

Abstract
Background
progression of chronic cholestatic disorders towards ductopenia results from the dysregulation of cholangiocyte survival, with cell death by apoptosis prevailing over compensatory proliferation. Currently, no therapy is available to sustain cholangiocyte survival in the course of those disorders. We have recently shown that cholangiocytes express the Glucagon-Like Peptide-1 receptor (GLP-1R); its activation results in enhanced proliferative reaction to cholestasis. The GLP-1R selective agonist exendin-4 sustains pancreatic β-cell proliferation and prevents cell death by apoptosis. Exendin-4 is now employed in humans as a novel therapy for diabetes.
Aim
to verify whether exendin-4 is effective in preventing cholangiocyte apoptosis.
Methods
in vitro, we tested if exendin-4 is able to prevent apoptosis of cholangiocytes isolated from normal rats induced by glycochenodeoxycholic acid (GCDCA); in vivo, animals subjected to 1 week bile duct ligation (BDL) and to a single IP injection of CCl4 were treated with exendin-4 for 3 days.
Results
exendin-4 prevented the GCDCA-induced Bax mitochondrial translocation, cytochrome c release and increase in caspase 3 activity. PI3K, but not cAMP/PKA or Ca2+-CamKinase inhibitors neutralized the effects of exendin-4. In vivo, exendin-4 administration prevented the increase in TUNEL positive cholangiocytes and the loss of bile ducts observed in BDL rats treated with CCl4.
Summary/conclusion
exendin-4 prevents cholangiocyte apoptosis both in vitro and in vivo; such an effect is due to the ability of exendin-4 to counteract the activation of the mitochondrial pathway of apoptosis. These findings support the hypothesis that exendin-4 may be effective in relenting the progression of cholangiopathies towards ductopenia.
Keywords: cholangiocytes, apoptosis, GLP-1, exendin-4, cholangiopathies
INTRODUCTION
Cholangiopathies are a wide array of congenital or acquired disorders that share the feature of being chronic cholestatic conditions leading to liver failure [1]. Despite the different etiology, cholangiopathies share the common feature of primarily targeting cholangiocytes, the epithelial cells lining the intrahepatic biliary tree [1]. These diseases are characterized by the progressive vanishing of bile ducts (e.g. ductopenia), that results from an abnormal cholangiocyte homeostasis [1]. It is thought that ductopenia results from excessive cell death by apoptosis, that prevails over ineffective cholangiocyte compensatory proliferative response [1, 2].
Cholangiopathies are a challenge for clinicians: 20% of liver transplants among adults and 50% of those among pediatric patients are due to these disorders [3]. This is associated, at least in part, to the fact that there is no therapy effective in maintaining an adequate cholangiocyte survival [1]. The factors that regulate the balance between proliferation and death of cholangiocytes are undefined; such a lack of pathophysiological knowledge may contribute to relent the development of effective therapies for cholangiopathies.
Exendin-4 is a “long-lasting” analogue of Glucagon-like peptide-1 (GLP-1) [4]. GLP-1 is secreted by enteroendocrine L cells and modulates the biology of a number of cells, by interacting with a specific G-protein coupled receptor (GLP-1R) [4, 5, 6]. GLP-1 modulates glucose homeostasis, by preventing pancreatic β-cell death by apoptosis and by eliciting their proliferation [4, 5, 6]. As a consequence, exendin-4 is now successfully employed in humans as a novel anti-diabetic therapy [7].
GLP-1 serum levels are increased in the course of cholestasis [8]. We have recently shown that cholangiocytes are susceptible to the action of GLP-1. They express the GLP-1R, the activation of which (by GLP-1 or exendin-4) results in a marked increase in cell growth, both in vitro and in vivo [9]. GLP-1 action on cholangiocytes is required for their proliferative response to cholestasis [9]. Therefore, the aim of this study was to verify if the GLP-1 analogue exendin-4 is also able to prevent cholangiocyte cell death by apoptosis. Thus, we asked the following questions: (i) Does exendin-4 prevent cholangiocyte apoptosis in vitro? (ii) Which are the intracellular pathways that mediate the anti-apoptotic effects of exendin-4 in cholangiocytes? And (iii) is exendin-4 effective in sustaining cholangiocyte survival in vivo?
METHODS
Materials
Reagents were purchased from Sigma-Aldrich (Milan, Italy) unless differently indicated. Antibodies for immunoblotting were purchased from Santa Cruz Biotechnologies Inc. (Santa Cruz, CA), unless differently indicated. The antibody anti-Cytokeratin (CK)-19 was purchased from Novocastra (Milan, Italy); the antibody anti-Bax was purchased from Cell-Signalling (Milan, Italy); Exendin-4 was purchased from American Peptide Inc. (Sunnyvale, CA). PD98059, Rp-cAMPs, KN62 and BAPTA/AM were purchased from Calbiochem (Milan, Italy). APO-ONE Homogeneous Caspase-3/7 Assay was purchased from Promega (Milan, Italy).
Experimental design
In vitro studies
Experiments were performed in cholangiocytes purified from normal rats, after a 1-hour incubation at 37°C to regenerate membrane proteins damaged by proteolytic enzymes during the purification [10, 11, 12]. During incubation, cells were kept in suspension in an RPMI medium [9, 13, 14], added with 5% FBS to limit the constitutive apoptosis.
To verify whether exendin-4 protects cholangiocytes from cell death by apoptosis, cholangiocytes were incubated for 4 hours at 37°C[13] with: (i) 0.2 % BSA (control); (ii) glycochenodeoxycholic acid (GCDCA, 400 μmol/L) [15, 16], in the absence or presence of a 30 minute pre-incubation with exendin-4 (100 nmol/L) [9]. To verify the eventual cytoprotective effects of exendin-4 against bile acids with different cytotoxicity, cells were incubated with taurochenodeoxycholic acid (TCDCA, 50 μmol/L) [17, 18, 19], in the absence or presence of a pre-incubation with exendin-4, as above described.
To identify the intracellular mechanisms that mediate the anti-apoptotic effects of exendin-4 the same experiments were also performed by pre-incubating cells for 30 minutes at 37°C with either (iii) Rp-cAMPs (100 μmol/L, a cAMP-dependent PKA inhibitor) [9, 13] or wortmannin (100 nmol/L, a PI3K inhibitor) [9, 13]. In addition, to establish whether the Ca2+ signaling plays any role in mediating exendin-4 cytoprotective effects, cells were also pre-incubated with (iv) either BAPTA/AM (an intracellular Ca2+ chelator, 5 μmol/L) [9, 13] or KN62 (10 μmol/L, a CamKinaseII inhibitor) [9, 13].
In vivo studies
Male Fischer rats (150–175 g), purchased from Charles River (Milan, Italy), were maintained in a temperature-controlled environment (20–22°C) with a 12-hour light-dark cycle and with free access to drinking water and to standard rat chow.
