

Abstract
During the life cycle of retroviruses, establishment of a productive infection requires stable joining of a DNA copy of the viral RNA genome into host cell chromosomes. Retroviruses are thus promising vectors for the efficient and stable delivery of genes in therapeutic protocols. Integration of retroviral DNA is catalyzed by the viral enzyme integrase (IN), and one salient feature of retroviral DNA integration is its lack of specificity, as many chromosomal sites can serve as targets for integration. Despite the promise for success in the clinic, one major drawback of the retrovirus-based vector is that any unintended insertion events from the therapy can potentially lead to deleterious effects in patients, as demonstrated by the development of malignancies in both animal and human studies. One approach to directing integration into predetermined DNA sites is fusing IN to a sequence-specific DNA-binding protein, which results in a bias of integration near the recognition site of the fusion partner. Encouraging results have been generated in vitro and in vivo using fusion protein constructs of human immunodeficiency virus type 1 IN and E2C, a designed polydactyl zinc-finger protein that specifically recognizes an 18-base pair DNA sequence. This review focuses on the method for preparing infectious virions containing the IN fusion proteins and on the quantitative PCR assays for determining integration site specificity. Efforts to engineer IN to recognize specific target DNA sequences within the genome may lead to development of effective retroviral vectors that can safely deliver gene-based therapeutics in a clinical setting.
Keywords: designed polydactyl zinc-finger protein, E2C, gene therapy, HIV-1 integrase, IN fusion proteins, insertional mutagenesis, quantitative PCR, retroviral vectors, retroviral DNA integration, site-directed integration
1. Introduction
Efficient transgene delivery is critical for genetic manipulation and therapeutic intervention of mammalian cells. Both virus- and non-virus-based vectors have been studied extensively in vitro and in animal models to optimize gene delivery and expression [1–3]. Among the virus-based vectors, one type of vehicle commonly employed in many gene therapy protocols is derived from retroviruses.
During the life cycle of retroviruses, the single-stranded viral RNA genome is reverse-transcribed into double-stranded DNA within the cytoplasm of a susceptible host cell. The viral cDNA is then transported to the nucleus and integrated into the host chromosome to form a provirus, which then allows the retrovirus to exploit the host cellular transcriptional and translational machineries for synthesizing new viral RNAs and proteins, ultimately producing progeny. Integration is therefore essential for retroviral replication, and results in the permanent insertion of the viral genome into the chromosome of the host cell [4]. The ability to permanently and stably insert transgenes and to maintain the transgene during cell division, as well as the efficiency of the integration reaction, are attractive features for developing retroviruses as gene delivery vehicles [3]. In addition, the ability of Lentiviruses, one of seven retroviral genera, to infect non-dividing cells is an appealing feature that is particularly suited for certain gene therapy applications, such as transduction of neurons or hematopoietic stem cells [5, 6].
Integration of a cDNA copy of the retroviral genome into the host cellular DNA is a highly ordered process, similar to reactions catalyzed by other members of the family of polynucleotidyl transferases [7, 8]. For integration to occur, two factors are absolutely required: the viral enzyme integrase (IN) and sequences present at the ends of the viral DNA genome [4, 9]. Although IN can specifically recognize and process viral DNA ends, host DNA that serves as a target substrate for integration shows little or no sequence consensus [10–13]. The non-specificity of retroviral DNA integration has been studied in further detail by recent genome-wide analyses of integration sites using different retroviruses in various cells types [14–17]. The mechanism of target site recognition and selection by IN is still not well understood, but one determining factor is the interaction between IN and host DNA-binding proteins [18].
Because the insertion events into chromosomes are largely non-specific, retroviral DNA integration is inherently mutagenic, and a wayward integration event can lead to disastrous consequences to the infected cell and host organism. Studies on murine mammary tumor virus and other slow-acting retroviruses establish that unregulated integration events can cause tumors either by directly activating proto-oncogenes or by producing a new chimeric protein product that stimulates cellular transformation [19–21]. In a gene therapy trial, four children suffering from X-linked severe combined immunodeficiency (SCID)-XI disease received autologous hematopoietic stem cells, which had been transduced ex vivo using a murine leukemia virus (MLV) vector bearing the gene for the γ-c chain cytokine receptor, have developed leukemia-like symptoms [22]. The observed complications in three of these four children stem from vector-mediated insertional mutagenesis events near the LIM Domain Only 2 (LMO2) oncogene [23, 24]. These tragic results highlight the need for continued development of improved gene transfer technologies that avoid undesired side effects and ensure the safety of the patient [25, 26].
One means of improving current gene therapy protocols is to develop gene delivery vehicles that integrate therapeutic genes at “safe sites” within the human genome, and thereby avoid insertional mutagenesis and improve transgene expression. We and others have tested in vitro the feasibility of directing integration into specific DNA sites by use of a fusion protein composed of a full-length HIV-1 or avian sarcoma virus (ASV) IN and a sequence-specific DNA-binding protein. The fusion proteins direct integration by recognizing and binding to their cognate target sites on the DNA, causing integration to be mediated into immediately adjacent regions [27, 28]. Sequence-specific DNA-binding proteins tested previously include the Escherichia coli LexA repressor and the DNA-binding domain of bacteriophage λ repressor [29–31]. Although the described IN fusion proteins can bias integration near their cognate binding sites in vitro, one major limitation in pursuing the strategy in vivo is that the DNA-binding sequences for the particular fusion proteins may not be present at the desired chromosomal site for integration. In addition, both E. coli LexA and phage λ repressors may recognize other DNA sequences that are closely-related (either by sequence or by structure) to their consensus binding sequence, and the number of contiguous nucleotides present in the cognate DNA binding site may be insufficient for specifying a unique address within a mammalian genome [32–34]. For example, the LexA protein binds to a 16-base pair (bp) sequence that has approximate twofold rotational symmetry, with only three nucleotides at each end of the palindromic LexA target sequence highly conserved in the consensus binding site [33].
One class of DNA-binding proteins that offers several advantages in conferring site specificity to retroviral IN is the synthetic polydactyl proteins derived from the Cys2-His2 zinc-finger proteins [35, 36]. Analysis of the three-zinc finger protein Zif268-DNA complex revealed that the α-helix of each zinc finger fits directly into the major groove of the target DNA, and the amino acid side chains make specific contacts with a 3-bp DNA sub-site. Most of the base contacts involve the G-rich strand of the binding site [34, 37]. Studies directed at modifying the sequence specificity of the zinc-finger DNA-binding domains have shown that these motifs can be selected to bind specifically to a wide array of DNA sequences [38]. Therefore, designing a zinc-finger protein with specificity for any desired sequence may be possible. In addition, many selected zinc-finger domains exhibit sufficient modularity in their recognition of DNA triplets, and can be combined with other such domains to create polydactyl proteins that recognize extended sequences of DNA [39–47]. One example of such a protein is the six-zinc finger E2C, constructed by grafting the amino acid residues of each zinc finger involved in specific DNA recognition into the framework of the designed consensus protein Sp1C, a derivative of Sp1 [48]. E2C binds with high affinity (in the sub-nanomolar range) and recognizes a contiguous 18-bp sequence, 5′-GGGGCCGGAGCCGCAGTG-3′. The E2C-binding site, referred to as e2c, is unique in the human genome, located within the 5′ untranslated region of the erbB-2 gene on human chromosome 17 [39, 49].
