

Abstract
Hepatic ischemia/reperfusion (I/R) leads to liver injury and dysfunction through the initiation of a biphasic inflammatory response that is regulated by the transcription factor, NF-κB. We have previously shown that there is an age-dependent difference in the injury response to hepatic I/R in mice that correlates with divergent activation of NF-κB such that young mice have greater NF-κB activation, but less injury than old mice. In the present study, we investigated the mechanism by which age alters the activation of NF-κB in the liver during I/R. Young (4-5 weeks) and old (12-14 months) mice underwent partial hepatic ischemia/reperfusion. Livers were obtained for RNA microarray analysis and protein expression assays. Using microarray analysis, we identified age-dependent differences in the expression of genes related to protein ubiquitinylation and the proteasome. In old mice, genes that are involved in the ubiquitin-proteasome pathway were significantly down-regulated during I/R. Consistent with these findings, expression of a critical proteasome subunit, non-ATPase 4 (PSMD4), was reduced in old mice. Expression of the NF-κB-inhibitory protein, IκBα, was increased in old mice and was greatly phosphorylated and ubiquitinylated. The data provide strong evidence that the age-related defect in hepatic NF-κB signaling during I/R is a result of decreased expression of PSMD4, a proteasome subunit responsible for recognition and recruitment of ubiquitinylated substrates to the proteasome. It appears that decreased PSMD4 expression prevents recruitment of phosphorylated and ubiquitinylated IκBα to the proteasome, resulting in a defect in NF-κB activation.
Keywords: liver injury, inflammation, ageing, genomics, microarray
Ischemia/reperfusion (I/R) injury of the liver is a major complication of transplantation, liver resection and hypovolemic shock (1-5). Extended periods of hepatic ischemia and subsequent reperfusion lead to liver injury and dysfunction through the initiation of a biphasic inflammatory response (6, 7). The acute phase of injury is related to the generation of reactive oxygen species which cause mild hepatocellular injury (8-10). The subacute phase of injury is characterized by the production of inflammatory mediators that culminate in the recruitment of neutrophils to the injured liver (11). The neutrophils then directly damage hepatocytes and vascular endothelial cells through their release of oxidants and proteases (12).
Recently, our laboratory reported that there is a distinct difference in the injury response to hepatic I/R in mice of different ages (13). Subsequently, other laboratories reported similar results.(14). These studies demonstrated that the degree of liver injury was far worse in older mice. Interestingly, trauma and critical care physicians have long noted a significant divergence in the responses of pediatric and adult populations to severe trauma (15). There is also evidence that age is an important factor in liver transplantation as well as liver resection with or without ischemic-preconditioning (16, 17). Our earlier work discovered that activation of the transcription factor, NF-κB, was abrogated after liver I/R in old mice (13). Since NF-κB is critical for hepatocellular proliferation and survival (18, 19), reduced activation of NF-κB in old mice may represent a mechanism contributing to increased liver dysfunction after I/R.
NF-κB activation in the liver during I/R is initiated with the stimulation of hepatic parenchymal and non-parenchymal cells by reactive oxygen species and proinflammatory mediators. This stimulation leads to activation of the IκB kinase complex (IKK) which phosphorylates the NF-κB inhibitory protein, IκBα (20). Phosphorylated IκBα then becomes the target of ubiquitin ligase which polyubiquitinylates the protein for subsequent degradation by the proteasome (21). Degradation of IκBα by the proteasome exposes nuclear localization sequences and allows nuclear translocation of the NF-κB complex (22).
In the present study, we investigated the mechanism by which age alters the activation of NF-κB in the liver during I/R. Using microarray-based genome wide expression analysis, we found that gene expression of NF-κB signaling proteins were not different in young versus old mice. However, we found a marked reduction in the expression of a critical proteasome subunit in old mice that was associated with defective degradation of the NF-κB inhibitory protein, IκBα. The data suggest that reduced expression of proteasome-related genes contributes to defective processing of IκBα and therefore subsequent activation of NF-κB in the livers of old mice.
Methods
Hepatic I/R injury
Male C57BL/6 mice (4-5 weeks of age or 12-14 months of age) were obtained from Harlan Sprague Dawley (Indianapolis,IN) and Charles River Laboratories, Inc. (Wilmington, WA). In this study, 4-5 week old mice were called “young” and 12-14 month old mice were called “old.” This project was approved by the University of Cincinnati Animal Care and Use Committee and conforms to the National Institutes of Health guidelines. Partial hepatic ischemia was induced as described previously (23). Briefly, mice were anaesthetized with pentobarbital (100mg/kg, i.p.). A midline laparotomy was performed and an atraumatic clip was used to interrupt blood supply to the left and median lobe of the liver. After different time points of ischemia (30 min, 60 min, or 90 min) or 90 min of ischemia followed by 1 hour of reperfusion, mice were sacrificed. Sham control mice underwent the same protocol without vascular occlusion. Liver tissue from the median and left lobes were sampled upon sacrifice and immediately submerged into RNAlater™ (Qiagen, Valencia, CA) solution according to the manufactures protocol and stored at -80°C until RNA extraction. Additional liver samples were obtained for protein analysis by Western blot.
RNA preparation and analysis of whole mouse genome microarrays
The data and protocols described in this manuscript have been deposited in the National Center for Biotechnology Information Gene Expresion Omnibus (GEO. http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series number GSE10657. RNA was extracted from the liver tissue of each mouse (n=3) using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. The quality of RNA was assessed by the Agilent 2100 Bioanalyzer using the RNA 6000 Nano Assay (Ambion/Applied Biosystems, Foster City, CA). Affymetrix Mouse Genome MOE 430 2.0 GeneChip® arrays were hybridized with labeled, non-pooled total RNA extracts, using one chip per animal (n=3 for each group). Arrays containing 45,101 well-characterized mouse genes/ESTs were used to examine the ischemic-liver transcriptom. Target preparation, hybridization, and initial data collection were done at the Affymetrix Genechip® Microarray Core, at the Cincinnati Children’s Hospital Research Foundation, according to standard protocols (Affymetrix, Inc., Santa Clara, CA).