To study the effects of exendin-4 activation on cholangiocyte survival, our studies were performed in rats subjected to bile duct ligation (BDL). After 7 days, animals received vehicle or a single IP injection of CCl4, 0.4 ml per 100 g body weight (50% mineral oil:CCl4), that triggers cholangiocyte cell death by apoptosis [20]. Subsequently, animals were treated with either (i) exendin-4 (0.1 μg/kg b.w. twice/day, I.P., n = 8) or (ii) control injections (n = 8) [9, 21]. Rats subjected to BDL, vehicle injected and control solution treated were used as internal control (n = 4). No death in each experimental group were counted; at the end of the treatment, animals were sacrificed for liver sections. Treatment options and schedule are depicted in Figure 1.
Figure 1.

outline of the treatment protocol of our in vivo model. All the animals were subjected to BDL at day 0. After 7 days, the group named “BDL + CCl4” received a single IP injection of CCl4 and then received control injection treatment for 3 days, as in previous studies (n = 8) [20]. The group named “BDL + CCl4 + Exendin-4” was subjected to the same surgery and CCl4 intoxication, but was treated for 3 days with exendin-4 (0.1 μg/kg b.w. twice/day, I.P. n = 8) [9]. The group named “BDL” employed, as in previous studies [20], as a control for CCl4 intoxication, received a single IP injection of a control vehicle and, afterwards control injections for 3 days (n = 4).
The animals were fasted overnight before each experiment [9, 13]. Before each procedure, animals were anesthetized with sodium pentobarbital (50 mg/Kg IP). Study protocols were performed in compliance with the institution guidelines.
Cholangiocyte purification and assessment of cell viability
Purification of cholangiocytes from rat liver was performed using a monoclonal antibody (IgM, kindly provided by Dr. R. Faris, Brown University, Providence, RI) against an unidentified membrane antigen expressed by all rat intrahepatic cholangiocytes [22]. At the end of each procedure, purity of cholangiocytes was assessed by cytochemistry for γ-GT [9, 13], a specific marker for cholangiocytes [23]. Cell viability at the end of the purification procedure was determined by trypan blue exclusion and was found greater than 96%.
Caspase 3 activity
Changes in caspase 3 activation were measured by the APO-ONE Homogeneous Caspase-3/7, according to the instruction provided by the vendor. Briefly, at the end of each experiment, 10,000 cells were resuspended with and incubated in a 1/100 dilution of the substrate Z-DEVD for 1 hour. Fluorescence was measured by a 96 multiwell plate reader.
Subcellular fractionation and mitochondria purification
Cholangiocytes were lysed with a Dounce homogenizer in 10 mmol/L Hepes/KOH (pH 7.6), 10 mmol/L KCl, 1 mmol/L MgCl2, 1 mmol/L dithiothreitol, containing aprotinin, 0.5 mmol/L phenylmethylsulfonyl and complete protease inhibitor mixture. Immediately after homogenization sucrose was added to 250 mmol/L. Nuclei and unbroken cells were removed by centrifugation at 3000 × g for 3 min, and the heavy membrane fraction (mitochondrial rich) was sedimented by centrifugation at 9000 × g for 20 min [24].
Study of changes in Bax and Cytochrome c expression and AKT phosphorylation
Proteins obtained from mitochondrial and cytosolic extracts (10 μg/lane) were resolved by SDS-12% polyacrylamide gel electrophoresis and then transferred onto a nitrocellulose membrane. After blocking, membranes were incubated overnight at 4°C with either anti-Bax or anti-cytochrome c antibodies, followed by incubation with the corresponding secondary antibody. Similarly, proteins obtained from whole cell lysates (10 μg/lane) were resolved by SDS-12% polyacrylamide gel electrophoresis and then transferred onto a nitrocellulose membrane. Membranes were then incubated with anti-phospho-AKT and anti-AKT, followed by incubation with the corresponding secondary antibody. Equal loading was evaluated by incubating membranes with the anti-β-actin antibody [25, 26]. Proteins were visualized using chemiluminescence (ECL Plus kit; Amersham). The intensity of the bands was determined by scanning video densitometry using the Chemi Doc imaging system (Bio Rad, Milan, Italy).
Assessment of changes in liver injury and morphology
Serum levels of alanine aminostrasferase (ALT), alkaline phosphatase (ALP) and bilirubin were measured in rats from the different experimental groups, by commercially available kits (Sigma), as previously shown [20, 27]. Changes in liver injury and inflammation were assessed in liver sections after staining with hematoxylin and eosin, as previously reported [28].
In vivo changes in cell death by apoptosis were assessed by the terminal deoxynucleotidyl transferase-mediated triphosphate end-labeling (TUNEL) analysis, as previously described (ApopTag Kit, Oncor, Gaithersburg, MD) [10]. After counterstaining with hematoxylin solution, liver sections (four for each group of treatment) were examined by light microscopy. Approximately 100 cells per slide were counted in a coded manner in 7 non-overlapping fields.
Changes in bile duct mass were assessed by the computerized analysis of the immunohistochemistry for CK-19, as previously shown by us [9, 13, 14, 20]. After examination of the staining with a microscope, photographs of seven different fields per group (selected in a blinded, random fashion) were taken. The volume percent of liver occupied by ducts was calculated from the total number of points over hepatic tissue and the number of points over CK-19 positive ducts, as previously reported [9, 13, 14, 20].
Statistical analysis
Data are expressed as mean ± SE. Data obtained from the in vitro experiments are expressed as % of basal value, with the exception of results of caspase 3 activity that are expressed as arbitrary units. 95% Confidence Interval (CI) was calculated. Differences between groups were analyzed by analysis of variance (ANOVA). Differences between groups were considered significant when the p value was lower than 0.05. We considered principal indicators of induction of cell death by apoptosis: (i) caspase 3 activity for in vitro studies [16, 29] and (ii) TUNEL staining for the in vivo studies [10, 20, 29, 30, 31]. Changes in Bax mitochondrial expression and cytochrome c cytosolic expression were taken as indexes of activation of the “mitochondrial pathway” of induction of apoptosis [32].
RESULTS
Exendin-4 protects cholangiocytes from cell death by apoptosis in vitro
Incubation with GCDCA induced apoptosis in cholangiocytes isolated from normal rats. GCDCA markedly increased the activity of caspase 3 compared to control (Figure 2A; mean 16672 ± 407, 95% CI 15873–17470). In comparison with untreated cells, GCDCA also enhanced Bax mitochondrial translocation and cytochrome c release, as suggested by increased expression of Bax in mitochondria (Figure 2B; mean 257.06 ± 8.16, 95% CI 241.04–273.07 % of basal value) and cytochrome c in cytosol (Figure 2C; mean 454.88 ± 45.87, 95% CI 364.98–544.79 % of basal value) fractions, respectively. Pre-incubation with exendin-4 limited the GCDCA-induced increases in caspase 3 activity (Figure 2A; mean 3793 ± 800, 95% CI 2224–5361), Bax mitochondrial expression (Figure 2B; mean 112.27 ± 11.84, 95% CI 89.05–135.49 % of basal value) and cytochrome c in cytosol (Figure 2C; mean 227.76 ± 38.83, 95% CI 151.65–303.86 % of basal value).
Figure 2.
