

Abstract
Desminopathy is a genetically heterogeneous disorder with autosomal dominant pattern of inheritance in most affected families; the age of disease onset is on average 30 years. We studied a patient with a history of recurrent episodes of syncope from infancy who later developed second-degree AV block and restrictive cardiomyopathy; she subsequently suffered several episodes of ventricular tachyarrhythmia requiring implantation of bicameral defibrillator. Neurological examination revealed rapidly progressive bilateral facial weakness, winging of the scapulae, symmetric weakness and atrophy of the trunk muscles, shoulder girdle and distal muscles of both upper and lower extremities. Muscle biopsy demonstrated signs of myofibrillar myopathy with prominent subsarcolemmal desmin-reactive aggregates. Molecular analysis identified a homozygous deletion in DES resulting in a predicted in-frame obliteration of seven amino acids (p.R173_E179del) in the 1B domain of desmin. We describe the youngest known desminopathy patient with severe cardiomyopathy and aggressive course leading to the devastation of cardiac, skeletal and smooth musculature at an early age.
Keywords: Desmin mutation, restrictive cardiomyopathy, heart failure, skeletal myopathy, autosomal recessive inheritance
1. Introduction
Myofibrillar myopathy is a familial or sporadic disorder caused by mutations in DES, CRYAB, MYOT, ZASP, FLNC and BAG3 genes [1]. The most frequent of these five subtypes, desminopathy, is a disease that usually manifests between the second and fourth decades of live with skeletal muscle weakness often followed by cardiac conduction disturbances and cardiomyopathy [2,3]. Desminopathy is inherited with an autosomal dominant pattern or results from de novo mutations; autosomal recessive mutations are rare [2].
Desmin is a member of the intermediate filament family of cytoskeletal proteins expressed in striated, cardiac and smooth muscle. In mature striated muscle, desmin filaments surround the Z discs, interconnect them to each other and link the contractile apparatus to the sarcolemma, cytoplasmic organelles and the nuclear membrane [4]. In cardiac muscle, desmin is increased at intercalated discs and represents the major component of the Purkinje fibers [5]. Human desmin is encoded by a single copy gene (DES) located in chromosome 2q35 band [6]; it encompasses nine exons within an 8.4 kb region and codes for 476 amino acids [7]. The gene is highly conserved among vertebrate species. In accordance with its function, desmin protein is organized into three domains: a highly conserved alpha-helical core of 308 amino acid residues flanked by globular N- and C-terminal (“head” and “tail”) structures [4]. The alpha-helical core maintains a seven-residue (heptad) repeat pattern that guides two polypeptides into formation of a homopolymeric coiled-coil dimer, the elementary unit of desmin filament [8].
Multiple mutations in the desmin gene, point substitutions, insertion, small in-frame deletions and a larger exon-skipping deletion have been identified [2]. The majority of these mutations are located in conserved alpha-helical segments [2]. Filament and network assembly studies indicate that most but not all disease-causing mutations make desmin assembly-incompetent and able to disrupt a pre-existing filamentous network in dominant-negative fashion [9].
We provide a clinical, myopathological and molecular description of a patient suffering from an infantile onset severe cardiac, skeletal and smooth muscle myopathy due to a homozygous DES deletion.
2. Patient and methods
The patient was studied at the Neurology Division, Hospital Clínico Universitario de Zaragoza. Routine laboratory tests, echocardiography, cardiac MRI, coronary angiography, cardiac electrophysiological study, brain MRI, motor and sensory nerve conduction studies and electromyography were performed before initiation of muscle biopsy and molecular analysis. An open muscle biopsy was performed on the left deltoid muscle and biopsy tissue processed by routine histological, histochemical techniques, and desmin immunohistochemistry as previously described [10]. A small sample was used for ultrastructural examination using standard methods.
Sequence analysis of desmin and alphaB-crystallin genes was performed in the patient and her unaffected parents after informed consent was obtained from each individual. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki. Genomic DNA was isolated from peripheral lymphocytes and used for amplification in polymerase chain reaction (PCR) with intronic primers. The resulting PCR-produced fragments were purified by QIAquick PCR Purification Kit (Qiagen, Valencia, CA) and directly sequenced using BigDyeTerminator™ sequencing protocol on an automated 3100 ABI Prism® Genetic Analyzer (Applied Biosystems, Foster City, CA). Data was extracted and analyzed using Sequencing Analysis software (Applied Biosystems, Foster City, CA).