We have constructed several fusion proteins consisting of HIV-1 IN and E2C. The fusion proteins are catalytically active and bias integration of viral DNA near the e2c site in vitro [50]. The results demonstrate that the IN-E2C fusion proteins offer an efficient approach and a versatile framework for directing the integration of retroviral DNA into a predetermined DNA site. The characteristic features of the integrations events observed with IN-E2C are consistent with our working model (Fig. 1) in which the fusion protein binds to its cognate recognition site and mediates integration of viral DNA into nearby regions. Notwithstanding the ability of purified fusion proteins to select a specific target site in vitro, a major challenge in studying directed integration has been to determine the efficacy of the fusion protein strategy in mediating site-directed integration in vivo. To this end, we have successfully incorporated the fusion protein into infectious virions in trans [51–54]. The IN-E2C-containing viruses are functional and capable of biasing integration near the e2c site about 10-fold higher than those viruses containing WT IN, indicating that the IN-E2C fusion protein strategy is capable of directing integration of retroviral DNA into a predetermined chromosomal site in the human genome [53]. This review is focused on preparation of infectious viruses containing the fusion protein of interest, and development of a quantitative PCR assay for measuring integration site specificity in a host cell genome.
Fig. 1.
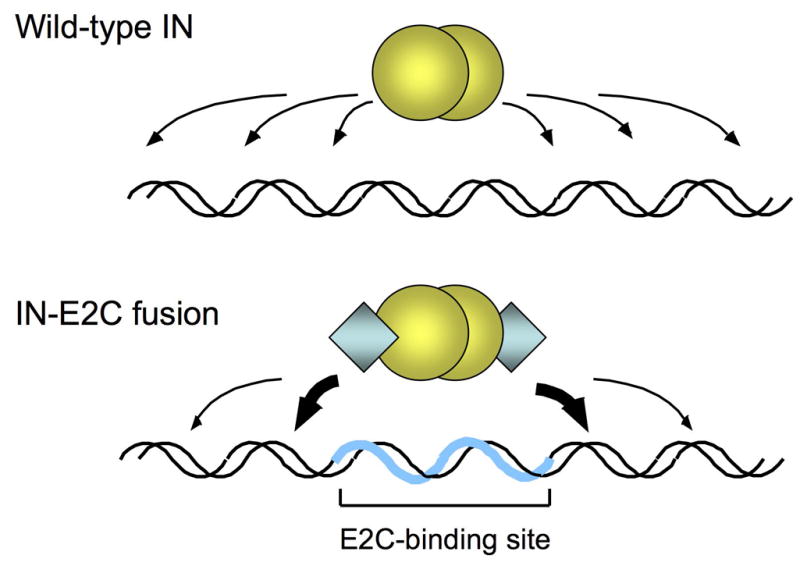
Site-directed integration catalyzed by engineered IN fusion proteins. Integration of retroviral DNA catalyzed by wild-type IN (yellow spheres) is largely non-specific and can occur at various positions along the length of a target DNA (upper panel). In reactions catalyzed by fusion proteins consisting of IN and the designed polydactyl zinc-finger protein E2C (blue diamonds; lower panel), the fusion protein binds specifically to the E2C-recognition sequence e2c (thick blue line) and thereby biases integration in the nearby regions (thick arrows). The e2c site is non-palindromic, and most of the amino acid-DNA contacts are made with the G-rich strand of the target sequence [34, 37]. The IN is depicted as a dimer, but the multimeric state of active IN has not been firmly established.
2. Description of methods
2.1 Virus preparation
All viral stocks are prepared by standard calcium phosphate transfection of monolayers of 293T (obtained from American Type Culture Collection) cells with 20 μg of DNA in 75 cm2 flasks [55]. The cells are grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL) with 10% fetal calf serum (HyClone), 100 U of penicillin (Cellgro) per ml, and 0.1 mg of streptomycin (Sigma) per ml. For the hygromycin resistance infection assay, viruses provided with wild-type (WT) IN or IN-E2C fusion proteins in trans are generated by triple transfection of an HIV-1 expression construct (pHIV-IN64-Hygro), an envelope expression construct (pMD. G), and an appropriate fusion protein construct at a ratio of 5:1:14 (Fig. 2).
Fig. 2.
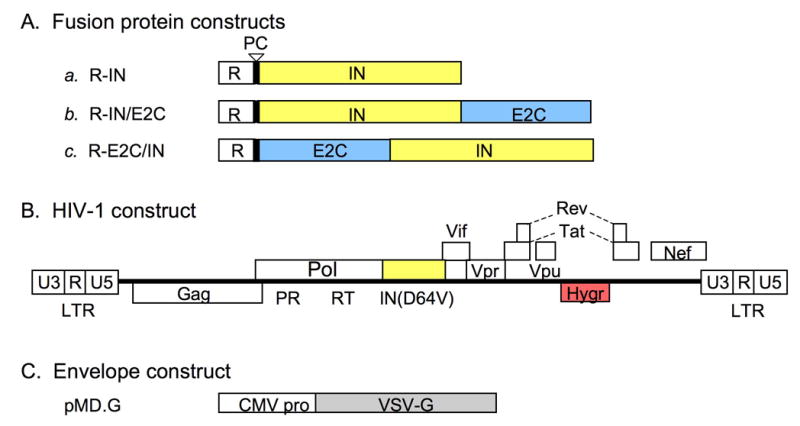
DNA constructs used for trans incorporation of IN-E2C fusion proteins. (A) Fusion protein constructs. Each construct encodes the Vpr protein at the N-terminus (R, open boxes) for packaging of IN (a), IN/E2C (b), or E2C/IN (c) proteins into the viruses. The IN and E2C coding segments are denoted by yellow and blue boxes, respectively. The vertical bars with an open arrowhead denote HIV-1 protease cleavage (PC) sites that are required for removal of Vpr from the IN proteins after packaging [56]. All constructs are encoded in the pLR2P expression plasmid [54]. (B) HIV-1 construct. The plasmid HXB-IN64-Hygro is derived from the HXB2 strain of HIV-1, and contains a defective vpr gene due to an insertion of a T nucleotide at position 5771 [57], and a hygromycin resistance gene (Hygr; red box) in place of env. In addition, IN (yellow box) contains the inactivating Asp to Val substitution at amino acid position 64 (D64V). (C) Envelope expression construct. Each virus was pseudotyped with VSV-G (gray box) driven by the cytomegalovirus (CMV) early promoter [58]. Constructs are not drawn to scale.