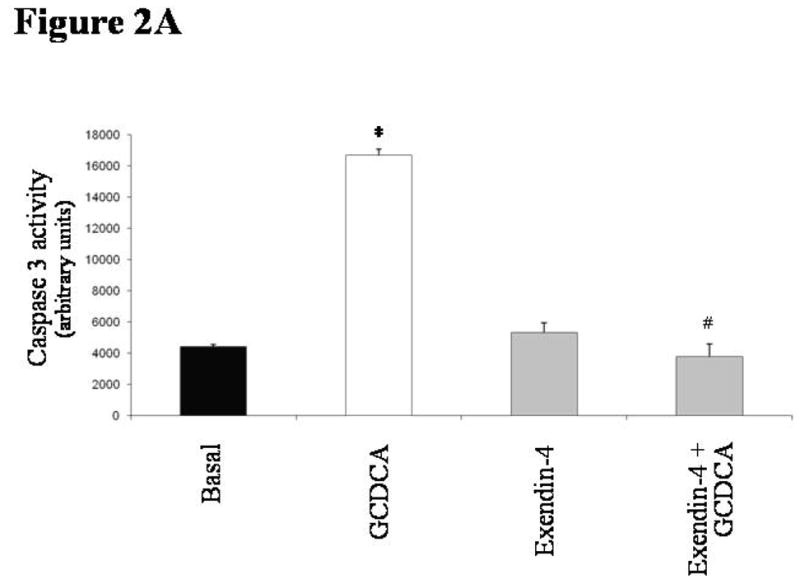
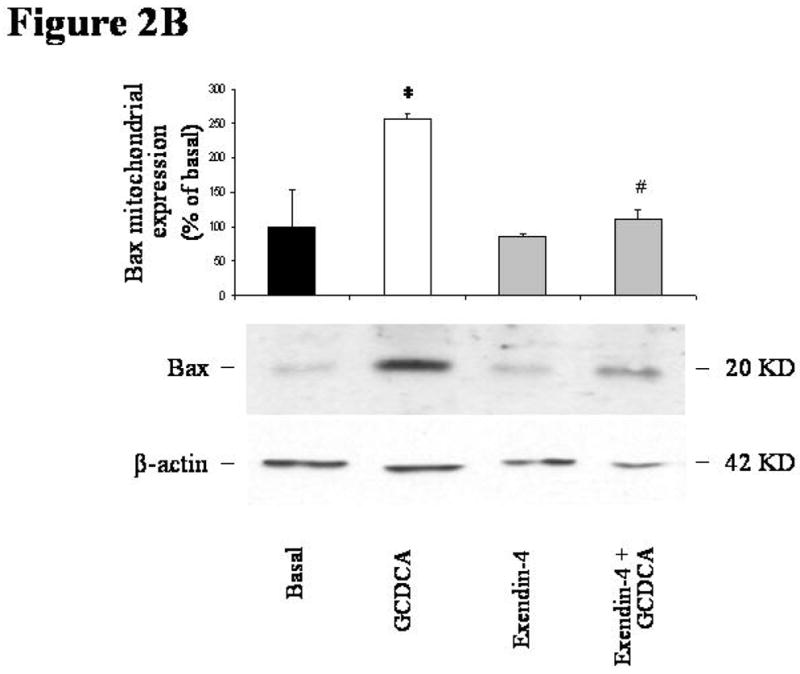
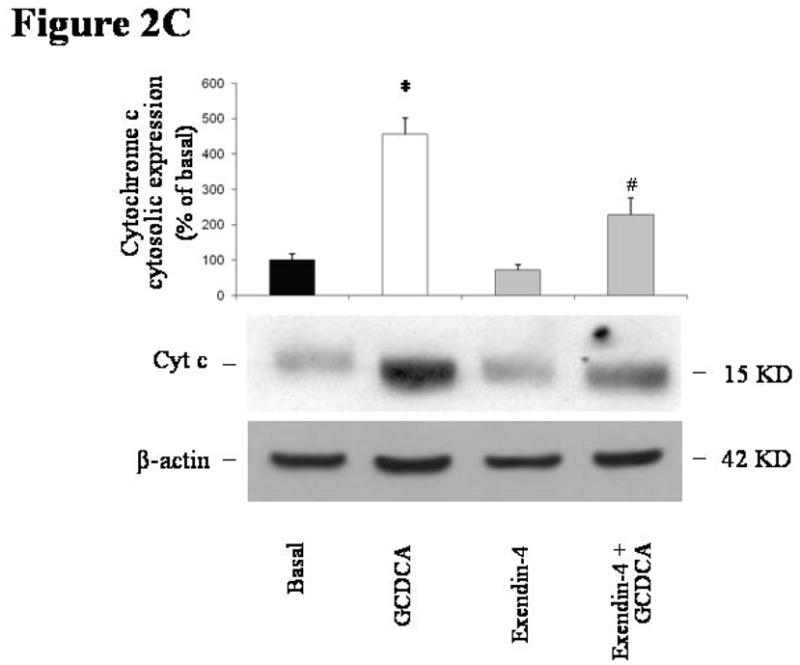
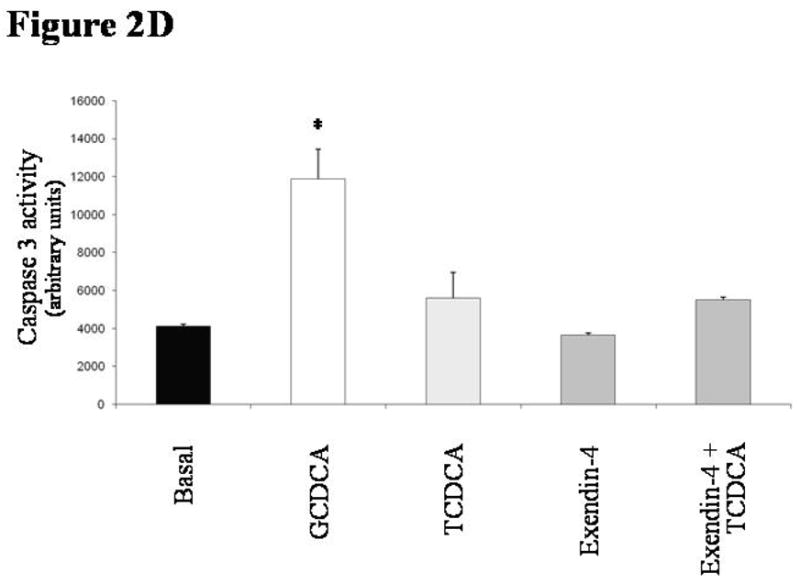
in vitro effects of exendin-4 on cholangiocyte apoptosis. (A) GCDCA (400 μmol/L) induced a significant increase in caspase 3 activity, that was prevented by the pre-incubation with exendin-4 (100 nmol/L). Similarly, expression of Bax in mitochondria (B) and of cytochrome c in cytosol (C) was markedly enhanced by GCDCA. Both those changes were prevented by the preincubation with exendin-4. (D) TCDCA (50 μmol/L) slightly but not significantly increased caspase 3 activity compared to untreated cells. Data are mean ± SE of at least 3 experiments. *: p < 0.05 vs. basal; #: p < 0.05 vs. GCDCA.
In contrast to GCDCA, TCDCA did not induce apoptosis in cholangiocytes, since it did not elicit any significant increase in caspase 3 activity (Figure 2D; mean 5590 ± 1353, 95% CI 2937–8243).