Analysis and Statistics
Analyses were performed using one liver sample per chip. Image files were captured using an Affymetrix GeneChip Scanner 3000. The data were analyzed with GeneSpring GX 7.3 software (Silicon Genetics, Agilent Technologies, Palo Alto, CA) using Robust Multiple-Array Average (RMA) normalization with a custom CDF file (Mm430_Mm_REFSEQ_8), and were then normalized to median values of sham controls. Differential regulation was defined as 1.5-fold difference from sham control mice (P<0.05; False Detection Rate P<0.05 using Benjamini Hochberg multiple testing correction).
Gene lists of differentially expressed genes were primarily analyzed using the Ingenuity Pathway Analysis (IPA) application (Ingenuity Systems, Redwood City, CA) that provides a tool for discovery of canonical signaling pathways and gene networks within the uploaded gene lists (24). Additional analyses for derivation of functional gene annotations were preformed by uploading specific gene expression lists to two distinct web-based applications that allow public access to relational databases of functional gene annotations: D.A.V.I.D. (Database for Annotation, Visualization and Integrated Discovery) (25) and PANTHER Classification System (Protein Analysis Through Evolutionary Relationships) (26, 27). These applications use specific approaches to estimate significance (p-values) based on non-redundant representations of the microarray chip and to convert the uploaded gene lists to gene lists containing a single value for each gene. The p-values for a given category and term provide an estimate of the likelihood that a given annotation in enriched in a given gene list by chance alone.
Western Blot and Immunoprecipitation
Hepatocytes and Kupffer cells were isolated and processed as previously described (28). Liver samples were homogenized in RIPA buffer (50 mM Tris HCl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate (NaDOC), 1% SDS) on ice. An appropriate amount of protease inhibitors was added (Calbiochem, Merck KGaA, Darmstadt, Germany) before homogenates were sonicated and centrifuged 3 times at 10,000 × g at 4°C to remove cellular debris. Protein concentrations were determined and samples containing equal amounts of protein in equal volumes of sample buffer were separated in a denaturating 10% polyacrylamid gel, transferred to a 0.1-μm pore nitrocellulose membrane and probed with antibodies against IκBα, Ser-32, Ser-36 phosphorylated-IκBα, PSMA5, or S5a-PSMD4 (cell signaling, Boston, MA) overnight at 4°C. After washing and incubating with a secondary antibody conjugated to horseradish peroxidase, immunoreactive proteins were detected by enhanced chemiluminescence. IκBα proteins were immunoprecipitated from cytoplasmic extracts with A/G agarose together with an IκBα antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Protein blots of the immunoprecipitates were prepared, probed with a polyubiquitin antibody (Abcam, Cambridge, UK), and processed for enhanced chemiluminescence detection as described above.
Proteasome activity assay
Proteasome activity in liver extracts was determined using a fluorogenic assay (Chemicon, Billerica, MA) which is based on the detection of the fluorophore, 7-amino-4-metylcoumarin (AMC) after cleavage from the labeled substrate LLVY-AMC.
siRNA Knockdown of PSMD4
AML-12 hepatocytes were grown in 6-well flat-bottomed plates at a density of 3 × 105 cells/well. siRNA and transfection reagents were obtained from Qiagen (Valencia, CA). Monolayers of AML-12 cells were transfected 24 hours after plating with 20 nM of non-specific or PSMD4 siRNA using 12ml HiPerFect Transfection Reagent according to the manufacturers protocol. Seventy-two hours later, cells were treated with media or 10 ng/ml TNFα (R&D Systems, Minneapolis, MN) for 15 minutes. Cells were then lysed and prepared for Western blot analysis.
Results
Age-Specific Expression of Genes Related to Protein Ubiquitinylation and the Proteasome
To determine the effect of age on the expression of genes related to the NF-κB signaling pathway during hepatic I/R, RNA isolated from post-ischemic liver tissue was analyzed using Affymetrix gene chips. To eliminate unreliable data, raw expression data was normalized and pre-filtered as described in Materials and Methods. We assessed genes that had significantly reduced or increased expression (1.5-fold cut-off, P < 0.05) when compared to sham-operated controls. To focus on the significance of the NF-κB signaling pathway in young and old mice, the gene lists representing decreased as well as increased gene expression were analyzed further using the Ingenuity Pathway Analysis (IPA) application (24). Each list of up- or down-regulated genes during different points of ischemia or I/R was individually uploaded to the IPA application and analyzed with regard to the pathways of interest. Surprisingly, the analysis did not reveal any age-related differences in the NF-κB pathway. However, genes corresponding to the protein ubiquitinylation pathway were consistently down-regulated after 60 and 90 minutes of ischemia, as well as after 90 minutes of ischemia and 1 hour of reperfusion exclusively in old mice (Table 1). The differences observed in this pathway were highly significant, with p-values ≤ 3.4E-9 Furthermore, not a single gene in this pathway was significantly down-regulated in young mice. The gene lists generated by these analyses are shown in Table 2.
Table 1.