2.1.1 Fusion protein constructs
The genes encoding WT IN and the fusion proteins IN/E2C and E2C/IN (Fig. 2A) are each cloned into a pLR2P expression plasmid (provided by Beatrice Hahn of the University of Alabama, Birmingham). Important features of this in-trans expression construct include an HIV-2 long terminal repeat (LTR) at the 5′ end of the gene of interest to drive its expression, an HIV-2 Rev-response element (RRE) located 3′ to the gene to ensure Rev-mediated nuclear export of the expressed mRNA, and a polyadenylation signal that stabilizes the transcript and increases translational efficiency [54]. The sequence encoding the Vpr (R)-fusion protein, situated between the HIV-2 elements, contains an HIV-1 protease cleavage (PC) site following Vpr [56]. IN/E2C and E2C/IN (Fig. 2A, constructs b and c) contain WT HIV-1 IN at the N and C termini of E2C, respectively. To construct the R-IN/E2C clone, an Nsi I-Sal I fragment of pIN1-288/E2C, which is derived from pT7-7 expression vector and contains the coding sequence of full-length HIV-1 IN fused with E2C [50], was inserted in-frame into pLR2P previously digested with Nsi I and Xho I. For the R-E2C/IN clone, an Nde I (blunted)-Sal I fragment of pE2C/IN1-288, which contains the coding sequence of E2C/IN fusion protein [50], was ligated with pLR2P previously cut with Xho I and partially digested with Sca I. For simplicity, the term “IN-E2C” refers to both IN/E2C and E2C/IN.
2.1.2 HIV-1 genome construct
The plasmid HXB-IN64-Hygro (obtained from Andrew D. Leavitt at the University of California, San Francisco) is derived from the HXB2 strain of HIV-1, which carries a defective vpr gene due to an insertion of a T nucleotide at position 5771 [57]. pHXB-IN64-Hygro encodes a substitution in one of the catalytic core triad residues of IN, Asp at position 64 to Val (D64V), and contains a hygromycin resistance gene inserted in place of the viral env gene (Fig. 2B) [51].
2.1.3 Envelope glycoprotein construct
The expression plasmid pMD. G (Fig. 2C), provided by Didier Trono at the University of Geneva [58], encodes the G-glycoprotein of vesicular stomatitis virus (VSV-G). Pseudotyping HIV-1 with VSV-G expands the tropism of the resulting virus.
2.1.4 Detailed method
Culture supernatants are collected 48 h after transfection. Virions are treated with 10–20 U of RNase-free DNase I (Amersham Pharmacia) per ml of viral stock at room temperature for 1 h, and then pelleted by ultracentrifugation at 125,000 × g for 2 h at 4°C through a cushion of 20% sucrose in phosphate-buffered saline (PBS). Viral pellets are resuspended in culture medium without serum and stored at −80°C until use. The virus titer is estimated by an enzyme-linked immunosorbent assay (Coulter Inc.) that detects the HIV-1 capsid (p24) protein.
With the in-trans method, the IN-E2C fusion protein is incorporated into infectious virions by linking to Vpr, an HIV-1 accessory protein that is packaged into viruses by interacting with the p6 protein of Gag (Fig. 3). During maturation, the Vpr component is removed by HIV-1 protease cleavage within the virion to produce IN/E2C or E2C/IN, which can then mediate integration [56]. Inclusion of the various IN-E2C fusion proteins into the virion does not interfere with virus packaging and maturation. The presence of the correctly processed IN-E2C fusion proteins in viral particles can be confirmed by immunoblotting using rabbit polyclonal antibodies raised against synthetic HXB2 IN peptides (amino acids 23–34 and 142–153). The anti-IN antibodies can be obtained from Duane Grandgenett at the St. Louis University through the AIDS Research and Reference Reagent Program. The presence of other viral proteins can be verified using sera from HIV-1-infected patients. The sera can be purchased from The Scripps Research Institute.
Fig. 3.
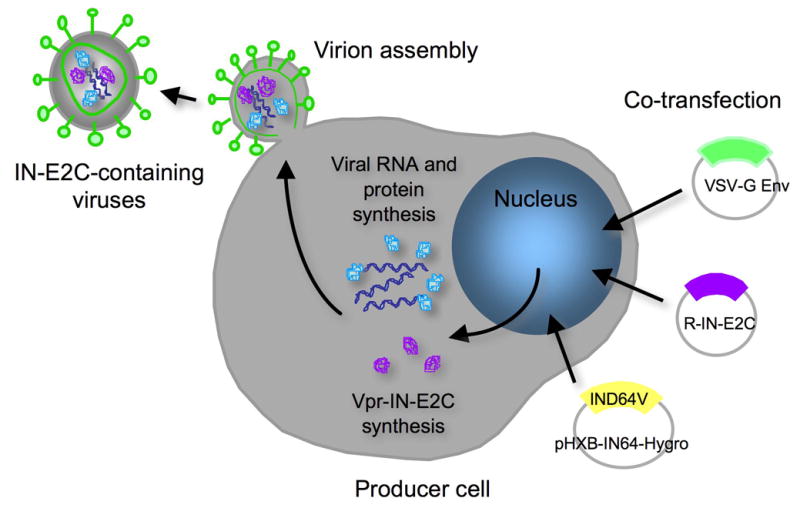
Trans incorporation of IN-E2C fusion proteins into infectious virions. Producer cells are co-transfected with an HIV expression construct (pHXB-IN64-Hygro) that encodes an IN catalytic mutant (D64V) and hygromycin resistance gene, a fusion protein construct (pLR2P-R-PC-IN or IN-E2C), and an envelope expression construct (pMD.G). After protein expression, the Vpr-fusion protein is packaged into the virus particles via the interaction of Vpr with the p6 portion of Gag.
2.2 HIV-1 infectivity assay
This assay has been used to test the functionality of an IN-containing fusion protein provided in trans to an HIV-1 genome containing a catalytic-inactivating mutation (D64V in this case) in IN. The infectivity of various virus stocks is measured using an assay that quantifies the acquisition of the hygromycin resistance gene from HXB-IN64-Hygro as described previously [51].
2.2.1 Hygromycin resistance infectivity assay
Infect 1×106 HeLa cells (American Type Culture Collection catalog number CCL-121) in 10-cm plates with five, twenty, or one hundred nanograms p24 equivalent of virus collected from the triple plasmid transfections (pHXB-IN64-Hygro, pMD.G, and a pLR2P expression construct). Remove the virus-containing medium 4 h post-infection and replace with 10 ml fresh DMEM. Maintain the cells in nonselective media for an additional 48 h, and then exchange the DMEM for medium containing 200 μg/ml hygromycin B (Sigma). Continue the selection for 21 days. Remove the culture medium and wash the hygromycin-selected cell colonies gently and thoroughly with 10 ml of PBS twice. Add 1 ml stain solution (0.2% crystal violet in 10% phosphate-buffered formalin, pH 7.0) and agitate to distribute the solution evenly. Incubate 1 min at room temperature, wash the cells with 10 ml PBS twice, and count the stained, viable cell colonies.