The in vitro anti-apoptotic effects of exendin-4 are mediated by the PI3K pathway
As shown in Figure 3A, only the pre-incubation with wortmannin (a PI3K inhibitor) neutralized the ability of exendin-4 to inhibit the activation of caspase 3 by GCDCA (GCDCA: mean 7047 ± 389, 95% CI 6285–7810; exendin-4+GCDCA: mean 3826 ± 834, 95% CI 2191–5461; wortmannin+exendin-4+GCDCA: mean 6406 ± 985, 95% CI 4475–8337). In contrast, no effects were observed when cells were pre-incubated with a cAMP-dependent PKA inhibitor (Rp-cAMPs), a CamKinaseII inhibitor (KN62) or an intracellular Ca2+ chelator (BAPTA/AM).
Figure 3.
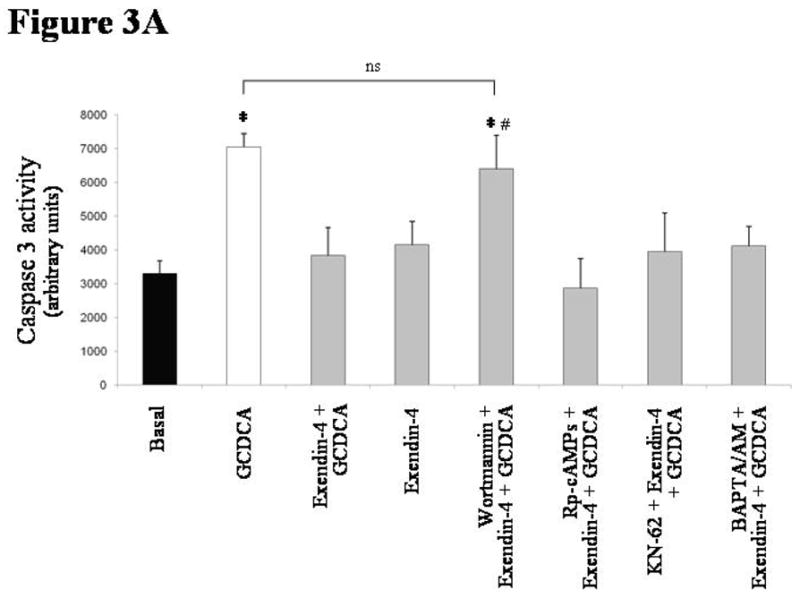


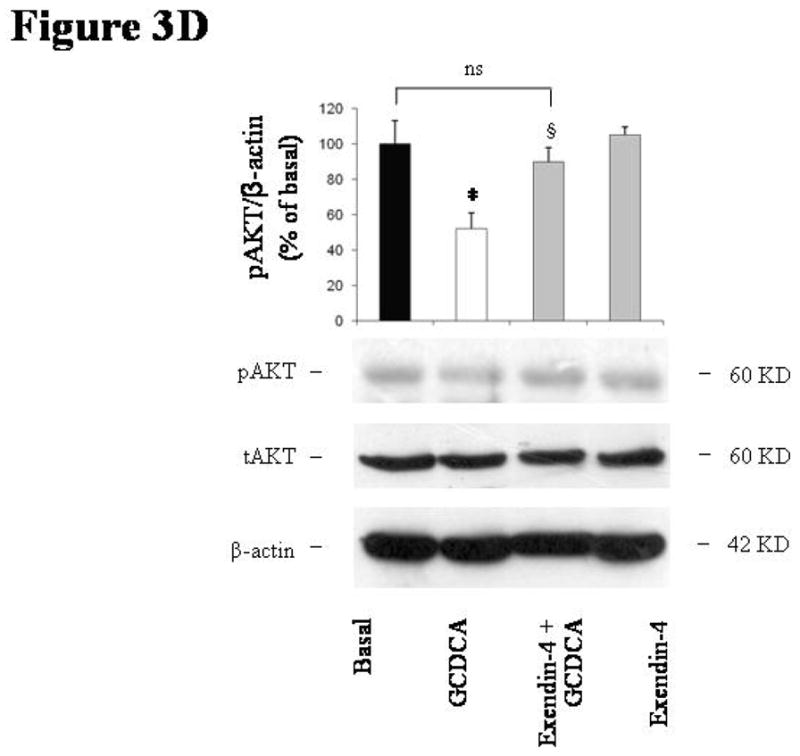
intracellular signals mediating exendin-4 anti-apoptotic effects on cholangiocytes. (A) only the PI3K inhibitor wortmannin (100 nmol/L) was able to neutralize the effect of exendin-4 (100 nmol/L) on GCDCA (400 μmol/L) induced increase in caspase 3 activity. In contrast, no effects were observed when cells were pre-incubated with Rp-cAMPs (a cAMP-dependent PKA inhibitor, 100 μmol/L), KN62 (a CamKinaseII inhibitor, 10 μmol/L) or BAPTA/AM (an intracellular Ca2+ chelator, 5 μmol/L). Similarly, wortmannin neutralized exendin-4 effects on GCDCA induced increase in Bax mitochondrial expression (B) and cytochrome c cytosolic expression (C). (D) Exendin-4 limited AKT dephosphorylation induced by GCDCA. Data are mean ± SE of at least 3 experiments. *: p < 0.05 vs. basal; #: p < 0.05 vs. exendin-4 + GCDCA; §: p < 0.05 vs. GCDCA; ns = not significant.
The blockage of the PI3K signaling by wortmannin also abolished the ability of exendin-4 to prevent the GCDCA-induced increase in Bax mitochondrial expression (Figure 3B; GCDCA: mean 270.34 ± 49.84, 95% CI 172.63–368.04; exendin-4+GCDCA: mean 131.01 ± 17.45, 95% CI 96.79–165.22; wortmannin+exendin-4+GCDCA: mean 245.74 ± 24.87, 95% CI 196.99–294.48 % of basal). Similarly, wortmannin neutralized the effects of exendin-4 on GCDCA induced increase in cytochrome c expression in cytosol (Figure 3C; GCDCA: mean 272.84 ± 53.99, 95% CI 167.00–378.67; exendin-4+GCDCA: mean 94.51 ± 23.25, 95% CI 47.92–139.09; wortmannin+exendin-4+GCDCA: mean 296.13 ± 39.32, 95% CI 219.04–373.21 % of basal).
As a confirmation, cell pre-incubation with wortmannin prevented the reduction in AKT phosphorylation observed in cells exposed to GCDCA when compared to untreated cells (Figure 3D: GCDCA: mean 51.97 ± 8.96, 95% CI 34.40–69.55; exendin-4+GCDCA: mean 89.87 ± 8.01, 95% CI 95.86–114.14 % of basal).
Exendin-4 ameliorates indexes of hepatocellular injury and cholestasis and reduces inflammation an in vivo model of cholestasis and cell death
As previously shown [20], in vivo, a single CCl4 injection to 1 week BDL rats determined a significant increase in serum levels of ALT, ALP and bilirubin and in liver inflammatory infiltrate [20] (Table 1). In contrast, administration of exendin-4 to rats determined a significant reduction of levels of hepatocellular injury and cholestasis and of the degree of liver inflammation (Table 1).