Gene signaling pathways and networks down-regulated in old mice.
| Condition | Signaling Pathway (p value) |
Network Score |
Database | Functional annotations (p value) |
|---|---|---|---|---|
| 60 min ischemia | Protein ubiqutination (3.8E-18) |
44 | D.A.V.I.D. | SP_PIR_KEYWORDS, proteasome (1.3E-31) |
|
|
||||
| PANTHER |
Pathway: Ubiquitin proteasome pathway (8.6E-18) |
|||
|
Biological Process: Proteolysis (5.8E-13) |
||||
|
Molecular Function: Other proteases (1.4E-6) |
||||
|
| ||||
| 90 min ischemia | Protein ubiqutination (3.0E-12) |
50 | D.A.V.I.D. | INTERPRO_NAME, proteasome α subunit (1.3E-3) |
|
|
||||
| PANTHER |
Pathway: Parkinsons disease (3.4E- 5) |
|||
|
Biological Process: Protein metabolism and modification (6.8E- 6) |
||||
|
Molecular Function: Chaperonin (5.7E-6) |
||||
|
|
||||
| 45 | D.A.V.I.D. | SP_PIR_KEYWORDS, proteasome (2.2E-15) |
||
|
|
||||
| PANTHER |
Pathway: Ubiquitin proteasome pathway (1.2E-9) |
|||
|
Biological Process: Proteolysis (1.3E-4) |
||||
|
Molecular Function: Nucleic acid binding (1.0E-2) |
||||
|
| ||||
| 90 min ischemia + 1 h reperfusion |
Protein ubiqutination (3.4E-9) |
38 | D.A.V.I.D. | SP_PIR_KEYWORDS, proteasome (4.3E-18) |
|
|
||||
| PANTHER |
Pathway: Ubiquitin proteasome pathway (2.0E-9) |
|||
|
Biological Process: Protein metabolism and modification (9.7E- 8) |
||||
|
Molecular Function: Other proteases (7.4E-3) |
||||
Table 2.
Down-regulated genes of the protein-ubiquitination pathway in old mice.
| Ischemia (60 minutes) | ||
|---|---|---|
| Gene | Description | GenBank |
| BAG1 | BCL2-associated athanogene | NM_009736 |
| HSP90AA1 | heat shock protein 90kDa alpha, class A member 1 | NM_010480 |
| HSPA5 | heat shock 70kDa protein 5 | NM_022310 |
| PSMA1 | proteasome subunit, alpha type, 1 | NM_011965 |
| PSMA2 | proteasome subunit, alpha type, 2 | NM_008944 |
| PSMA4 | proteasome subunit, alpha type, 4 | NM_011966 |
| PSMA5 | proteasome subunit, alpha type, 5 | NM_011967 |
| PSMA7 | proteasome subunit, alpha type, 7 | NM_011969 |
| PSMC1 | proteasome 26S subunit, ATPase, 1 | NM_008947 |
| PSMC2 | proteasome 26S subunit, ATPase, 2 | NM_011188 |
| PSMC4 | proteasome 26S subunit, ATPase, 4 | NM_011874 |
| PSMC5 | proteasome 26S subunit, ATPase, 5 | NM_008950 |
| PSMC6 | proteasome 26S subunit, ATPase, 6 | NM_025959 |
| PSMD2 | proteasome 26S subunit, non-ATPase, 2 | NM_134101 |
| PSMD4 | proteasome 26S subunit, non-ATPase, 4 | NM_008951 |
| PSMD6 | proteasome 26S subunit, non-ATPase, 6 | NM_025550 |
| PSMD7 | proteasome 26S subunit, non-ATPase, 7 | NM_010817 |
| PSMD11 | proteasome 26S subunit, non-ATPase, 11 | NM_178616 |
| PSMD12 | proteasome 26S subunit, non-ATPase, 12 | NM_025894 |
| PSMD14 | proteasome 26S subunit, non-ATPase, 14 | NM_021526 |
| SKP1A | S-phase kinase-associated protein 1 | NM_011543 |
| TCEB1 | transcription elongation factor B (SIII), polypeptide 1 | NM_026456 |
| UBE2E1 | ubiquitin-conjugating enzyme E2E 1 | XM_975886 |
| UBE2L6 | ubiquitin-conjugating enzyme E2L 6 | NM_019949 |
| UBE2V2 | ubiquitin-conjugating enzyme E2 variant 2 | NM_023585 |
| UBE3A | ubiquitin protein ligase E3A | NM_173010 |
| UCHL3 | ubiquitin carboxyl-terminal esterase L3 | NM_016723 |
| UCHL5 | ubiquitin carboxyl-terminal hydrolase L5 | NM_019562 |
| USP1 | ubiquitin specific peptidase 1 | NM_146144 |
| USP14 | ubiquitin specific peptidase 14 | NM_001038589 |
| USP33 | ubiquitin specific peptidase 33 | NM_133247 |
| VDP | USO1 homolog, vesicle docking protein | NM_019490 |
| Ischemia (90 minutes) | ||
|---|---|---|
| Gene | Description | GenBank |
| HSP90AA1 | heat shock protein 90kDa alpha, class A member 1 | NM_010480 |
| HSPA5 | heat shock 70kDa protein 5 | NM_022310 |
| NEDD4 | neural precursor cell expressed, developmentally down-regulated | NM_010890 |
| PSMA1 | proteasome subunit, alpha type, 1 | NM_011965 |
| PSMA2 | proteasome subunit, alpha type, 2 | NM_008944 |
| PSMA4 | proteasome subunit, alpha type, 4 | NM_011966 |
| PSMA5 | proteasome subunit, alpha type, 5 | NM_011967 |
| PSMA7 | proteasome subunit, alpha type, 7 | NM_011969 |
| PSMC3 | proteasome 26S subunit, ATPase, 3 | NM_008948 |
| PSMC6 | proteasome 26S subunit, ATPase, 6 | NM_025959 |
| PSMD2 | proteasome 26S subunit, non-ATPase, 2 | NM_134101 |
| PSMD4 | proteasome 26S subunit, non-ATPase, 4 | NM_008951 |
| PSMD7 | proteasome 26S subunit, non-ATPase, 7 | NM_010817 |
| PSMD10 | proteasome 26S subunit, non-ATPase, 10 | NM_016883 |
| PSMD11 | proteasome 26S subunit, non-ATPase, 11 | NM_178616 |
| PSMD12 | proteasome 26S subunit, non-ATPase, 12 | NM_025894 |
| PSMD14 | proteasome 26S subunit, non-ATPase, 14 | NM_021526 |
| PSME2 | proteasome activator subunit 2 (PA28 beta) | NM_001029855 |
| TCEB1 | transcription elongation factor B (SIII), polypeptide 1 | NM_026456 |
| UBE2B | ubiquitin-conjugating enzyme E2B | NM_009458 |
| UBE2E1 | ubiquitin-conjugating enzyme E2E 1 | NM_009455 |
| UCHL5 | ubiquitin carboxyl-terminal hydrolase L5 | NM_019562 |
| USP1 | ubiquitin specific peptidase 1 | NM_146144 |
