

Abstract
Depending on its cellular localization, p120 catenin (p120ctn) can participate in various processes, such as cadherin-dependent cell-cell adhesion, actin cytoskeleton remodeling, and intracellular trafficking. Recent studies also indicate that p120ctn could regulate cell proliferation and contact inhibition. This report describes a new function of p120ctn in the regulation of cell cycle progression. Overexpression of the p120ctn isoform 3A in human colon adenocarcinoma cells (HT-29) results in cytoplasmic accumulation of the protein, as observed in many tumors. This cytoplasmic increase is correlated with a reduction in proliferation and inhibition of DNA synthesis. Under these conditions, experiments on synchronized cells revealed a prolonged S phase associated with cyclin E stabilization. Both confocal microscopy and biochemical analysis showed that cyclin E and cyclin-dependent kinase 2 colocalized with p120ctn in centrosomes during mitosis. These proteins are associated in a functional complex evidenced by coimmunoprecipitation experiments and the emergence of Thr199-phosphorylated nucleophosmin/B23. Such post-translational modification of this centrosomal target has been shown to trigger the initiation of centrosome duplication. Therefore, p120ctn-mediated accumulation of cyclin E in centrosomes may participate in abnormal amplification of centrosomes and the inhibition of DNA replication, thus leading to aberrant mitosis and polyploidy. Because these modifications are often observed in cancer, p120ctn may represent a new therapeutic target for future therapy.
Keywords: Cell Adhesion Molecules, biosynthesis, genetics, metabolism, Cell Cycle, physiology, Cell Growth Processes, physiology, Centrosome, metabolism, Colonic Neoplasms, genetics, metabolism, pathology, Cyclin E, metabolism, Cyclin-Dependent Kinase 2, metabolism, Cytoplasm, metabolism, Disease Progression, Gene Amplification, Genomic Instability, Green Fluorescent Proteins, biosynthesis, genetics, HT29 Cells, Humans, Phosphoproteins, biosynthesis, genetics, metabolism, Phosphorylation, Recombinant Fusion Proteins, biosynthesis, genetics, Up-Regulation
Introduction
E-cadherin is the main epithelial cell-cell adhesion molecule. Its loss of function or expression is observed during tumor progression and contributes to tumor cell invasion and metastasis (1, 2). The molecular basis for this epithelial phenotypic “gatekeeper” function of E-cadherin remains unclear. The intracellular domain of E-cadherin interacts directly with β-catenin and p120 catenin (p120ctn) via distinct conserved binding domains. These interactions are necessary to dynamically regulate cell-cell adhesion via the modulation of events, such as cadherin clustering and connection with the actin cytoskeleton, or by the control of cadherin endocytosis and metabolic stability (reviewed in ref. 3). Loss of E-cadherin may induce signaling by catenins released from the cell membrane. Indeed, β-catenin released from the membrane is either rapidly degraded in the cytoplasm or acts as an oncogenic transcription cofactor under the activation of the Wnt signaling pathway (4). In contrast, the level of p120ctn is not altered in cadherin-deficient cells but the protein mislocalizes to the cytoplasm and/or the nucleus (5, 6). Abnormal localization of p120ctn in breast or colon carcinomas is prognostic for aggressive diseases (7, 8). In the cytoplasm, p120ctn has key roles in modulating Rho-GTPases and thereby cytoskeletal and cell cycle functions (9). In the nucleus, p120ctn can interact with the transcription factor Kaiso and relieves its gene repression activity (10, 11). Mechanisms underlying the trafficking of p120ctn to and from these various locations have not been elucidated. However, the recently described binding of p120ctn to kinesin and microtubules would affect both the targeting and the activity of p120ctn (12–14). Consistent with its interphasic microtubule localization, endogenous p120ctn was also found localized to the centrosomes and the mitotic spindle during mitosis in MDA-MB-231 breast adenocarcinoma cells (13). This peculiar localization suggests an implication of this catenin in cell cycle regulation.
The interaction of transiently expressed cyclins with cyclin-dependent kinases (cdk) permits cell cycle progression. It has been shown that cyclin E is involved in both S-phase entry and centrosome duplication. Cyclin E interacts with cdk2 and the resulting complex phosphorylates several targets including nucleophosmin, the phosphorylated form of which is required for centrosome duplication (15). Cyclin E overexpression in human cancer cells increases the frequency of centrosome hyperamplification. This thereby favors chromosome instability (CIN) and hence emphasizes the importance of understanding the regulatory mechanism that governs faithful centrosome duplication in cells (16).