Table 1.
| BDL (95% CI) | BDL + CCl4 (95% CI) | BDL + CCl4 Exendin-4 (95% CI) | |
|---|---|---|---|
| ALT (IU/I) | 337 ± 23.8 (290–333) | 705.7 ± 41.8 (613–737)* | 494.6 ± 27.1 (441–547) # |
| ALP (IU/I) | 767.5 ± 89.6 (591–943) | 1425.7 ± 82.6 (1263–1587)* | 693.6 ± 34.8 (624–761) # |
| Bilirubin (mg/dI) | 5.8 ± 0.7 (4.4–7.2) | 9.35 ± 0.8 (7.7–10.3)* | 63 ± 0.6 (5.1–74) # |
| Inflammation (score) | 1.12 ± 0.3 (0.4–1.8) | 2.43 ± 0.4 (1.9–2.9)* | 1.06 ± 0.26 (0.5–25) # |
Effect of extendin-4 in vivo administration on biochemical parameters of cholestasis and hepatocellular injury and on inflammation. CCl4 intoxication of BDL rats markedly increased indexes of hepatocellular injury (ALT) and of cholestasis (ALP and bilirubin); CCl4 also increased the inflammation score. Extendin-4 in vivo administration was effective in significantly diminishing the effects of CCl4 on the indexes of heptocellular injury and of cholestasis and on the inflammation score.
: p < 0.05 vs. BDL;
: p < 0.05 vs BDL + CCl4.
Exendin-4 prevents cholangiocyte apoptosis in an in vivo model of cholestasis and cell death
In vivo, a single CCl4 injection to 1 week BDL rats induced cell death by apoptosis, as suggested by the relevant increase in the number of TUNEL positive cells [20] (BDL: mean 1.15 ± 0.29, 95% CI 0.58–1.71; BDL + CCl4: mean 6.51 ± 0.94, 95% CI 4.66–8.35). In contrast, administration of exendin-4 to rats determined a significant reduction of the apoptotic cholangiocytes (mean 2.31 ± 0.25, 95% CI 1.81–2.80; Figure 4).
Figure 4.
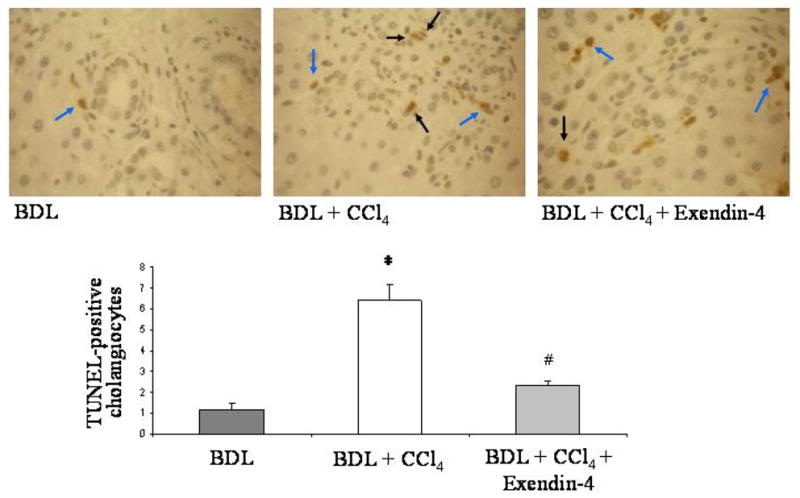
effect of exendin-4 in vivo administration on cholangiocyte apoptosis. CCl4 intoxication induced cholangiocyte death in BDL rats; treatment with exendin-4 markedly reduced the number of TUNEL positive cholangiocytes. Black arrows indicate cholangiocytes positive to the TUNEL staining. Blue arrows indicate other cells positive to TUNEL staining (internal control). Four slices per each rat were considered; seven fields per slice were considered. *: p < 0.05 vs. BDL; #: p < 0.05 vs BDL + CCl4.
Exendin-4 prevents the loss of bile ducts in an in vivo model of cholestasis and cell death
Loss of bile ducts was induced in vivo by a single CCl4 injection to 1 week BDL rats, as witnessed by the strong reduction of the bile duct mass (BDL: mean 8.01 ± 1.12, 95% CI 5.81–10.22; BDL + CCl4: mean 3.36 ± 0.50, 95% CI 2.38–4.34). In contrast, administration to rats of exendin-4 prevented the CCL4 induced reduction of bile duct mass (mean 6.33 ± 0.78, 95% CI 4.79–7.87; Figure 5).
Figure 5.
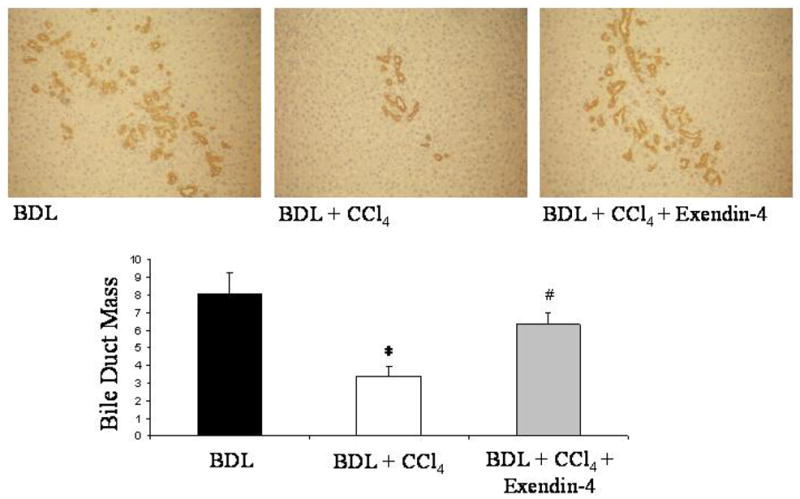
effect of exendin-4 in vivo administration on bile duct mass. CCl4 intoxication markedly diminished, in BDL rats, the bile duct mass, estimated by the computerized analysis of the CK-19 staining. Exendin-4 treatment prevented the CCl4 induced loss of bile ducts. Four slices per each rat were considered; seven fields per slice were considered. *: p < 0.05 vs. BDL; #: p < 0.05 vs BDL + CCl4.
DISCUSSION
The current study shows that exendin-4 protects the biliary epithelium from cell death by apoptosis, both in vivo and in vitro. Specifically, our study demonstrates that: (i) exendin-4 prevents cholangiocyte apoptosis induced, in vitro, by GCDCA; (ii) the anti-apoptotic effect of exendin-4 depends on the PI3K inhibition of Bax mitochondrial translocation, cytochrome c release and caspase 3 activation; (iii) exendin-4 prevents cholangiocyte apoptosis and loss of bile ducts in an in vivo model of cholestasis and ductopenia.