| USP7 | ubiquitin specific peptidase 7 | NM_001003918 |
| Ischemia/Reperfusion (90 minutes/1 hour) | ||
|---|---|---|
| Gene | Description | GenBank |
| PSMA1 | proteasome subunit, alpha type, 1 | NM_011965 |
| PSMA2 | proteasome subunit, alpha type, 2 | NM_008944 |
| PSMA4 | proteasome subunit, alpha type, 4 | NM_011966 |
| PSMA5 | proteasome subunit, alpha type, 5 | NM_011967 |
| PSMA7 | proteasome subunit, alpha type, 7 | NM_011969 |
| PSMC1 | proteasome 26S subunit, ATPase, 1 | NM_008947 |
| PSMC3 | proteasome 26S subunit, ATPase, 3 | NM_008948 |
| PSMC6 | proteasome 26S subunit, ATPase, 6 | NM_025959 |
| PSMD4 | proteasome 26S subunit, non-ATPase, 4 | NM_008951 |
| PSMD7 | proteasome 26S subunit, non-ATPase, 7 | NM_010817 |
| PSMD10 | proteasome 26S subunit, non-ATPase, 10 | NM_016883 |
| PSMD11 | proteasome 26S subunit, non-ATPase, 11 | NM_178616 |
| PSMD12 | proteasome 26S subunit, non-ATPase, 12 | NM_025894 |
| PSMD14 | proteasome 26S subunit, non-ATPase, 14 | NM_021526 |
| UBE3A | ubiquitin protein ligase E3A | NM_173010 |
| UBR1 | ubiquitin protein ligase E3 component n-recognin 1 | NM_009461 |
| UCHL3 | ubiquitin carboxyl-terminal esterase L3 | NM_016723 |
| UCHL5 | ubiquitin carboxyl-terminal hydrolase L5 | NM_019562 |
| USP1 | ubiquitin specific peptidase 1 | NM_146144 |
| USP3 | ubiquitin specific peptidase 3 | NM_144937 |
| USP7 | ubiquitin specific peptidase 7 | NM_001003918 |
| USP15 | ubiquitin specific peptidase 15 | NM_027604 |
Gene Network Analysis of Ubiquitin-Proteasome Pathway during I/R
To explore the potential biologically relevance of the gene lists defined in Table 2, these lists were uploaded into the IPA application for network analysis. The resulting networks had significant scores (Table 1) which exclude the likelihood that focus genes in these given networks are found together due to random chance. A score of 2 or higher has a greater than 99% confidence of not being generated by random chance alone. We performed additional analyses for derivation of functional gene annotations by uploading the specific network gene lists to two distinct web-based applications, the D.A.V.I.D. (25) and PANTHER Classification System (26, 27). As shown in Table 1, both analyses yielded functional annotations involved in the ubiquitin proteasome pathway. Figure 1 shows the focus genes that are involved in the down-regulation of the ubiquitin-proteasome pathway after 60 minutes of ischemia in old mice. Decreased genes were found to be related to the PA700 (also called the 19S regulatory complex)/20S, the proteasome 26S subunit, non-ATPase (PSMD), as well as the proteasome 26S subunit ATPase (PSMC). The networks constructed with genes that are decreased after 90 minutes of ischemia revealed two networks. The first network contained focus genes concentrated around the proteasome 19S/20S subunit as well as the immunoproteasome Pa28 (also called 11S regulatory complex)/20S (Figure 2A). The second network contained genes that were focused on the PSMD as well as ubiquitin signaling (Figure 2B). After ischemia and 1 hour of reperfusion the network contained focus genes that were assembled around PA700/20S, PSMD, and the PSMC (Figure 3). These results suggest that the down-regulation of genes in the ubiquitin-proteasome pathway are specifically controlled during different times of ischemia or I/R in old mice, but not in young mice.
Figure 1.

Ingenuity pathway analysis identified a network of genes down regulated in response to 60 minutes of ischemia in aged mouse liver. The network is displayed graphically as nodes (genes/gene products) and edges (the biological relationships between the nodes). Different shapes of nodes represent the functional class of the gene product. Edges describe the nature of the relationship between the nodes. A total of 17 differentially expressed focus genes involved in the proteasome pathway are identified: proteasome subunit, alpha type (PSMA) 1, 2, 4, 5, and 7; proteasome 26S subunit, ATPase (PSMC) 1,2,4,5, and 6; proteasome 26S subunit, non-ATPase (PSMD) 2, 4, 6, 7, 11, and 12; and phosducin-like (PDCL).
Figure 2.


Ingenuity pathway analysis identified two networks of genes down regulated in response to 90 minutes of ischemia in aged mouse liver. (A) Network 1 shows 6 differentially expressed focus genes involved in the proteasome ubiquitin pathway which include proteasome subunit, alpha type (PSMA) 1, 4, 5, and 7; cytoplasmic FMR1 interacting protein 1 (CYFIP1); and ring finger protein 14 (RNF14). (B) Network 2 classified 14 differentially expressed focus genes involved in the proteasome ubiquitinylation pathway. Proteasome 26S subunit, non- ATPase (PSMD) 2, 4, 7, 10, 11, 12 and 14; N-myc (and STAT) interactor (NMI); histidyl-tRNA synthetase (HARS); proteasome subunit, alpha type 2 (PSMA2); proteasome 26S subunit, non-ATPase (PSMD) 3 and 6; eukaryotic translation initiation factor 3, subunit E (EIF3S6); and COP9 (constitutive photomorphogenic) homolog, subunit 6 (Arabidopsis thaliana) (COPS6).