In the present study, we used the HT-29 human colon carcinoma cell line to examine the expression and function of p120ctn when the balance between E-cadherin and p120ctn was modified. We show that mislocalized p120ctn can interact with the cdk2/cyclin E complex within the centrosome leading to the stabilization of cyclin E levels. This effect correlated with the inhibition of DNA replication, a delayed S phase, hyperamplification of centrosomes, and, finally, polyploidy. Altogether, our observations reveal a new functional interaction and mechanism by which p120ctn may participate in tumor initiation and/or progression of colorectal carcinoma.
Material and Methods
Cell culture
The human colon adenocarcinoma HT-29 cell line was cultured at 37°C in a 5% CO2 atmosphere in DMEM containing 25 mmol/L glucose (Invitrogen/Life Technologies) and supplemented with 10% FCS, penicillin, and streptomycin.
Antibodies and reagents
Monoclonal antibody (mAb) against p120ctn (clone 98) and green fluorescent protein (GFP) were purchased from BD Biosciences/Transduction Laboratories. Anti-actin polyclonal antibodies, anti-bromodeoxyuridine (BrdUrd; clone BU-33), β-tubulin (clone 2.1), and γ-tubulin (clone GTU-88) mAbs were obtained from Sigma-Aldrich. Anti-human E-cadherin (clone HECD1) mAb was obtained from Takara Biochemicals (Cambrex Bioscience). Polyclonal antibody directed against Aurora A kinase was purchased from Calbiochem (VWR International), and monoclonal anti-human cyclin E (clone HE12) was from Upstate Biotechnology (Chemicon International). Polyclonal anti-cdk2 antibody was purchased from BD Biosciences/PharMingen and polyclonal antibody recognizing nucleophosmin phosphorylated on Thr199 was from Cell Signaling (Ozyme). Alexa-conjugated goat anti-mouse secondary antibody was obtained from Molecular Probes. Horseradish peroxidase–conjugated goat anti-mouse (1:3,000) was from Bio-Rad, and donkey anti-rabbit (1:20,000) and control IgG2A antibodies were from Jackson ImmunoResearch (Immunotech). Mouse and rabbit TrueBlot antibodies used as secondary antibodies (1:1,000) in immunoprecipitation experiments were from eBioscience (Cliniscience). Nocodazole and aphidicolin were purchased from Sigma-Aldrich. MG132 was from Calbiochem.
Western blot analysis
Cells were lysed and extracts were processed as described previously (17). Primary antibodies dilutions were 1:1,000 for p120ctn, GFP, cdk2, E-cadherin, cyclin E, and nucleophosmin and 1:250 for Aurora A and actin.
Plasmids and cell transfections
The p120-3A cDNA (gift of Prof. Albert B. Reynolds, Vanderbilt University, Nashville, TN) was extracted from PRc/RSV p120-3A plasmid by EcoRI and KpnI digestions and subcloned in pEGFP-C3 plasmid (BD Biosciences). GFP-p120-3A cDNA and control GFP cDNA were amplified by PCR from pEGFP-C3 constructs and cloned into pEF6/V5/His-TOPO plasmid (Invitrogen). HT-29 cells were transfected with 1 μg of plasmid DNA using LipofectAMINE 2000 (Invitrogen) according to the manufacturer’s instructions and seeded at one cell per well in a 96-well plates before selection with 5 μg/mL blasticidin. In this study, the HT-29 GFP-p120ctn cells represent the average of two independent clones.
Immunofluorescence microscopy
Cells grown on glass coverslips were treated as described previously (17). Antibody dilutions were 1:200 for anti-β-tubulin, 1:500 for anti-γ-tubulin, and 1:100 for anti-cyclin E, anti-cdk2, and anti-nucleophosmin, and 1:1,000 for secondary antibodies (see Supplementary Materials and Methods).
Immunoprecipitations
Cells were lysed in 10 mmol/L PIPES (pH 6.8), 100 mmol/L NaCl, 300 mmol/L sucrose, 3 mmol/L MgCl2, 0.5% NP40, and protease inhibitor cocktail for 10 min on ice. Extracts were immunoprecipitated using anti-cdk2 antibody and processed as described previously (ref. 17; see Supplementary Material and Methods).
Cell numeration
Cells were seeded in 24-wells plates at 2 × 104 cells per well, trypsinized after the indicated times, and mixed with trypan blue solution (1:1; Sigma-Aldrich) to count living cells. All experiments were reproduced at least thrice.