Apoptosis is a highly organized type of cell death that ensures proper organogenesis and health of adult organs [32]. For years, the dogma depicting apoptosis as an “innocuous” event has been accepted [33]. Recent evidences suggest this concept to be true for “physiologic” but not for “pathologic” apoptosis [33]. In physiologic conditions, apoptosis is limited to a small number or subset of cells, both in number and over time [32]. Pathologic apoptosis involves a wide number of cells in a non-selective fashion, is sustained over time and is typically associated with inflammatory conditions [32]. Therapeutic modulation of apoptosis may represent a valid strategy for the treatment of several human diseases [32].
Several liver diseases (including cholangiopathies) are considered due to dysregulation of cell survival [1, 32]. Cholangiopathies target cholangiocytes and progress as a result of enhanced apoptosis that prevails over compensatory proliferation, thus leading to ductopenia [1]. In addition to that, in the course of primary biliary cirrhosis (PBC, the most common of cholangiopathies) [1] apoptotic cholangiocytes process some mitochondrial proteins differently compared to other cells [34]. This amplifies the attraction of immune cells and enhances liver injury [34]. Liver cell apoptosis is also thought to promote fibrogenesis [33, 35], a typical feature of liver injury in late stage cholangiopathies [1]. Hepatic stellate cells engulf apoptotic debris, an event that triggers their activation, e.g. the transdifferentiation towards a myofibroblast-like cell [33, 35].
Currently, there is no molecule known to be effective in maintaining an adequate survival of the biliary epithelium. In this study we demonstrate that exendin-4 prevents cholangiocyte death by apoptosis, both in vitro and in vivo. In vitro, we found that the cytotoxic bile acid GCDCA but not TCDCA significantly induced apoptosis, in a similar fashion to what observed in hepatocytes [15, 16, 17, 25]. Exendin-4 was able to prevent the induction of apoptosis by GCDCA (Figure 2). In vivo, the administration of exendin-4 to rats subjected to BDL and CCl4 intoxication resulted in a significant reduction of TUNEL-positive cells and in the maintenance of the bile duct mass (Figures 4 and 5). Thus, exendin-4 was found to be effective in two distinct models of cholangiocyte apoptosis, both reproducing important features of cholangiopathies, such as the over-exposure to cytotoxic bile acids and the simultaneous presence of cholestasis and loss of bile ducts [1, 20, 34, 36]. To this extent, in its effects exendin-4 exhibited features similar to ursodeoxycholic acid (UDCA), the only compound that has a certain degree of clinical effectiveness at least in some cholangiopathies (like PBC) [1, 34]. We recently found UDCA to be anti-apoptotic for cholangiocytes when tested in an in vivo model of cholestasis and bile duct loss [10]. However, a specific anti-apoptotic effect of UDCA on cholangiocytes in in vitro systems has never been demonstrated. In addition, the outcome of UDCA administration in vivo in different experimental sets resulted in heterogeneous changes in duct mass [10, 37]. UDCA may even amplify liver injury in the course of chronic cholestatic injury [38]. In contrast, exendin-4 is effective in maintaining cholangiocyte proliferative and functional responses and duct mass in the course of cholestasis [9] as well as being beneficial even in other types of liver injury [39]. Together, these findings suggest that exendin-4 has the potential to be effective in relenting the progression of cholangiopathies, since it selectively acts on cholangiocytes reverting the dysregulated balance between cell survival and growth [1].
Apoptosis can occur substantially because of the activation of two defined molecular pathways, the extrinsic or intrinsic pathways [32]. The extrinsic pathway is triggered at the plasma membrane following the activation of specific death receptors; the intrinsic pathway is triggered by different extra- or intra-cellular stimuli that cause mitochondrial dysfunction [32]. The activation of either one or the other pathway may depend on the kind of cell injury or on the cell type [32]. However, the extrinsic and intrinsic pathways are not mutually exclusive [32]; in some cells, hepatocytes and cholangiocytes included, the death receptor pathway requires the mitochondrial signals to be amplified enough to effectively deliver the pro-apoptotic message [32]. Thus, we wanted to investigate how exendin-4 affects the last steps of the apoptotic cascade in cholangiocytes. The in vitro incubation of cholangiocytes with GCDCA was associated with increased Bax expression in mitochondria and increased cytochrome c expression in cytosol (Figure 2). Bax is a member of a family of the Bcl-2 proteins, involved in the regulation of cell death [32]. Upon the activation of the apoptotic machinery (either extrinsic or intrinsic), Bax migrate from cytosol to mitochondria, where it causes membrane permeabilization; as a consequence, cytochrome c is released from mitochondria into cytosol, where it activates proteases mediators and effectors of the programmed cell death, e.g. caspases [32]. One of the main executor caspases is caspase 3. A similar sequence of events has also been shown to occur in hepatocytes upon incubation with GCDCA or other cytotoxic bile acids [25, 40, 41]. Our in vitro system, therefore, reproduced the same cascade of events. Interestingly, when cells were pre-incubated with exendin-4 such a sequence of events was abolished (Figure 2). These data suggest that exendin-4 is able to counteract the apoptotic machinery in cholangiocytes. In addition to that, we also showed that the anti-apoptotic effects of GLP-1R activation are mediated by the activation of the PI3K (Figure 3). Indeed, blocking the PI3K pathway neutralized the effect of exendin-4 on Bax mitochondrial translocation and, as a consequence, cytochrome c release and caspase 3 activation. In several cell types, translocation of Bax can be modulated by the PI3K signalling [42, 43, 44]. As a confirmation, exendin-4 limited the dephosphorylation of AKT, an immediate downstream of PI3K [45], induced by GCDCA. PI3K is a major determinant of cholangiocyte survival [2, 29, 30, 31, 46, 47]; the current study strengthens such a concept, since amongst the different intracellular pathways that allow the GLP-1R to enhance cholangiocyte proliferation (Figure 6), PI3K is the only one to be involved in its anti-apoptotic effects.
Figure 6.

proposed sequence of events that mediate the anti-apoptotic effects of GLP-1R activation by exendin-4 in cholangiocytes. GLP-1R enhances the activation state of PI3K, which reduces Bax translocation to mitochondria. As consequence, cytochrome c release from mitochondria to cytosol and the consequent activation of caspase cascade is reduced. cAMP/PKA and Ca2+/CamKII pathways, that mediate the pro-proliferative effects, do not participate to the anti-apoptotic actions of GLP-1R activation.
Overall, our current findings are consistent with the action of GLP-1R activation on pancreatic β-cell survival [4, 5, 6]. GLP-1R is known to maintain β-cell mass not only by enhancing cell proliferation but also by preventing apoptosis [4, 5, 6]. In parallel to what we observed in cholangiocytes, the key molecule that mediates such a dual property of GLP-1R activation is PI3K [5, 6, 46]. Interestingly, the administration of exendin-4 to Zucker diabetic rats resulted also in a reduction of the number of apoptotic pancreatic ductal cells, cells that share several features in common with cholangiocytes [48, 49, 50, 51]. Counteraction of apoptosis upon GLP-1R activation has been also observed in rat neurons [6, 52].
In summary, we demonstrated that activation of the GLP-1R in cholangiocytes by its selective agonist exendin-4 prevents cholangiocyte apoptosis. Overall, our findings suggest that exendin-4 is a molecule that is able to correct the dysregulated balance between cholangiocyte proliferation and death. Besides being relevant for the understanding of the pathophysiology of chronic cholestasis, our study supports exendin-4 trials in patients with cholangiopathies [4, 5, 6, 7].