Viruses encoding the IN D64V alone cannot integrate, thereby the hygromycin resistance gene cannot be expressed and no antibiotic-resistant colonies will be formed (Fig. 4A). If the IN-containing fusion protein is functional for catalyzing integration, viruses containing the INfusion protein provided in trans are able to express the hygromycin resistance gene after infecting target cells and forming antibiotic-resistant colonies. This is illustrated by the formation of hygromycin-resistant colonies by the IN mutant virus supplied with either IN/E2C or E2C/IN (Figs. 4C and D). The efficiency of integration catalyzed by the fusion protein can be determined by comparing the number of hygromycin-resistant colonies produced by viruses containing IN-E2C with the number of colonies produced by viruses supplied with WT IN (Fig. 4B). For the HXB-IN64V-Hygro virus supplied with WT IN in trans, the number of hygromycin-resistant colonies under the described reaction condition ranges from 10 – 50/ng p24 equivalent of virus.
Fig. 4.

Integration efficiency of viruses containing various IN-E2C fusion proteins. One million HeLa cells were infected for 4 h with equal amounts (ranging from 5–100 ng) of p24 equivalent of the indicated virus (panels A–D). Representative plates of the hygromycin resistance assay are shown. Dark spots on each plate are colonies that grew after selection with 200 μg/ml of hygromycin B for three weeks, beginning two days post-infection. The colonies result from provirus formation and stable expression of the hygromycin resistance gene. The number of resistant colonies per ng p24 for each virus was determined and expressed as a percentage of the colonies produced by the virus containing the WT IN supplied in trans (panel B). The figure is reproduced from previously published data with permission from Copyright © 2006, American Society for Microbiology (doi:10.1128/JVI.80.4.1939-1948.2006).
2.3 Proviral DNA standards for the quantitative, two-step real-time PCR assay
Given the unique location of the 18-bp e2c site on chromosome 17 [39, 49], the number of specifically-targeted proviruses can be determined using a fluorescence-monitored, two-step PCR assay modified from those described previously [59, 60]. To assay for total integration events catalyzed by WT IN or the fusion proteins, the same DNA sample is subjected to nested Alu-PCR [53, 60]. The repetitive Alu element was chosen as a PCR “anchor” for genomic DNA given the prevalence of this element in the human genome, with over one million copies randomly distributed and oriented in each diploid cell [61]. The specificity of directed integration is then determined by comparing the number of proviruses near the e2c site to that of the whole genome. The sequences and annealing positions of primers and probes are listed in Table 1 and Figure 5, respectively.
TABLE 1.
DNA sequences of primers and probes used in real-time PCR assays
| Primer or probe | Sequence |
|---|---|
| λT | 5′-ATGCCACGTAAGCGAAACT |
| Alu1 | 5′-TCCCAGCTACTGGGGAGGCTGAGG |
| BGF | 5′-CAACCTCAAACAGACACCATG |
| BGR | 5′-TCCACGTTCACCTTGCCC |
| BGX-P | 5′-FAM-CTCCTGAGGAGAAGTCTGCCGTTACTGCC-TAMRA |
| EcF2 | 5′-GGCCCTTTACTGCGCCGCGC |
| EcR2 | 5′-GGTCCCGCCGCTGCTCCGT |
| LM652 | 5′-ATGCCACGTAAGCGAAACTCCCTGTTCGGGCGCCACTGCTAGAG |
| LM667 | 5′-ATGCCACGTAAGCGAAACTCTGGCTAACTAGGGAACCCACTG |
| LM667-mod | 5′-CTGGCTAACTAGGGAACCCACTG |
| LR | 5′-TCCACACTGACTAAAAGGGTCTGA |
| LRT-P | 5′-FAM-CAGTGGCGCCCGAACAGGGA-TAMRA |
| MH531 | 5′-TGTGTGCCCGTCTGTTGTGT |
| MH532 | 5′-GAGTCCTGCGTCGAGAGAGC |
| MH535 | 5′-AACTAGGGAACCCACTGCTTAAG |
| ZXF-P | 5′-FAM-TGTGACTCTGGTAACTAGAGATCCCTCAGACCC-TAMRA |
Fig. 5.

Fluorescence-monitored, real-time nested PCR assays for quantifying integration specificity. HIV-1 cDNA is represented as a thin line flanked by LTRs (open boxes). The arrows above and below the U3-R junction of the LTR indicate the direction of viral transcription. Alu-PCR quantifies proviral DNA integrated in the entire genome, whereas e2c-PCR quantifies proviral DNA integrated near the e2c site on chromosome 17. Proviral integration can be in either orientation and upstream or downstream relative to a specified locus, Alu (yellow box) or e2c (blue box). For simplicity, the diagram depicts only the scenario in which the proviral DNA is integrated in either orientation upstream of an Alu element or the e2c site. Green arrows represent the locations and orientations of the PCR primers for determining the proviral copy number of the Alu-PCR Standard. The quantitative Alu-PCR and e2c-PCR assays consist of two-rounds of PCR. Red arrows represent the locations and orientations of the first-round PCR primers. Alu1 is used for total integration and EcF2 and EcR2 are for e2c-specific integration. Blue arrows represent the locations and orientations of second-round PCR primers, whereas LRT-P (green) and ZXF-P (blue) denote the locations of the fluorescent probes used during real-time PCR. Primers LM652 and LM667 contain phage λ-specific sequences at their 5′ ends, and primer λT anneals specifically to the phage λ sequences.
To determine accurately the copy number of integrated proviruses in a cell sample, a DNA standard representative of a natural infection with a full distribution of distances between each proviral LTR and nearest Alu element is necessary. Cellular DNA containing proviruses integrated at many locations is generated by infecting HeLa cells with the HXB-IN64-Hygro virus containing Vpr-IN and pseudotyped with VSV-G at a multiplicity of infection (MOI) of 10. The infected cells are grown in the presence of 200 μg/ml hygromycin B for four weeks to select cells containing proviruses and to allow for the loss of unintegrated DNA through multiple rounds of cell division. Total genomic DNA is isolated using the DNeasy Tissue Kit (Qiagen). The copy number of proviral DNA per cell is determined by quantitative real-time PCR.
2.3.1 Determining the proviral copy number of the Alu-PCR Standard
For a 20-μl reaction:
10 μl of 2 × TaqMan Universal PCR Master Mix
MH531 primer (400 nM final concentration)
MH532 primer (400 nM final concentration)
TaqMan probe LRT-P (200 nM final concentration)
0.5 U Platinum Taq DNA polymerase (Invitrogen)
H2O to 20 μl
PCR is performed using an ABI Prism 7900 Sequence Detection System (PE-Applied Biosystems). The amplification condition includes a hot start of 50°C for 2 min, and 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and extension at 60°C for 1 min. A quantitative dilution series of plasmid pCR-5L-e2c (see below) is used as a copy number standard. The copy number of proviral DNA per cell is calculated, and the genomic DNA isolated from this standard cell line is referred as “Alu-PCR Standard”.
