's disease-relevant insults. Peptides. 2003;24:585–595. doi: 10.1016/s0196-9781(03)00106-2. [DOI] [PubMed] [Google Scholar]
- Yamatsuji T., et al. G protein-mediated neuronal DNA fragmentation induced by familial Alzheimer's disease-associated mutants of APP. Science. 1996;272:1349–1352. doi: 10.1126/science.272.5266.1349. [DOI] [PubMed] [Google Scholar]
- Yankner B. A., Dawes L. R., Fisher S., Villa-Komaroff L., Oster-Granite M., Nevé R. L. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989;45:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- Ying G., Iribarren P., Zhou Y., Gong W., Zhang N., Yu Z. X., Le Y., Cui Y., Wang J. M. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 2004;172:7078–7085. doi: 10.4049/jimmunol.172.11.7078. [DOI] [PubMed] [Google Scholar]
- Yoshida H., et al. WSX-1 is required for the initiation of Th1 responses and resistance to L. major infection. Immunity. 2001;15:569–578. doi: 10.1016/s1074-7613(01)00206-0. [DOI] [PubMed] [Google Scholar]
- Zhao B., Chrest F. J., Horton W. E., Jr, Sisodia S. S., Kusiak J. W. Expression of mutant amyloid precursor proteins induces apoptosis in PC12 cells. J. Neurosci. Res. 1997;47:253–263. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.