Acknowledgments
This work was supported by MIUR grant 2005067975_004 to Dr. Marzioni and by the Università Politecnica delle Marche intramural grants ATBEN00205 to Dr. Benedetti and ATMAR01105 to Dr. Marzioni; by a VA Merit Award, a VA Research Scholar Award, the Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology from Scott & White, and the NIH grant DK062975 to Dr. Alpini. The authors are thankful to Dr. Paolo Onori, Department of Experimental Medicine, University of L’Aquila, L’Aquila, Italy, for his precious support in the histochemical studies.
Abbreviations used
- BDL
bile duct ligation
- IP
intra-peritoneal
- cAMP
cyclic adenosine 3′, 5′-monophosphate
- BSA
bovine serum albumin
- IP3
D-myo-Inositol 1,4,5-triphosphate
- KRH
Krebs Ringer Henseleit
- PKA
Protein Kinase A
- PI3K
phosphatidyl-inositol-3-kinase
- GCDCA
glycochenodeoxycholic acid
- TCDCA
taurochenodeoxycholic acid
Footnotes
Statement of competing interests: none to declare
The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Gut editions and any other BMJPGL products to exploit all subsidiary rights, as set out in our licence http://gut.bmjjournals.com/ifora/licence.pdf.
References
- 1.Lazaridis KN, Strazzabosco M, LaRusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology. 2004;127:1565–77. doi: 10.1053/j.gastro.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Alvaro D, Mancino MG, Glaser S, et al. Proliferating cholangiocytes: a neuroendocrine compartment in the diseased liver. Gastroenterology. 2007;132:415–31. doi: 10.1053/j.gastro.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 3.Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry for Transplant Recipients: Transplant Data 1991–2000. 2001. Rockville, MD, Department of Health and Human Services, Health Resources and Services Administration, Office of Special Programs, Division of Transplantation.
- 4.Drucker DJ. Biological actions and therapeutic potential of the glucagon-like peptides. Gastroenterology. 2002;122:531–44. doi: 10.1053/gast.2002.31068. [DOI] [PubMed] [Google Scholar]
- 5.Brubaker PL, Drucker DJ. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology. 2004;145:2653–9. doi: 10.1210/en.2004-0015. [DOI] [PubMed] [Google Scholar]
- 6.Drucker DJ. Glucagon-like peptides: regulators of cell proliferation, differentiation, and apoptosis. Mol Endocrinol. 2003;17:161–71. doi: 10.1210/me.2002-0306. [DOI] [PubMed] [Google Scholar]
- 7.Fineman MS, Bicsak TA, Shen LZ, et al. Effect on glycemic control of exenatide (synthetic exendin-4) additive to existing metformin and/or sulfonylurea treatment in patients with type 2 diabetes. Diabetes Care. 2003;26:2370–7. doi: 10.2337/diacare.26.8.2370. [DOI] [PubMed] [Google Scholar]
- 8.Niwa T, Nimura Y, Niki I. Lack of effect of incretin hormones on insulin release from pancreatic islets in the bile duct-ligated rats. Am J Physiol Endocrinol Metab. 2001;280:E59–64. doi: 10.1152/ajpendo.2001.280.1.E59. [DOI] [PubMed] [Google Scholar]
- 9.Marzioni M, Alpini G, Saccomanno S, et al. Glucagon-like peptide-1 and its receptor agonist exendin-4 modulate cholangiocyte adaptive response to cholestasis. Gastroenterology. 2007;133:244–55. doi: 10.1053/j.gastro.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 10.Marzioni M, Francis H, Benedetti A, et al. Ca2+-dependent cytoprotective effects of ursodeoxycholic and tauroursodeoxycholic Acid on the biliary epithelium in a rat model of cholestasis and loss of bile ducts. Am J Pathol. 2006;168:398–409. doi: 10.2353/ajpath.2006.050126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LeSage G, Glaser S, Gubba S, et al. Regrowth of the rat biliary tree after 70% partial hepatectomy is coupled to increased secretin-induced ductal bile secretion. Gastroenterology. 1996;111:1633–44. doi: 10.1016/s0016-5085(96)70027-6. [DOI] [PubMed] [Google Scholar]
- 12.Kato A, Gores GJ, LaRusso NF. Secretin stimulates exocytosis in isolated bile duct epithelial cells by a Cyclic AMP-mediated mechanism. J Biol Chem. 1992;267:15523–9. [PubMed] [Google Scholar]
- 13.Marzioni M, Alpini G, Saccomanno S, et al. Endogenous opioids modulate the growth of the biliary tree in the course of cholestasis. Gastroenterology. 2006;130:1831–47. doi: 10.1053/j.gastro.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 14.Marzioni M, Glaser S, Francis H, et al. Autocrine/paracrine regulation of the growth of the biliary tree by the neuroendocrine hormone serotonin. Gastroenterology. 2005;128:121–37. doi: 10.1053/j.gastro.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Bucher BT, Feng X, Jeyabalan G, et al. Glycochenodeoxycholate (GCDC) inhibits cytokine induced iNOS expression in rat hepatocytes. J Surg Res. 2007;138:15–21. doi: 10.1016/j.jss.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 16.Higuchi H, Yoon JH, Grambihler A, et al. Bile acids stimulate cFLIP phosphorylation enhancing TRAIL-mediated apoptosis. J Biol Chem. 2003;278:454–61. doi: 10.1074/jbc.M209387200. [DOI] [PubMed] [Google Scholar]
- 17.Rust C, Bauchmuller K, Fickert P, et al. Phosphatidylinositol 3-kinase-dependent signaling modulates taurochenodeoxycholic acid-induced liver injury and cholestasis in perfused rat livers. Am J Physiol Gastrointest Liver Physiol. 2005;289:G88–94. doi: 10.1152/ajpgi.00450.2004. [DOI] [PubMed] [Google Scholar]
- 18.Schoemaker MH, Gommans WM, Conde de la Rosa L, et al. Resistance of rat hepatocytes against bile acid-induced apoptosis in cholestatic liver injury is due to nuclear factor-kappa B activation. J Hepatol. 2003;39:153–61. doi: 10.1016/s0168-8278(03)00214-9. [DOI] [PubMed] [Google Scholar]
- 19.Hirano F, Haneda M, Makino I. Chenodeoxycholic acid and taurochenodexycholic acid induce anti-apoptotic cIAP-1 expression in human hepatocytes. J Gastroenterol Hepatol. 2006;21:1807–13. doi: 10.1111/j.1440-1746.2006.04363.x. [DOI] [PubMed] [Google Scholar]
- 20.LeSage G, Glaser S, Marucci L, et al. Acute carbon tetrachloride feeding induces damage of large but not small cholangiocytes from bile duct ligated rat liver. Am J Physiol. 1999;276:G1289–G301. doi: 10.1152/ajpgi.1999.276.5.G1289. [DOI] [PubMed] [Google Scholar]
- 21.Ozyazgan S, Kutluata N, Afsar S, et al. Effect of glucagon-like peptide-1(7–36) and exendin-4 on the vascular reactivity in streptozotocin/nicotinamide-induced diabetic rats. Pharmacology. 2005;74:119–26. doi: 10.1159/000084277. [DOI] [PubMed] [Google Scholar]