PCR primers MH531 (anneals to viral nucleotide positions 556–576) and MH532 (anneals to nucleotides 699-680) are specific for late reverse transcription products as described previously [62]. The fluorescent TaqMan probe LRT-P used for quantifying the amplified U5-gag-containing product is modified at its 5′ and 3′ ends with 6-carboxy-fluoresein (FAM) and 6-carboxytetramethyl-rhodamine (TAMRA), respectively. The probe anneals to nucleotide positions 633–652 of HIV-1.
As an internal standard for normalizing the amount of cellular DNA, the level of human β-globin DNA is measured using the TaqMan PCR Reagent Kit (PE-Applied Biosystems) following the manufacturer’s instruction. The reaction is carried out using 400 nM of forward primer BGF, 400 nM of reverse primer BGR, and 200 nM of BGX-P as the fluorescence probe (Table 1). The standard curve for the human β-globin sequence is generated using genomic DNA isolated from uninfected HeLa cells.
Using the late-reverse transcription primer set for quantifying HIV-1 DNA and the β-globin set for determining the cell number, the “Alu-PCR Standard” under the described condition should contain 1–2 copies of proviral DNA per cell.
Given the impracticality of generating a DNA standard representative of HIV-1 DNA integrated randomly at a range of distances from the e2c site, plasmids pCR-5L-e2c and pCR-3L-e2c are used as copy number standards for quantifying proviruses integrated downstream and upstream, respectively, of the e2c site [53]. The pCR-5L-e2c plasmid contains a 336-bp e2c DNA fragment upstream of the left LTR and part of the gag gene of HIV-1, whereas pCR-3L-e2c contains the e2c DNA fragment downstream of the right LTR and part of the viral nef gene (Fig. 6). The 336-bp DNA fragment flanking the e2c site, located within the 5′ untranslated region of the erbB-2 gene in human chromosome 17 [49], was PCR-amplified using cellular DNA isolated from uninfected HeLa cells as the template.
Fig. 6.

DNA constructs for quantifying proviral DNA integrated upstream or downstream of the e2c site. A 336-bp DNA fragment (blue box) flanking the e2c site, located within the 5′ untranslated region of the erbB-2 gene on human chromosome 17 [49], was amplified by PCR using cellular DNA isolated from uninfected HeLa cells as the template. The PCR product was cloned into the pCR-Blunt II-Topo vector (Invitrogen), resulting in pCR-e2c. An HIV-1 DNA fragment (open box) containing the upstream LTR and part of the gag sequence or the downstream LTR and part of the nef sequence was obtained by PCR. The LTR-gag fragment was cloned into pCR-e2c downstream of the 336-bp e2c-containing fragment, resulting in pCR-5L-e2c. The LTR-nef fragment was cloned upstream of the e2c-containing fragment to form pCR-3L-e2c.
2.4 Quantitation of total integrated proviral DNA by Alu-PCR
HeLa cells are infected with viruses containing WT IN or IN-E2C fusion proteins in the presence of 20 μg/ml DEAE-dextran at a MOI of 1. Genomic DNA is extracted using the DNeasy Tissue Kit (Qiagen) three days after infection and diluted in TE buffer (10 mM Tris pH 7.5–1 mM EDTA). An Alu-LTR-based quantitative nested-PCR procedure is used to quantify the total number of HIV-1 integration events in the human genome (Alu-PCR; Fig. 5).
2.4.1 Alu-PCR
The technique is comprised of two PCR rounds. The first utilizes conventional exponential amplification to detect LTR sequences that lie within approximately 3 kbp upstream or downstream of any given Alu element in the human genome. The Alu primer, Alu1, anneals within conserved regions of Alu repeat elements and an HIV-1 LTR-specific primer, LM667, anneals to viral nucleotides 494–516). The 5′ end of the LTR-specific primer contains a phage lambda-specific heel sequence.
For a 20-μl reaction, add the following reagents to the indicated final concentrations:
10 mM Tris-HCl, pH 8.3
1.5 mM MgCl2
1 mM dNTPs (250 μM each dNTP)
50 mM KCl
100 nM Alu1 primer
300 nM LM667 primer
100 ng human genomic DNA (as assessed by A260)
0.5 U Platinum Taq DNA polymerase
The PCR cycle condition is as follows: a denaturation step of 5 min at 95°C and then 20 cycles of amplification (94°C for 15 s, 55°C for 30 s, and 70°C for 2 min). The second PCR round, referred to as real-time or kinetic, utilizes the fluorescent TaqMan probe ZXF-P, which anneals to viral nucleotides 574–606, to quantify the level of HIV sequences produced in the first PCR round. To enhance the specificity of this amplification, the lambda-specific primer λT, which anneals to the 5′ region of the LM667 sequence, is used in conjunction with the internal LTR primer LR, which anneals to viral nucleotides 622–599 (Fig. 5).
For a 20-μl reaction, add:
2 μl of the first-round PCR mixture
10 μl of 2 × TaqMan Universal PCR Master Mix
λT primer (400 nM final concentration)
LR primer (400 nM final concentration)
TaqMan probe ZXF-P (200 nM final concentration)
0.5 U Platinum Taq DNA polymerase
H2O to 20 μl
The thermal program is as follows: a hot start of 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and extension at 60°C for 1 min.
Run the Alu-PCR Standard and infected cell samples concomitantly. The total number of integrated proviruses in the infected cell samples is calculated in reference to the standard curve generated using 100 ng of Alu-PCR Standard serially diluted with genomic DNA isolated from uninfected cells. As the number of Alu elements vastly exceed the number of viral LTRs, it is important to maintain the same level of total cellular DNA across PCR samples. The regression obtained from the Alu-PCR Standard dilutions should be linear over a 3-log range from 18 to 1.8 × 104 provirus copies per reaction, equating to the detection of approximately 0.1 provirus within 1,000 cell equivalents.
The method requires a number of specificity controls to ensure for the accuracy of integrated virus values. For example, it is essential to control for linear amplification that arises from the HIV-specific LM667 primer. To accomplish this, run parallel reactions of each sample, but omit the Alu1 primer from the first-round PCR. The copy number of integrated proviruses is then adjusted by subtracting the copy number quantified in the absence of the Alu1 primer (ranging from <0.1 to 0.7 copies/1,000 cells) from the copy number measured in the presence of the Alu1 primer. To test the specificity of the first round PCR, amplification is carried out with genomic DNA extracted from uninfected HeLa cells as the template and Alu1 and LTR-specific LM667 as the primers. To control for the specificity of the second round PCR, the first round PCR is carried out with Alu-PCR Standard as the template and primers Alu1 and LM667-mod, a modified version of LM667 that lacks the phage lambda sequences at its 5′ end (Table 1). In both specificity controls, one-tenth of the first-round PCR product is used in the second round with the lambda-specific primer λT and an internal LTR primer LR under the identical condition as described earlier. The signal of both reactions should be <0.1 copy/1,000 cells.