Figure 3.

Ingenuity pathway analysis shows a network of 17 decreased focus genes involved in the proteasome pathway in old mice after 90 minutes of ischemia followed by 1 hour of reperfusion, which include proteasome subunit, alpha type (PSMA) 2, 4, and 7; proteasome 26S subunit, ATPase (PSMC) 1,3, and 6; proteasome 26S subunit, non-ATPase (PSMD) 4, 7, 10, 11, and 12; eukaryotic translation initiation factor 3, subunit E (EIF3S6); COP9 homolog, subunit (COPS) 2, 4 and 6; nuclear receptor subfamily 3, group C, member 1 (NR3C1); and glutamate-ammonia ligase (glutamine synthetase) (GLUL).
Accumulation of Phosphorylated and Ubiquitinylated IκBα in Old Mice
Because our genomic data demonstrated that both ubiquitinylation- and proteasome-related genes were downregulated in old mice, we next examined which process (ubiquitinylation or proteasome-mediated degradation) might be contributing to the defect in NF-κB activation observed in old mice (13). To answer this question, IκBα protein expression was determined during ischemia and I/R. Hepatic IκBα protein expression was similar in young and old mice undergoing sham operation (Figure 4). After 90 minutes of ischemia, IκBα expression in old mice remained similar to sham-operated controls, whereas expression in young mice was greatly decreased, which is likely due to degradation of IκBα (Figure 4). After 90 minutes of ischemia and 1 hour of reperfusion, hepatic IκBα expression decreased in old mice and remained was even lower in young mice (Figure 4). We next evaluated the phosphorylation state of IκBα. Interestingly, IκBα was phosphorylated in sham-operated old mice (Figure 4). This phosphorylation increased after 90 minutes of ischemia and 1 hour of reperfusion. In young mice, no phosphorylation of IκBα was detected (Figure 4). The lack of IκBα phosphorylation in young mice could be due to the increased degradation observed in this age group. Thus, these data suggest that IκBα degradation occurs much more rapidly and robustly in young mice compared to old mice.
Figure 4.

Effects of age on protein expression of total (upper panel) and phosphorylated (lower panel) IκBα. Cytoplasmic fractions of whole liver tissue from from young (4-5 weeks) and old (12-14 months) mice undergoing sham surgery, 90 minutes of ischemia (90/0), or ischemia and 1 hour of reperfusion (90/1) were immunoblotted for total IκBα or phosphorylated IκBα (p-IκBα).
We next examined the extent of IκBα polyubiquitinylation during I/R by immunoprecipitating IκBα and probing for polyubiquitin. Ubiquitinylation is a requisite step for degradation of IκBα by the proteasome (29). Similar to our results of IκBα phosphorylation, we found that sham-operated old mice had ubiquitinylated IκBα (Figure 5). Ubiquitinylation of IκBα in old mice was increased after ischemia or I/R. In contrast, IκBα in young sham controls was not ubiquitinylated and IκBα became ubiquitinylated only after I/R (Figure 5). These data demonstrate that ubiquitinylation of IκBα is not the limiting step in old mice for IκBα degradation.
Figure 5.

Effects of age on polyubiquitinylation of IκBα during hepatic I/R injury. Cytoplasmic fractions from young (4-5 weeks) and old (12-14 months) mice undergoing sham surgery, 90 minutes of ischemia (90/0), or ischemia and 1 hour of reperfusion (90/1) were immunoprecipited (IP) with anti-IκBα followed by immunoblotting (IB) with anti-polyubiquitin.
To examine the possibility that proteolytic activity of the proteasome might be the age-related defect in NF-κB signaling, we evaluated proteasome activity in liver extracts from young and old mice. This assay uses a peptide substrate that is not poly-ubiquitinylated and therefore would not be affected by any proteasome defect (ie, decreased PSMD4) affecting recruitment of ubiquitinylated substrates. At no time throughout the course of I/R did we observe any differences in proteasome activity between liver extracts from young and old mice (Figure 6).
Figure 6.
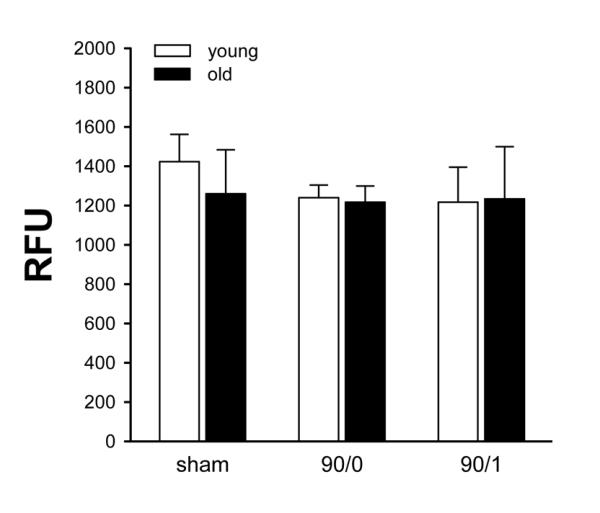
Effects of age on proteasome proteolytic activity. Proteasome activity was determined in liver extracts from young (4-5 weeks) and old (12-14 months) mice undergoing sham surgery, 90 minutes of ischemia (90/0), or ischemia and 1 hour of reperfusion (90/1). Data are expressed as mean ± SEM with n=3 per group.