3. Results
3.1. Case Report
The patient had repetitive episodes of syncope since the age of 6 months which were initially attributed to right posteroseptal accessory conduction pathway and subsequently diagnosed as Wolf-Parkinson-White (WPW) syndrome. Transthoracic echocardiography, cardiac MRI and coronary angiography performed during infancy were assessed as normal. At the age of 3 years, the patient developed progressive syncopal, pre-syncopal and cyanotic attacks. On examination at the age of 15 years, she had a long-narrow face with bilateral facial weakness, ptosis, symmetrical winging of the scapulae and poor muscle bulk. There was symmetric atrophy of the masseter, temporalis, sternocleidomastoid and trunk muscles, bilateral pes cavus, and mild muscle weakness involving the shoulder girdle and distal muscles of the upper and lower extremities. Deep tendon reflexes were all hypoactive. Sensation was intact. There was no clinical myotonia. Blood tests including serum electrolytes, lactate curve, albumin, vitamin B12, and anti-ganglioside antibody levels yielded normal results. Creatine kinase was elevated to twice-normal level. Sensory nerve conduction studies were normal, whereas motor nerve studies showed low amplitude of the motor potentials with normal latencies and conduction velocities. Needle electromyography demonstrated abundant complex repetitive discharges, some myotonic discharges and occasional fibrillation potentials. During voluntary contraction, a myopathic recruitment pattern was recorded. Cerebral MRI was normal. Respiratory function tests did not show any abnormalities.
Cardiac electrophysiological study failed to show accessory conduction pathway or induced ventricular or supraventricular dysrhythmia. Twenty-four-hour Holter monitoring revealed monomorphic and polymorphic ventricular tachycardia, occasional prolongation of the QT interval and second degree AV block (Figure 1). In view of these life-threatening arrhythmias and risk of progression to higher-degree AV block, an automatic implantable cardioverter defibrillator (AICD) and pacemaker device were implanted (Figure 2).
Figure 1.
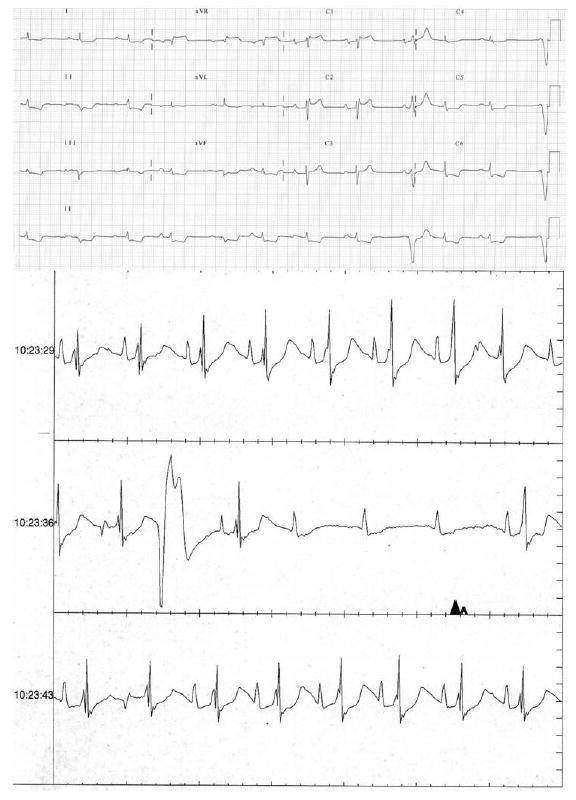
Basal electrocardiogram. Rhythm strip obtained with an insertable loop recorder (IRL) demonstrate high degree 4:1 atrioventricular block
Figure 2.