2.5 Quantitation of proviral DNA integrated near the e2c site by e2c-PCR
To quantify integration events near the e2c site in the human genome, a real-time nested-PCR procedure (e2c-PCR) similar to the Alu-PCR described above is carried out (Fig. 5). The quantitative, two-step PCR assay includes an initial non-kinetic pre-amplification step with one primer annealing to viral sequences and the other primer annealing near the e2c site, under the condition that the primers, dNTPs, and enzyme are not limiting. The pre-amplification is followed by real-time, quantitative PCR. The Alu1 primer of the first round PCR is replaced with e2c-specific primers EcF2 and EcR2, which anneal to sequences 67 bp upstream and 101 bp downstream, respectively, of the e2c site. The primers were chosen using the MacVector software program (Accelrys Inc.) and tested for low background signals in the quantitative PCR reaction using uninfected human genomic DNA as template. To measure the number of proviruses integrated in both orientations upstream of the e2c site, two primer sets are used in two separate first-round PCRs. One primer set includes LM652 (anneals to nucleotide positions 651-627) and EcR2 as the forward and reverse primers, respectively. The other set includes LM667 and EcR2 as the forward and reverse primers, respectively (Fig. 5). Both LM652 and LM667 contain the phage lambda-specific heel sequence and anneal to U5 and R regions of the HIV-1 LTR, respectively. Similarly, the number of proviruses integrated in both orientations downstream of the e2c site is measured by two separate first-round PCRs. The forward primer in both reactions is EcF2 and the reverse primer is LM652 or LM667. The PCR condition is identical to that described earlier for Alu-PCR (see section 2.4.1).
In the second-round real-time PCR, one-tenth of the first-round mixture is used as the template. In reactions where primer LM652 was used in the first-round PCR, the forward and reverse primers for the second-round PCR are λT and MH535, respectively; use both at the final concentration of 400 nM. MH535 anneals to nucleotide positions 500–522 of HIV-1 U5. In reactions where primer LM667 was used in the first-round PCR, the forward and reverse primers for the second-round are λT and LR, respectively; again use both at 400 nM. The ZXF-P fluorescence TaqMan probe is used in both second round reactions at the final concentration of 200 nM. Under the described PCR condition, we estimate that each primer set can amplify DNA fragments that are on average 3 kbp in length. Therefore, the e2c-PCR assay is capable of measuring proviruses integrated within an approximate 6-kbp region surrounding the e2c site.
The copy number of integrated DNA is determined in reference to a standard curve generated by a concomitant two-step PCR amplification of a serial dilution of the e2c-PCR DNA standard, pCR-5L-e2c, for quantifying proviruses downstream of the e2c site, and pCR-3L-e2c for quantifying proviruses upstream of the e2c site (Fig. 6). The amplification signals for the pCR-5L-e2c DNA standard are linear over a 7-log range from 20 to 2 × 108 provirus copies per reaction, whereas the pCR-3L-e2c DNA standard are linear over a 6-log range from 200 to 2 × 108 provirus copies per reaction.
As in Alu-PCR, linear amplification arising from LM667 and LM652 primers is determined for each virus-infected sample in the absence of the e2c-specific primers (ranging from <0.1 to 0.3 copies/1,000 cells), and subtracted from the copy number quantified in the presence of the e2c-specific primers. The quantitation of proviral DNA integrated near the e2c site of each sample is done concomitantly with the real-time PCR assay described earlier for human β-globin DNA.
The specificity of directed integration by the IN-E2C fusion proteins within the human genome can be quantified by comparing the number of proviruses integrated near the e2c site (determined by the e2c-PCR) to that of the whole genome (determined by the Alu-PCR). As a negative control for site-directed integration, proviral DNA copy numbers in the whole genome and near the e2c site are similarly quantified using the IND64V-encoding viruses supplied with WT IN in trans.
Although the described fluorescence-monitored, two-step PCR assay is sensitive and linear over a wide range of proviral DNA copies, the assay is also time-consuming and laborious to perform. With the advent of high-throughput DNA sequencing, the massively parallel sequencing method has been applied successfully to carry out genome-wide analysis of integration site preference [63, 64]. Efforts are currently underway to develop simpler assays that are based on these new high-throughput sequencing technologies for quantifying site-directed retroviral DNA integration.
3. Concluding remarks
The ability of retroviruses to stably introduce transgenes into cellular chromosomes has resulted in their wide use as vectors for both genetic engineering in higher eukaryotes and for gene therapy [3]. However, the nonspecific nature by which IN chooses integration sites presents a major drawback toward the safety of these vectors and limits the utility of retrovirus-based vectors in gene therapy. Although it is apparent that fusion of a sequence-specific DNA-binding protein, such as the E2C protein described in this review, to a retroviral IN can direct integration of transgenes into predetermined sites in vitro and in vivo, the success of this strategy depends on the feasibility of eliminating nonspecific DNA binding by the IN component and increasing the affinity and site-specific recognition by the sequence-specific DNA-binding component of the fusion protein. Structural and biochemical studies in identifying the IN domain responsible for binding nonspecific DNA and for target site selection are critical in suppressing integration at unwanted sites [18, 65–67]. Likewise, improved designs, selection, and engineering of DNA-binding proteins represent the other crucial parameter for providing an efficient and versatile system for site-specific integration [38, 68, 69].
In addition to the IN-fusion protein strategy, another approach to site-directed integration is prompted by the Ty family of retrotransposons found in Saccharomysces cerevisiae. The Ty retrotransposons mediate integration with a high specificity into the yeast genome through protein-protein interactions [28]. For instance, Ty3 IN interacts directly with the Pol III transcription factor B/C complex [70], and Ty5 IN recognizes Sir4p of the telomeric chromatin [71, 72]. An analogous strategy of targeting retroviral integration through specific protein-protein interactions is to explore the various cellular proteins that bind HIV-1 IN, such as the lens epithelial-derived growth factor (LEDGF/p75) and IN interactor 1 [73]. LEDGF/p75, a member of the hepatoma-derived growth factor related protein family, is a ubiquitous nuclear protein tightly associated with chromatin [74, 75] and is required for efficient HIV-1 replication [74, 76–78]. The possibility that the specific interaction between HIV-1 and LEDGF/p75 may be exploited for targeted integration has been raised [79]. One advantage of such an approach is to eliminate the necessity of fusing IN to a sequence-specific DNA-binding protein.
Integration is a critical component toward the use of retroviruses in gene therapy. Studies on testing site-directed integration using engineered IN may lead to a new approach for inserting transgenes at predetermined sites, which will have a wide application as an experimental tool and improve the therapeutic application of current retrovirus- and lentivirus-based vectors.