Reduced Proteasome Subunit Protein Expression in Hepatocytes from Old Mice
Because our IκBα data implicated the proteasome as the prime contributor to the defect in NF-κB activation, we examined the protein expression of two critical proteasome subunits. The proteasome subunit, type alpha 5 (PSMA5) is a subunit of the 20S proteasome, which comprises the proteolytic core particle of the 26S proteasome (30). The 19S proteasome subunit, non-ATPase 4 (PSMD4), has a quite different function, to recruit ubiquitinylated substrates to the proteasome for degradation (31, 32). We selected these two proteins based on their divergent function. Expression of PSMA5 was not different between age groups and was not affected by ischemia or I/R (Figure 7A). In contrast, expression of PSMD4 was significantly decreased in old mice compared to young mice (Figure 7A). These data validate our microarray-based data at the level of protein expression and suggest that the ability of the proteasome to recruit ubiquitinylated substrates, such as IκBα, impair its ability to process and degrade these substrates.
Figure 7.

Effects of age on protein expression of proteasome subunits. (A) Protein expression of PSMA5 (upper panel) and PSMD4 (lower panel) in cytoplasmic fractions of whole liver tissue from young (4-5 weeks) and old (12-14 months) mice undergoing sham surgery, 90 minutes of ischemia (90/0), or ischemia and 1 hour of reperfusion (90/1). PSMD4 expression was quantitated by image analysis. Data represent mean relative intensity units (RIU) ± SEM with n=3 per group. (B) Protein expression of PSMD4 in isolated Kupffer cells (KC) and hepatocytes (Hep) from young and old mice. Extracts from whole liver (WL) from young mice served as a control.
To further examine these effects and establish the cell-specificity of PSMD4 expression, we evaluated PSMD4 expression in isolated Kupffer cells and hepatocytes from young and old mice. We were unable to detect PSMD4 by Western blot in extracts of Kupffer cells from either young or old mice (Figure 7B). However, heptocytes from young mice expressed abundant PSMD4, whereas expression was very low in those from old mice (Figure 7B).
Decreased PSMD4 Expression in Old Mice is Causally Related to Decreased Degradation of IκBα in Hepatocytes
In order to confirm that the reduced hepatocyte PSMD4 expression observed in old mice was causative of the reduced IκBα degradation, we employed siRNA knockdown of PSMD4 in a murine hepatocyte cell line, AML-12. AML-12 cells transfected with siRNA to PSMD4 showed approximately 60% knockdown (Figure 8A). AML-12 cells that were not transfected, or those transfected with non-specific siRNA showed degradation of IκBα after treatment with TNFα (Figure 8B). In contrast, in cells transfected with siRNA to PSMD4, IκBα degradation was limited (Figure 8B). These data suggest that appropriate expression of PSMD4 is critical for degradation of IκBα by the proteasome.
Figure 8.
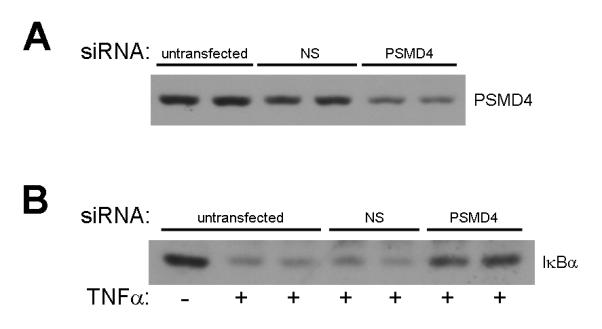
Effect of PSMD4 knockdown on TNFα-induced IκBα degradation in hepatocytes. (A) Transfection of AML-12 cells with non-specific (NS) or PSMD4 siRNA. (B) Effects of TNFα (10 ng/ml for 15 minutes) on untransfected AML-12 cells or cells transfected with NS or PSMD4 siRNA.
Discussion
We have previously demonstrated that NF-κB activation in the liver is greatly reduced in old mice compared to young mice after I/R (13). The objective of the current study was to examine the mechanism of this effect of age. We used a genomic analysis to guide our efforts and found no age-related differences in expression of genes of the NF-κB family (ie, p50, p65, IKKα/β, IκBα, etc.). We did, however, find substantial age-related differences in the expression of genes of the ubiquitin-proteasome pathway. NF-κB activation is initiated with phosphorylation of the inhibitory protein, IκBα. Phosphorylation of IκBα, generally regarded as the rate-limiting step in activation of the NF-κB pathway, targets this protein for polyubiquitinylation and subsequent degradation by the proteasome (21). We found that phosphorylated IκBα was accumulated in livers from old mice. Consistent with these findings were the demonstration that old mice had accumulation of polyubiquitinylated IκBα, implicating a blockade at the downstream degradation pathway. Our data indicate that there is not an age-related defect in the ability to ubiquitinylate IκBα, but rather a build-up of ubiquitinylated IκBα awaiting processing and degradation by the proteasome. It is important to note that proteasome inhibitors effectively prevent nuclear translocation of NF-κB, indicating that neither phosphorylation nor ubiquitinylation of IκBα is sufficient to cause its dissociation from NF-κB (33-35).
When we analyzed relevant proteasome subunits, we found decreased expression of genes of belonging to subunits of the 20S proteasome, which is the proteolytically active component (also called the core particle) of the 26S proteasome. In addition, we detected gene expression of subunits belonging to the 19S regulatory particle of the 26S proteasome. The ATPase subunits of the 19S proteasome exhibit a chaperone-like activity and are believed to unfold proteins prior to their entry in the proteolytic chamber (36, 37), whereas one of the non-ATPase regulatory subunits, the PSMD4, has been demonstrated to be responsible for the recognition of the polyubiquitin signal by binding multi-ubiquitin chains (31, 32). Expression of PSMD4 was decreased only in livers of old mice and we found no age-related difference in the proteolytic activity of the proteasome. Thus, our data suggest that in old mice the decreased expression of PSMD4, and perhaps related subunits, prevents the recruitment of ubiquitinylated IκBα leading to accumulation of IκBα in the cytoplasm where it continues to prevent NF-κB activation.