- 22.Ishii M, Vroman B, LaRusso NF. Isolation and morphological characterization of bile duct epithelial cells from normal rat liver. Gastroenterology. 1989;97:1236–47. doi: 10.1016/0016-5085(89)91695-8. [DOI] [PubMed] [Google Scholar]
- 23.Alpini G, Lenzi R, Sarkozi L, et al. Biliary physiology in rats with bile ductular cell hyperplasia. Evidence for a secretory function of proliferated bile ductules. J Clin Invest. 1988;81:569–78. doi: 10.1172/JCI113355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morishima N, Nakanishi K, Tsuchiya K, et al. Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J Biol Chem. 2004;279:50375–81. doi: 10.1074/jbc.M408493200. [DOI] [PubMed] [Google Scholar]
- 25.Webster CR, Usechak P, Anwer MS. cAMP inhibits bile acid-induced apoptosis by blocking caspase activation and cytochrome c release. Am J Physiol Gastrointest Liver Physiol. 2002;283:G727–38. doi: 10.1152/ajpgi.00410.2001. [DOI] [PubMed] [Google Scholar]
- 26.Tang HL, Le AH, Lung HL. The increase in mitochondrial association with actin precedes Bax translocation in apoptosis. Biochem J. 2006;396:1–5. doi: 10.1042/BJ20060241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Svegliati-Baroni G, Ghiselli R, Marzioni M, et al. Estrogens maintain bile duct mass and reduce apoptosis after biliodigestive anastomosis in bile duct ligated rats. J Hepatol. 2006;44:1158–66. doi: 10.1016/j.jhep.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 28.Fava G, Ueno Y, Glaser S, et al. Thyroid hormone inhibits biliary growth in bile duct-ligated rats by PLC/IP(3)/Ca(2+)-dependent downregulation of SRC/ERK1/2. Am J Physiol Cell Physiol. 2007;292:C1467–75. doi: 10.1152/ajpcell.00575.2006. [DOI] [PubMed] [Google Scholar]
- 29.Marzioni M, LeSage G, Glaser S, et al. Taurocholate prevents the loss of intrahepatic bile ducts due to vagotomy in bile duct ligated rats. Am J Physiol. 2003;284:G837–G52. doi: 10.1152/ajpgi.00398.2002. [DOI] [PubMed] [Google Scholar]
- 30.Marucci L, Alpini G, Glaser S, et al. Taurocholate feeding prevents CCl4-induced damage of large cholangiocytes through PI3-kinase-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2003;284:G290–301. doi: 10.1152/ajpgi.00245.2002. [DOI] [PubMed] [Google Scholar]
- 31.Marzioni M, Ueno Y, Glaser S, et al. Cytoprotective effects of taurocholic acid feeding on the biliary tree after adrenergic denervation of the liver. Liver Int. 2007;27:558–68. doi: 10.1111/j.1478-3231.2007.01443.x. [DOI] [PubMed] [Google Scholar]
- 32.Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54:1024–33. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–8. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 34.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–73. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 35.Canbay A, Higuchi H, Bronk SF, et al. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–30. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
- 36.Alpini G, Prall RT, LaRusso NF. The pathobiology of biliary epithelia. In: Arias IM, Boyer JL, Chisari FV, Fausto N, Jakoby W, Schachter D, Shafritz DA, editors. The Liver; Biology & Pathobiology, 4E. D Philadelphia, PA: Lippincott Williams & Wilkins; 2001. pp. 421–35. [Google Scholar]
- 37.Alpini G, Baiocchi L, Glaser S, et al. Ursodeoxycholate and tauroursodeoxycholate inhibit cholangiocyte growth and secretion of BDL rats through activation of PKC alpha. Hepatology. 2002;35:1041–52. doi: 10.1053/jhep.2002.32712. [DOI] [PubMed] [Google Scholar]
- 38.Fickert P, Zollner G, Fuchsbichler A, et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology. 2002;123:1238–51. doi: 10.1053/gast.2002.35948. [DOI] [PubMed] [Google Scholar]
- 39.Ding X, Saxena NK, Lin S, et al. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. 2006;43:173–81. doi: 10.1002/hep.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodrigues CM, Fan G, Wong PY, et al. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med. 1998;4:165–78. [PMC free article] [PubMed] [Google Scholar]
- 41.Rodrigues CM, Ma X, Linehan-Stieers C, et al. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 1999;6:842–54. doi: 10.1038/sj.cdd.4400560. [DOI] [PubMed] [Google Scholar]
- 42.Kim HJ, Oh JE, Kim SW, et al. Ceramide induces p38 MAPK-dependent apoptosis and Bax translocation via inhibition of Akt in HL-60 cells. Cancer Lett. 2008;260:88–95. doi: 10.1016/j.canlet.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 43.Tsuruta F, Masuyama N, Gotoh Y. The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem. 2002;277:14040–7. doi: 10.1074/jbc.M108975200. [DOI] [PubMed] [Google Scholar]
- 44.Kennedy SG, Kandel ES, Cross TK, et al. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19:5800–10. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1665–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 46.Hui H, Nourparvar A, Zhao X, et al. Glucagon-like peptide-1 inhibits apoptosis of insulin-secreting cells via a cyclic 5′-adenosine monophosphate-dependent protein kinase A- and a phosphatidylinositol 3-kinase-dependent pathway. Endocrinology. 2003;144:1444–55. doi: 10.1210/en.2002-220897. [DOI] [PubMed] [Google Scholar]
- 47.Glaser S, Alvaro D, Francis H, et al. Adrenergic receptor agonists prevent bile duct injury induced by adrenergic denervation by increased cAMP levels and activation of Akt. Am J Physiol Gastrointest Liver Physiol. 2006;290:G813–26. doi: 10.1152/ajpgi.00306.2005. [DOI] [PubMed] [Google Scholar]
- 48.Chu JY, Yung WH, Chow BK. Secretin: a pleiotrophic hormone. Ann N Y Acad Sci. 2006;1070:27–50. doi: 10.1196/annals.1317.013. [DOI] [PubMed] [Google Scholar]
- 49.Grapin-Botton A. Ductal cells of the pancreas. Int J Biochem Cell Biol. 2005;37:504–10. doi: 10.1016/j.biocel.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 50.Nagaya M, Kubota S, Isogai A, et al. Ductular cell proliferation in islet cell neogenesis induced by incomplete ligation of the pancreatic duct in dogs. Surg Today. 2004;34:586–92. doi: 10.1007/s00595-004-2789-2. [DOI] [PubMed] [Google Scholar]
- 51.Yang L, Li S, Hatch H, et al. In vitro trans-differentiation of adult hepatic stem cells into pancreatic endocrine hormone-producing cells. Proc Natl Acad Sci U S A. 2002;99:8078–83. doi: 10.1073/pnas.122210699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perry T, Haughey NJ, Mattson MP, et al. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther. 2002;302:881–8. doi: 10.1124/jpet.102.037481. [DOI] [PubMed] [Google Scholar]