Acknowledgments
This work was supported by National Institutes of Health Grant CA68859 to S.A.C. K.S. was supported by a predoctoral fellowship from the Ministry of Education of the People’s Republic of China.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Langer R. Nature. 1998;392 (6679 Suppl):5–10. [PubMed] [Google Scholar]
- 2.Kay MA, Glorioso JC, Naldini L. Nat Med. 2001;7:33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 3.Thomas CE, Ehrhardt A, Kay MA. Nat Rev Genet. 2003;4:346–58. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 4.Brown PO. In: Retroviruses. Coffin JM, Hughes SH, Varmus HE, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1997. pp. 161–203. [PubMed] [Google Scholar]
- 5.Kordower JH, Emborg ME, Bloch J, Ma SY, Chu Y, Leventhal L, McBride J, Chen EY, Palfi S, Roitberg BZ, Brown WD, Holden JE, Pyzalski R, Taylor MD, Carvey P, Ling ZD, Trono D, Hantraye P, Deglon N, Aebischer P. Science. 2000;290:767–73. doi: 10.1126/science.290.5492.767. [DOI] [PubMed] [Google Scholar]
- 6.Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. Science. 1999;283:682–86. doi: 10.1126/science.283.5402.682. [DOI] [PubMed] [Google Scholar]
- 7.Mizuuchi K. J Biol Chem. 1992;267:21273–76. [PubMed] [Google Scholar]
- 8.Rice P, Craigie R, Davies DR. Curr Opin Struct Biol. 1996;6:76–83. doi: 10.1016/s0959-440x(96)80098-4. [DOI] [PubMed] [Google Scholar]
- 9.Asante-Appiah E, Skalka AM. Antiviral Res. 1997;36:139–56. doi: 10.1016/s0166-3542(97)00046-6. [DOI] [PubMed] [Google Scholar]
- 10.Bor YC, Miller MD, Bushman FD, Orgel LE. Virology. 1996;222:283–88. doi: 10.1006/viro.1996.0422. [DOI] [PubMed] [Google Scholar]
- 11.Carteau S, Hoffmann C, Bushman F. J Virol. 1998;72:4005–14. doi: 10.1128/jvi.72.5.4005-4014.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzgerald ML, Grandgenett DP. J Virol. 1994;68:4314–21. doi: 10.1128/jvi.68.7.4314-4321.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pryciak PM, Sil A, Varmus HE. EMBO J. 1992;11:291–303. doi: 10.1002/j.1460-2075.1992.tb05052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bushman F, Lewinski M, Ciuffi A, Barr S, Leipzig J, Hannenhalli S, Hoffmann C. Nat Rev Microbiol. 2005;3:848–58. doi: 10.1038/nrmicro1263. [DOI] [PubMed] [Google Scholar]
- 15.Holman AG, Coffin JM. Proc Natl Acad Sci USA. 2005;102:6103–07. doi: 10.1073/pnas.0501646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S, Kim Y, Liang T, Sinsheimer JS, Chow SA. J Virol. 2006;80:11313–21. doi: 10.1128/JVI.01737-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewinski MK, Bisgrove D, Shinn P, Chen H, Hoffmann C, Hannenhalli S, Verdin E, Berry CC, Ecker JR, Bushman FD. J Virol. 2005;79:6610–19. doi: 10.1128/JVI.79.11.6610-6619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G, Collins F, Shinn P, Leipzig J, Hannenhalli S, Berry CC, Ecker JR, Bushman FD. PLoS Pathog. 2006;2:611–22. doi: 10.1371/journal.ppat.0020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z, Dullmann J, Schiedlmeier B, Schmidt M, von Kalle C, Meyer J, Forster M, Stocking C, Wahlers A, Frank O, Ostertag W, Kuhlcke K, Eckert HG, Fehse B, Baum C. Science. 2002;296:497. doi: 10.1126/science.1068893. [DOI] [PubMed] [Google Scholar]
- 20.Marchetti A, Buttitta F, Miyazaki S, Gallahan D, Smith GH, Callahan R. J Virol. 1995;69:1932–38. doi: 10.1128/jvi.69.3.1932-1938.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nusse R. Curr Top Microbiol Immunol. 1991;171:43–65. doi: 10.1007/978-3-642-76524-7_3. [DOI] [PubMed] [Google Scholar]
- 22.Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, Selz F, Hue C, Certain S, Casanova JL, Bousso P, Le Deist F, Fischer A. Science. 2000;288:669–72. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 23.Cavazzana-Calvo M, Thrasher A, Mavilio F. Nature. 2004;427:779–81. doi: 10.1038/427779a. [DOI] [PubMed] [Google Scholar]
- 24.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, Fischer A. N Engl J Med. 2003;348:255–56. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 25.Baum C, Dullmann J, Li Z, Fehse B, Meyer J, Williams DA, von Kalle C. Blood. 2003;101:2099–114. doi: 10.1182/blood-2002-07-2314. [DOI] [PubMed] [Google Scholar]
- 26.Verma IM, Weitzman MD. Annu Rev Biochem. 2005;74:711–38. doi: 10.1146/annurev.biochem.74.050304.091637. [DOI] [PubMed] [Google Scholar]
- 27.Bushman FD. Curr Top Microbiol Immunol. 2002;261:165–77. doi: 10.1007/978-3-642-56114-6_8. [DOI] [PubMed] [Google Scholar]
- 28.Sandmeyer S. Proc Natl Acad Sci USA. 2003;100:5586–88. doi: 10.1073/pnas.1031802100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bushman FD. Proc Natl Acad Sci USA. 1994;91:9233–37. doi: 10.1073/pnas.91.20.9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goulaouic H, Chow SA. J Virol. 1996;70:37–46. doi: 10.1128/jvi.70.1.37-46.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katz RA, Merkel G, Skalka AM. Virology. 1996;217:178–90. doi: 10.1006/viro.1996.0105. [DOI] [PubMed] [Google Scholar]
- 32.Jordan SR, Pabo CO. Science. 1988;242:893–99. doi: 10.1126/science.3187530. [DOI] [PubMed] [Google Scholar]
- 33.Lewis LK, Harlow GR, Gregg-Jolly LA, Mount DW. J Mol Biol. 1994;241:507–23. doi: 10.1006/jmbi.1994.1528. [DOI] [PubMed] [Google Scholar]
- 34.Pavletich NP, Pabo CO. Science. 1991;252:809–17. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 35.Alwin S, Gere MB, Guhl E, Effertz K, Barbas CF, 3rd, Segal DJ, Weitzman MD, Cathomen T. Mol Ther. 2005;12:610–17. doi: 10.1016/j.ymthe.2005.06.094. [DOI] [PubMed] [Google Scholar]
- 36.Beerli RR, Barbas CF., III Nat Biotechnol. 2002;20:135–41. doi: 10.1038/nbt0202-135. [DOI] [PubMed] [Google Scholar]
- 37.Elrod-Erickson M, Rould MA, Nekludova L, Pabo CO. Structure. 1996;4:1171–80. doi: 10.1016/s0969-2126(96)00125-6. [DOI] [PubMed] [Google Scholar]
- 38.Segal DJ, Beerli RR, Blancafort P, Dreier B, Effertz K, Huber A, Koksch B, Lund CV, Magnenat L, Valente D, Barbas CF., III Biochemistry. 2003;42:2137–48. doi: 10.1021/bi026806o. [DOI] [PubMed] [Google Scholar]