The function of NF-κB in the liver during I/R injury is complex and likely cell-specific. Clearly, NF-κB is involved in the regulation of proinflammatory mediators that contribute to the inflammatory response to this insult (7). However, NF-κB is also important for hepatocyte survival (38), and our prevous studies indicate that hepatocyte NF-κB activation is hepatoprotective after I/R (28). The most compelling evidence in this regard comes from the finding that NF-κB p65 knockout mice die in utero due to massive hepatic apoptosis (39). Interference with NF-κB activation by knockout of different components of the IKK complex has differential effects on hepatic I/R injury (40, 41). Our previous work has shown that old mice have more liver injury after I/R, but greatly reduced NF-κB activation (13). However, these studies also showed that there was far less inflammation in the livers of old mice. Treatment of hepatocytes with proteasome inhibitors prevents NF-κB activation and induces apoptosis (42). As such, it is plausible to suggest that the defect in proteasome function that we observe in old mice resulting in reduced NF-κB activation, may induce apoptotic signaling within the hepatic parenchyma. Others have shown that ischemic hepatocytes respond to apoptotic stimuli by undergoing necrosis as a result of low ATP availability (43). This is consistent with the pattern of injury observed in old mice after I/R (13, 14).
Decreased proteasome function is known to occur with ageing (44, 45). However, the linkage of age-related alterations in the proteasome and activation of NF-κB in an injury response have not been previously studied. The current report provides strong evidence that the age-related defect in NF-κB signaling in the liver after I/R is a result of decreased expression of PSMD4, a proteasome subunit responsible for recognition and recruitment of ubiquitinylated substrates to the proteasome. These findings are relevant to clinical observations of increased organ injury and dysfunction in older individuals after trauma, surgery and transplantation (15-17).
Acknowledgments
Grant Support: This work was supported by National Institutes of Health grants AG025881 and DK56029 to A.B.L. and GM064619 to H.R.W.
Abbreviations
- D.A.V.I.D.
Database for Annotation, Visualization and Integrated Discovery
- I/R
ischemia/reperfusion
- IκBα
inhibitor of NF-κB-alpha
- IPA
Ingenuity Pathway Analysis
- NF-κB
nuclear factor-κB
- PANTHER
Protein Analysis Through Evolutionary Relationships
- PSMA
proteasome subunity, type alpha
- PSMC
proteasome 26S subunit, ATPase
- PSMD
proteasome 26S subunit, non-ATPase.
Footnotes
Financial Disclosures: None of the authors have any financial or other conflicts of interest.
References
- 1.Delva E, Camus Y, Nordlinger B, Hannoun L, Parc R, Deriaz H, Lienhart A, et al. Vascular occlusions for liver resections. Operative management and tolerance to hepatic ischemia: 142 cases. Ann Surg. 1989;209:211–218. doi: 10.1097/00000658-198902000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hawker F. Liver dysfunction in critical illness. Anaesth Intensive Care. 1991;19:165–181. doi: 10.1177/0310057X9101900203. [DOI] [PubMed] [Google Scholar]
- 3.Huguet C, Gavelli A, Bona S. Hepatic resection with ischemia of the liver exceeding one hour. J Am Coll Surg. 1994;178:454–458. [PubMed] [Google Scholar]
- 4.Lemasters JJ, Thurman RG. Reperfusion injury after liver preservation for transplantation. Annu Rev Pharmacol Toxicol. 1997;37:327–338. doi: 10.1146/annurev.pharmtox.37.1.327. [DOI] [PubMed] [Google Scholar]
- 5.Liu DL, Jeppsson B, Hakansson CH, Odselius R. Multiple-system organ damage resulting from prolonged hepatic inflow interruption. Arch Surg. 1996;131:442–447. doi: 10.1001/archsurg.1996.01430160100022. [DOI] [PubMed] [Google Scholar]
- 6.Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:G15–26. doi: 10.1152/ajpgi.00342.2002. [DOI] [PubMed] [Google Scholar]
- 7.Okaya T, Lentsch AB. Cytokine cascades and the hepatic inflammatory response to ischemia and reperfusion. J Invest Surg. 2003;16:141–147. [PubMed] [Google Scholar]
- 8.Jaeschke H. Reactive oxygen and ischemia/reperfusion injury of the liver. Chem Biol Interact. 1991;79:115–136. doi: 10.1016/0009-2797(91)90077-k. [DOI] [PubMed] [Google Scholar]
- 9.Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Physiol. 1991;260:G355–362. doi: 10.1152/ajpgi.1991.260.3.G355. [DOI] [PubMed] [Google Scholar]
- 10.Rymsa B, Wang JF, de Groot H. O2-. release by activated Kupffer cells upon hypoxia-reoxygenation. Am J Physiol. 1991;261:G602–607. doi: 10.1152/ajpgi.1991.261.4.G602. [DOI] [PubMed] [Google Scholar]
- 11.Jaeschke H, Farhood A, Smith CW. Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. FASEB J. 1990;4:3355–3359. [PubMed] [Google Scholar]
- 12.Jaeschke H, Smith CW. Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol. 1997;61:647–653. doi: 10.1002/jlb.61.6.647. [DOI] [PubMed] [Google Scholar]
- 13.Okaya T, Blanchard J, Schuster R, Kuboki S, Husted T, Caldwell CC, Zingarelli B, et al. Age-dependent responses to hepatic ischemia/reperfusion injury. Shock. 2005;24:421–427. doi: 10.1097/01.shk.0000181282.14050.11. [DOI] [PubMed] [Google Scholar]