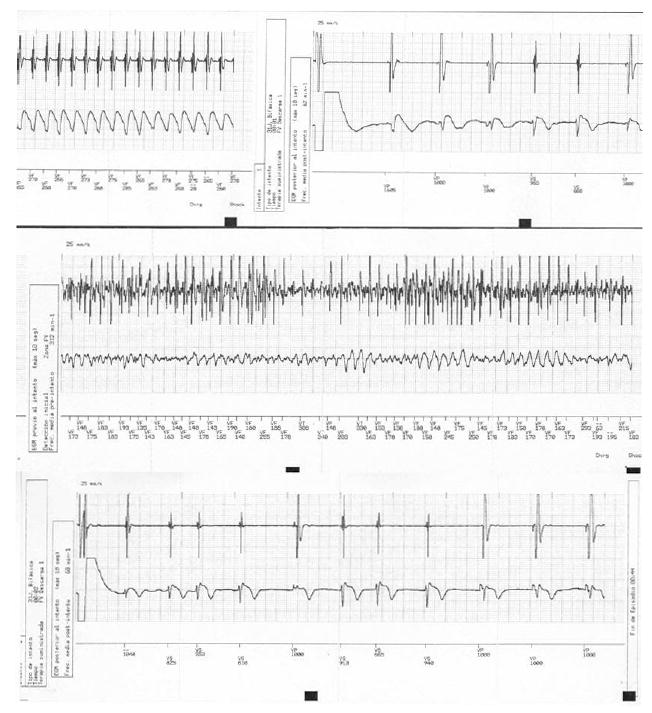
Electrogram rhythm strip obtained with the automatic implantable cardioverter-defibrillator (AICD). Top strip: ventricular flutter, bottom strip: ventricular fibrillation. Both were recognized and successfully treated by the AICD.
Three years later, at age 18 years, the patient suffered several consecutive episodes of ventricular tachyarrhythmia requiring implantation of bicameral defibrillator. Echocardiography showed normal morphology of the left ventricle, mild left systolic dysfunction (left ventricular ejection fraction, LVEF 45%) and slight right ventricular dilatation with mild systolic dysfunction. The atria were markedly enlarged. Doppler analysis of mitral flow revealed abnormal filling indicative of restrictive cardiomyopathy.
Skeletal muscle weakness rapidly progressed. Developing diarrhea, vomiting and weight loss suggested involvement of smooth intestinal muscles. No cardiac or intestinal biopsy was performed at this time due to patient’s further decline. She died from cardiac failure before her 21st birthday. Post-mortem examination was not performed.
The patient’s parents are in a consanguineous marriage (uncle and niece), in which the patient was the only child. The patient’s mother is in good health, the father has been diagnosed with acquired immuno-mediated peripheral neuropathy, currently in remission. No other first or second degree family member was affected with cardio- or skeletal myopathy like disease.
3.2. Myopathology
Muscle biopsy showed an excessive variation in fiber size, ranging from 15 to 40 microns. More than 90% of fibers had well-defined peripheral and often multiple deposits of homogeneous eosinophilic material (Figure 3A) that stained different shades of green with Engel trichrome, appeared Congo red positive and was strongly immunoreactive with antibodies to desmin (Figure 3B). These inclusions showed slight PAS positivity, were non-reactive against alpha-glycerophosphate dehydrogenase with and without substrate, and devoid of oxidative enzyme and ATPase activities. Several muscle fibers contained small vacuoles rimmed with granular basophilic material. Ultrastructural examination revealed a matrix of granulofilamentous material with amorphous, granular and electron-dense deposits forming a thick reticular network with interspersed, disorganized thin filaments (Figure 3C).
Figure 3.
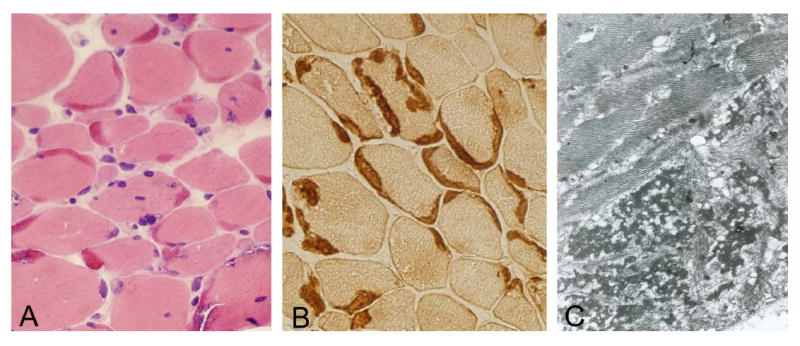
Patient’s muscle biopsy. Prominent crescent-shaped eosinophilic masses located under the sarcolemma, sometimes associated with basophilic granular material (A), and displaying strong desmin immunoreactivity (B). Under electron microscopy, a matrix of dense granulofilamentous material is observed among the myofibrils (C). Cryostat sections, original magnification in A and B x200; C x5000. Modified from reference 2, with permission from Landes Bioscience and Springer Science+Business Media.