- 39.Beerli RR, Segal DJ, Dreier B, Barbas CF., III Proc Natl Acad Sci USA. 1998;95:14628–33. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choo Y, Klug A. Proc Natl Acad Sci USA. 1994;91:11163–67. doi: 10.1073/pnas.91.23.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dreier B, Segal DJ, Barbas CF., III J Mol Biol. 2000;303:489–502. doi: 10.1006/jmbi.2000.4133. [DOI] [PubMed] [Google Scholar]
- 42.Kim JS, Pabo CO. Proc Natl Acad Sci USA. 1998;95:2812–17. doi: 10.1073/pnas.95.6.2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Q, Segal DJ, Ghiara JB, Barbas CF., 3rd Proc Natl Acad Sci USA. 1997;94:5525–30. doi: 10.1073/pnas.94.11.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu PQ, Rebar EJ, Zhang L, Liu Q, Jamieson AC, Liang Y, Qi H, Li PX, Chen B, Mendel MC, Zhong X, Lee YL, Eisenberg SP, Spratt SK, Case CC, Wolffe AP. J Biol Chem. 2001;276:11323–34. doi: 10.1074/jbc.M011172200. [DOI] [PubMed] [Google Scholar]
- 45.Rebar EJ, Greisman HA, Pabo CO. Methods Enzymol. 1996;267:129–49. doi: 10.1016/s0076-6879(96)67010-4. [DOI] [PubMed] [Google Scholar]
- 46.Rebar EJ, Huang Y, Hickey R, Nath AK, Meoli D, Nath S, Chen B, Xu L, Liang Y, Jamieson AC, Zhang L, Spratt SK, Case CC, Wolffe A, Giordano FJ. Nat Med. 2002;8:1427–32. doi: 10.1038/nm1202-795. [DOI] [PubMed] [Google Scholar]
- 47.Segal DJ, Dreier B, Beerli RR, Barbas CF., III Proc Natl Acad Sci USA. 1999;96:2758–63. doi: 10.1073/pnas.96.6.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Desjarlais JR, Berg JM. Proc Natl Acad Sci USA. 1994;91:11099–103. doi: 10.1073/pnas.91.23.11099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beerli RR, Dreier B, Barbas CF., III Proc Natl Acad Sci USA. 2000;97:1495–500. doi: 10.1073/pnas.040552697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan W, Zhu K, Segal DJ, Barbas CF, 3rd, Chow SA. J Virol. 2004;78:1301–13. doi: 10.1128/JVI.78.3.1301-1313.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holmes-Son ML, Chow SA. J Virol. 2000;74:11548–56. doi: 10.1128/jvi.74.24.11548-11556.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kondo E, Mammano F, Cohen EA, Gottlinger HG. J Virol. 1995;69:2759–64. doi: 10.1128/jvi.69.5.2759-2764.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan W, Dong Z, Wilkinson TA, Barbas CF, 3rd, Chow SA. J Virol. 2006;80:1939–48. doi: 10.1128/JVI.80.4.1939-1948.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu X, Liu H, Xiao H, Kim J, Seshaiah P, Natsoulis G, Boeke JD, Hahn BH, Kappes JC. J Virol. 1995;69:3389–98. doi: 10.1128/jvi.69.6.3389-3398.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ausubel FA, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. Wiley; New York: 1999. [Google Scholar]
- 56.Fletcher TM, III, Soares MA, McPhearson S, Hui H, Wiskerchen M, Muesing MA, Shaw GM, Leavitt AD, Boeke JD, Hahn BH. EMBO J. 1997;16:5123–38. doi: 10.1093/emboj/16.16.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wong-Staal F, Chanda PK, Ghrayeb J. AIDS Res Hum Retroviruses. 1987;3:33–39. doi: 10.1089/aid.1987.3.33. [DOI] [PubMed] [Google Scholar]
- 58.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. Science. 1996;272:263–67. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 59.Brussel A, Sonigo P. J Virol. 2003;77:10119–24. doi: 10.1128/JVI.77.18.10119-10124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Doherty U, Swiggard WJ, Jeyakumar D, McGain D, Malim MH. J Virol. 2002;76:10942–50. doi: 10.1128/JVI.76.21.10942-10950.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, Szustakowki J, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 62.Butler SL, Hansen MS, Bushman FD. Nature Med. 2001;7:631–34. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 63.Bushman FD, Hoffmann C, Ronen K, Malani N, Minkah N, Rose HM, Tebas P, Wang GP. Aids. 2008;22:1411–15. doi: 10.1097/QAD.0b013e3282fc972e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. Genome Res. 2007;17:1186–94. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Appa RS, Shin CG, Lee P, Chow SA. J Biol Chem. 2001;276:45848–55. doi: 10.1074/jbc.M107365200. [DOI] [PubMed] [Google Scholar]
- 66.Harper AL, Sudol M, Katzman M. J Virol. 2003;77:3838–45. doi: 10.1128/JVI.77.6.3838-3845.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ren G, Gao K, Bushman FD, Yeager M. J Mol Biol. 2007;366:286–94. doi: 10.1016/j.jmb.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chevalier BS, Kortemme T, Chadsey MS, Baker D, Monnat RJ, Jr, Stoddard BL. Mol Cell. 2002;10:895–905. doi: 10.1016/s1097-2765(02)00690-1. [DOI] [PubMed] [Google Scholar]
- 69.Porteus MH, Carroll D. Nat Biotechnol. 2005;23:967–73. doi: 10.1038/nbt1125. [DOI] [PubMed] [Google Scholar]
- 70.Kirchner J, Connolly CM, Sandmeyer SB. Science. 1995;267:1488–91. doi: 10.1126/science.7878467. [DOI] [PubMed] [Google Scholar]
- 71.Xie W, Gai X, Zhu Y, Zappulla DC, Sternglanz R, Voytas DF. Mol Cell Biol. 2001;21:6606–14. doi: 10.1128/MCB.21.19.6606-6614.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zou S, Voytas DF. Proc Natl Acad Sci USA. 1997;94:7412–16. doi: 10.1073/pnas.94.14.7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Turlure F, Devroe E, Silver PA, Engelman A. Front Biosci. 2004;9:3187–208. doi: 10.2741/1472. [DOI] [PubMed] [Google Scholar]
- 74.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. Science. 2006;314:461–64. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- 75.Turlure F, Maertens G, Rahman S, Cherepanov P, Engelman A. Nucleic Acids Res. 2006;34:1653–65. doi: 10.1093/nar/gkl052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cherepanov P, Sun ZY, Rahman S, Maertens G, Wagner G, Engelman A. Nat Struct Mol Biol. 2005;12:526–32. doi: 10.1038/nsmb937. [DOI] [PubMed] [Google Scholar]
- 77.Llano M, Vanegas M, Fregoso O, Saenz D, Chung S, Peretz M, Poeschla EM. J Virol. 2004;78:9524–37. doi: 10.1128/JVI.78.17.9524-9537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. Genes Dev. 2007;21:1767–78. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ciuffi A, Diamond TL, Hwang Y, Marshall HM, Bushman FD. Hum Gene Ther. 2006;17:960–67. doi: 10.1089/hum.2006.17.960. [DOI] [PubMed] [Google Scholar]