- 14.Park Y, Hirose R, Coatney JL, Ferrell L, Behrends M, Roberts JP, Serkova NJ, et al. Ischemia-reperfusion injury is more severe in older versus young rat livers. J Surg Res. 2007;137:96–102. doi: 10.1016/j.jss.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Calkins CM, Bensard DD, Moore EE, McIntyre RC, Silliman CC, Biffl W, Harken AH, et al. The injured child is resistant to multiple organ failure: a different inflammatory response? J Trauma. 2002;53:1058–1063. doi: 10.1097/00005373-200212000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Clavien PA, Selzner M, Rudiger HA, Graf R, Kadry Z, Rousson V, Jochum W. A prospective randomized study in 100 consecutive patients undergoing major liver resection with versus without ischemic preconditioning. Ann Surg. 2003;238:843–850. doi: 10.1097/01.sla.0000098620.27623.7d. discussion 851-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markmann JF, Markmann JW, Markmann DA, Bacquerizo A, Singer J, Holt CD, Gornbein J, et al. Preoperative factors associated with outcome and their impact on resource use in 1148 consecutive primary liver transplants. Transplantation. 2001;72:1113–1122. doi: 10.1097/00007890-200109270-00023. [DOI] [PubMed] [Google Scholar]
- 18.Luedde T, Trautwein C. Intracellular survival pathways in the liver. Liver Int. 2006;26:1163–1174. doi: 10.1111/j.1478-3231.2006.01366.x. [DOI] [PubMed] [Google Scholar]
- 19.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 20.Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 22.Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, Baldwin AS., Jr. I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: a mechanism for cytoplasmic retention. Genes Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 23.Lentsch AB, Yoshidome H, Cheadle WG, Miller FN, Edwards MJ. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and KC. Hepatology. 1998;27:1172–1177. doi: 10.1002/hep.510270440. [DOI] [PubMed] [Google Scholar]
- 24.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 25.Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 26.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mi H, Guo N, Kejariwal A, Thomas PD. PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res. 2007;35:D247–252. doi: 10.1093/nar/gkl869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuboki S, Okaya T, Schuster R, Blanchard J, Denenberg A, Wong HR, Lentsch AB. Hepatocyte NF-kappaB activation is hepatoprotective during ischemia-reperfusion injury and is augmented by ischemic hypothermia. Am J Physiol Gastrointest Liver Physiol. 2007;292:G201–207. doi: 10.1152/ajpgi.00186.2006. [DOI] [PubMed] [Google Scholar]
- 29.Hatakeyama S, Kitagawa M, Nakayama K, Shirane M, Matsumoto M, Hattori K, Higashi H, et al. Ubiquitin-dependent degradation of IkappaBalpha is mediated by a ubiquitin ligase Skp1/Cul 1/F-box protein FWD1. Proc Natl Acad Sci U S A. 1999;96:3859–3863. doi: 10.1073/pnas.96.7.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Groll M, Huber R. Substrate access and processing by the 20S proteasome core particle. Int J Biochem Cell Biol. 2003;35:606–616. doi: 10.1016/s1357-2725(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 31.Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem. 1994;269:7059–7061. [PubMed] [Google Scholar]
- 32.Wang Q, Young P, Walters KJ. Structure of S5a bound to monoubiquitin provides a model for polyubiquitin recognition. J Mol Biol. 2005;348:727–739. doi: 10.1016/j.jmb.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 33.Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1995;92:10599–10603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DiDonato JA, Mercurio F, Karin M. Phosphorylation of I kappa B alpha precedes but is not sufficient for its dissociation from NF-kappa B. Mol Cell Biol. 1995;15:1302–1311. doi: 10.1128/mcb.15.3.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin YC, Brown K, Siebenlist U. Activation of NF-kappa B requires proteolysis of the inhibitor I kappa B-alpha: signal-induced phosphorylation of I kappa B-alpha alone does not release active NF-kappa B. Proc Natl Acad Sci U S A. 1995;92:552–556. doi: 10.1073/pnas.92.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Braun BC, Glickman M, Kraft R, Dahlmann B, Kloetzel PM, Finley D, Schmidt M. The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat Cell Biol. 1999;1:221–226. doi: 10.1038/12043. [DOI] [PubMed] [Google Scholar]
- 37.Glickman MH, Rubin DM, Fu H, Larsen CN, Coux O, Wefes I, Pfeifer G, et al. Functional analysis of the proteasome regulatory particle. Mol Biol Rep. 1999;26:21–28. doi: 10.1023/a:1006928316738. [DOI] [PubMed] [Google Scholar]
- 38.Shin T, Kuboki S, Lentsch AB. Roles of nuclear factor-kappaB in postischemic liver. Hepatol Res. 2008;38:429–440. doi: 10.1111/j.1872-034X.2007.00303.x. [DOI] [PubMed] [Google Scholar]
- 39.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 40.Luedde T, Assmus U, Wustefeld T, zu Vilsendorf A Meyer, Roskams T, Schmidt-Supprian M, Rajewsky K, et al. Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion injury. J Clin Invest. 2005;115:849–859. doi: 10.1172/JCI23493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beraza N, Ludde T, Assmus U, Roskams T, Vander Borght S, Trautwein C. Hepatocyte-specific IKK gamma/NEMO expression determines the degree of liver injury. Gastroenterology. 2007;132:2504–2517. doi: 10.1053/j.gastro.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 42.Bellas RE, FitzGerald MJ, Fausto N, Sonenshein GE. Inhibition of NF-kappa B activity induces apoptosis in murine hepatocytes. Am J Pathol. 1997;151:891–896. [PMC free article] [PubMed] [Google Scholar]
- 43.Lemasters JJ. V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am J Physiol. 1999;276:G1–6. doi: 10.1152/ajpgi.1999.276.1.G1. [DOI] [PubMed] [Google Scholar]
- 44.Hayashi T, Goto S. Age-related changes in the 20S and 26S proteasome activities in the liver of male F344 rats. Mech Ageing Dev. 1998;102:55–66. doi: 10.1016/s0047-6374(98)00011-6. [DOI] [PubMed] [Google Scholar]
- 45.Shibatani T, Nazir M, Ward WF. Alteration of rat liver 20S proteasome activities by age and food restriction. J Gerontol A Biol Sci Med Sci. 1996;51:B316–322. doi: 10.1093/gerona/51a.5.b316. [DOI] [PubMed] [Google Scholar]