3.3 Mutation Detection Studies
Analysis of the patient’s genomic DNA for mutations in desmin and alphaB-crystallin genes identified a homozygous deletion of 21 nucleotides predicting an in-frame loss of 7 amino acids (RARVDVE) from Arg173 through Glu179 (p.R173_E179del) in the 1B helix domain of the desmin molecule. The mutation abolishes part of protein structure representing a full heptad. The entire desmin rod normally contains 35 full heptads, and the 1B helix domain consists of 14. The patient’s mother and father was each heterozygous for this mutation.
4. Discussion
We report the youngest known molecularly identified patient suffering of severe desminopathy characterized by infantile onset of cardiac arrhythmia followed by skeletal weakness and cardiac failure due to a homozygous desmin p.Arg173_Glu179del mutation. The mutation identified in our patient is located at the beginning of the 1B domain of the desmin alpha-helical core, the site known to be critically involved in filament assembly [8] and interactions with other cellular proteins [11].
The only closely related observation reported in the literature was the case of Muñoz-Mármol et al. [12], in which an identical homozygous deletion of 21 nucleotides in the DES gene caused a severe clinical syndrome. Their patient was a 15-year-old boy who developed generalized muscular weakness and atrophy, predominantly in distal muscles of the upper extremities, AV block requiring implantation of a permanent pacemaker, respiratory failure, and intestinal malabsorption [12,13]. The patient died at the age of 28 years. Echocardiography showed dilatation of the right cardiac chambers. Abundant subsarcolemmal crescent-shaped strongly eosinophilic masses in the skeletal myofibers and centrally located eosinophilic bodies in the cardiomyocytes were immunoreactive for desmin and ubiquitin [13]. Myopathological characteristics of the inclusions observed in our case match this description, but at the same time they were different from the amorphous patches previously identified in the majority of patients with dominant or de novo desmin mutations [2].
Atrioventricular conduction abnormalities requiring urgent implantation of a permanent pacemaker is a frequent feature of desminopathy. In a quarter of desminopathy patients the illness presents with cardiomyopathy, and heart disease is seen in more than 60% of patients in the advanced stages of illness [2]. Restrictive, dilated and hypertrophic cardiomyopathy have been documented [1-3,10]. The age of disease onset in cases associated with cardiomyopathy tends to be earlier than in patients with isolated skeletal myopathy, and the illness more severe.
Sudden death occurring in a significant number of desminopathy patients is the consequence of ventricular arrhythmia; this condition has previously been diagnosed and associated with desminopathy in a living patient [14]. The current observation additionally confirms that patients with desmin-related cardiomyopathy are at high risk of developing ventricular arrhythmia and therefore should be considered candidates for AICD implantation even before ventricular dysfunction becomes evident. The identification of the genetic cause of pediatric cardiomyopathy has therefore tremendous significance in patient management and genetic counseling. Ventricular arrhythmias have also been documented as an associated sign in several inherited myopathies including myotonic dystrophy [15] and laminopathy [16].
In about 6% of patients with desminopathy, the inheritance pattern was autosomal recessive; patients with recessive mutations develop the disease in their childhood (average 5 years) [2]. Our observation is the first known case of infantile-onset desmin mutation-associated cardiomyopathy and the first case exhaustively proven to be inherited as an autosomal recessive trait. The homozygous p.Arg173_Glu179del mutation prevented assembly of desmin filaments and compromised the ability of desmin to form an intermediate filament network in transiently transfected MCF-7 cells [12], providing a confirmation of high pathogenic potential. At the same time, the mutation that is clearly changing protein structure in a highly conserved region did not cause pathogenic effects in the patient’s parents who are heterozygous for the p.Arg173_Glu179del alteration.
Acknowledgments
The authors are grateful to the patient and members of her family for participation in the study. This research was supported in part by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health. FJR is a participant of the Gobierno de Aragón Research Groups Program (Ref. B20), Spain. MO was the recipient of the Fondo de Investigación Sanitaria PI08-574 grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Selcen D, Engel AG. Myofibrillar myopathy. Gene Reviews. 2008 http://www.genereviews.org/
- 2.Goldfarb LG, Olivé M, Vicart P, et al. Intermediate filament diseases: desminopathy. In: Laing N, editor. The Sarcomere and Skeletal Muscle Disease. Austin, New York: Landes Bioscience and Springer Science+Business Media; 2008. pp. 131–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arbustini E, Pasotti M, Pilotto A, et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail. 2006;8:477–483. doi: 10.1016/j.ejheart.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Fuchs E, Weber K, Cleveland DW. Intermediate filaments: structure, dynamics, function, and disease. Ann Rev Biochem. 1994;63:345–382. doi: 10.1146/annurev.bi.63.070194.002021. [DOI] [PubMed] [Google Scholar]
- 5.Price MG. Molecular analysis of intermediate filament cytoskeleton – putative load-bearing structure. Am J Physiol. 1984;246:566–572. doi: 10.1152/ajpheart.1984.246.4.H566. [DOI] [PubMed] [Google Scholar]
- 6.Viegas-Péquignot E, Li Z, Dutrillaux B, et al. Assignment of human desmin gene to band 2q35 by non-radioactive in situ hybridization. Hum Genet. 1989;83:33–36. doi: 10.1007/BF00274143. [DOI] [PubMed] [Google Scholar]
- 7.Li Z, Lilienbaum A, Butler-Browne G, Paulin D. Human desmin-coding gene: complete nucleotide sequence, characterization and regulation of expression during myogenesis and development. Gene. 1989;78:243–254. doi: 10.1016/0378-1119(89)90227-8. [DOI] [PubMed] [Google Scholar]
- 8.Herrmann H, Haner M, Brettel M, et al. Structure and assembly properties of the intermediate filament protein vimentin: the role of its head, rod and tail domains. J Mol Biol. 1996;264:933–953. doi: 10.1006/jmbi.1996.0688. [DOI] [PubMed] [Google Scholar]
- 9.Bar H, Mucke N, Kostareva A, et al. Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc Natl Acad Sci U S A. 2005;102:15099–15104. doi: 10.1073/pnas.0504568102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olive M, Armstrong J, Miralles F, et al. Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromusc Disord. 2007;17:443–450. doi: 10.1016/j.nmd.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bang ML, Gregorio C, Labeit S. Molecular dissection of the interaction of desmin with the C-terminal region of nebulin. J Struct Biol. 2002;137:119–127. doi: 10.1006/jsbi.2002.4457. [DOI] [PubMed] [Google Scholar]
- 12.Muñoz-Mármol AM, Strasser G, Isamat M, et al. A dysfunctional desmin mutation in a patient with severe generalised myopathy. Proc Natl Acad Sci USA. 1998;95:11312–13117. doi: 10.1073/pnas.95.19.11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ariza A, Coll J, Fernández-Figueras T, et al. Desmin myopathy: a multisystem disorder involving skeletal, cardiac, and smooth muscle. Hum Pathol. 1995;26:1032–1037. doi: 10.1016/0046-8177(95)90095-0. [DOI] [PubMed] [Google Scholar]
- 14.Luethje LG, Boennemann C, Goldfarb L, Goebel HH, Halle M. Prophylactic implantable cardioverter defibrillator placement in a sporadic desmin related myopathy and cardiomyopathy. Pacing Clin Electrophysiol. 2004;27:559–560. doi: 10.1111/j.1540-8159.2004.00484.x. [DOI] [PubMed] [Google Scholar]
- 15.Groh WJ, Groh MR, Saha C, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med. 2008;358:2688–2697. doi: 10.1056/NEJMoa062800. [DOI] [PubMed] [Google Scholar]
- 16.Meune C, Van Berlo JH, Anselme F, et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354:209–210. doi: 10.1056/NEJMc052632. [DOI] [PubMed] [Google Scholar]