Caspase activity test
Cells (1.6 × 103) were seeded in each well of a 96-well plate and cell death was analyzed after the indicated times using Apo-One homogeneous Caspase-3/7 assay (Promega) according to the manufacturer’s instructions. Fluorescence emission was quantified using a Fluoroskan Ascent (Thermo Fischer Scientific). All experiments were done in triplicates.
BrdUrd incorporation and staining
Cells were exposed to BrdUrd (5 μmol/L) for 15 min at 37°C before trypsinization. After centrifugation, the cell pellet was resuspended in 100 μL glucose-1 g/L in PBS, brought to 106/mL with 70% ethanol, and fixed for 30 min at 4°C. After centrifugation (1,000 × g, 10 min), cells were resuspended in 1 mL PBS-glucose and allowed to rehydrate 4 h at 4°C and HCl 4N (1:1) was added for 15 min at room temperature. After centrifugation (1,000 × g, 10 min), cells were washed successively in 2 mL PBS containing 0.1 mol/L sodium tetraborate, 0.5% Tween 20, and 0.5% bovine serum albumin (BSA), and in 2 mL PBS-BSA-Tween (PBT). The pellet was incubated with anti-BrdUrd antibody (1:10) for 30 min at room temperature and then with Alexa Fluor 546–coupled antibody (1:500) for 30 min at room temperature. After washes with PBT, cells were analyzed by flow cytometry (Facstar, BD Biosciences).
Cell cycle analysis
Cells (2 × 106) were fixed in 1 mL of cold absolute ethanol for 30 min at room temperature and left overnight in ethanol at 4°C. After centrifugation, pellet was resuspended for 20 min at 37°C in 1 mL propidium iodide staining solution (50 μg/mL propidium iodide, 0.1% NP40, and 0.1 mg/mL RNase A in PBS) and stored at 4°C. Cells were then centrifuged (260 × g, 5 min) and resuspended in PBS before flow cytometry analysis (Facstar, BD Biosciences).
Centrosome preparation
Cell centrosomal fractions were purified from 108 cells pretreated 16 h with 50 ng/mL nocodazole using a sucrose gradient according to the original protocol developed by Bornens and Moudjou (18). Fractions (500 μL) were collected from the bottom and numbered from 1 to 13; the remaining solution was collected as fraction 14.
Statistical analyses
All experiments were done at least thrice and Student’s t test was used for statistical analysis. Values are expressed as mean ± SE and were considered statistically significant with P < 0.05.
Results
Cytoplasmic localization of p120ctn after overexpression in HT-29 cells
Although having no significant effect on p120ctn levels, cadherin deficiency does release p120ctn into the cytoplasm and/or the nucleus where it is thought to promote cell migration, invasion, and metastasis (19). To investigate the cellular roles aside for cell adhesion of p120ctn, we modified the E-cadherin/p120ctn balance by overexpressing p120ctn in a HT-29 human colon adenocarcinoma cell line.
HT-29 cells were stably transfected with the isoform 3A of p120ctn fused to GFP. We next determined the expression level of the construct by Western blotting (Fig. 1A). In addition to the endogenous p120ctn (band 120 kDa), the anti-p120ctn antibody revealed an additional band of 140 kDa in the GFP-p120ctn–transfected HT-29 cells, corresponding to the expected size of the fusion protein (Fig. 1A, top). Although transfection only resulted in a weak increase in the expression of p120ctn, GFP detection indicated that the GFP-p120ctn construction was not degraded (Fig. 1A, bottom). Previous studies have described that p120ctn overexpression can stabilize cadherins at the plasma membrane, thereby increasing their expression (5). Surprisingly, E-cadherin expression levels showed no difference between control, GFP, and GFP-p120ctn HT-29 cells. These results were further confirmed by fluorescence-activated cell sorting (FACS) analysis (data not shown). Analysis of GFP fluorescence by confocal microscopy indicated that whereas GFP protein was evenly distributed in the nucleus and cytoplasm in HT-29 GFP cells, GFP-p120ctn protein was preferentially localized at the plasma membrane with enhanced signals at cell-cell contacts and in the cytoplasm of HT-29 GFP-p120ctn cells (Fig. 1B). A low level of fluorescence was detected in the nucleus. We further confirmed the increased cytoplasmic distribution of both endogenous and exogenous p120ctn in HT-29 GFP-p120 cells by Western blotting after cell fractionation (Fig. 1C).
Figure 1.
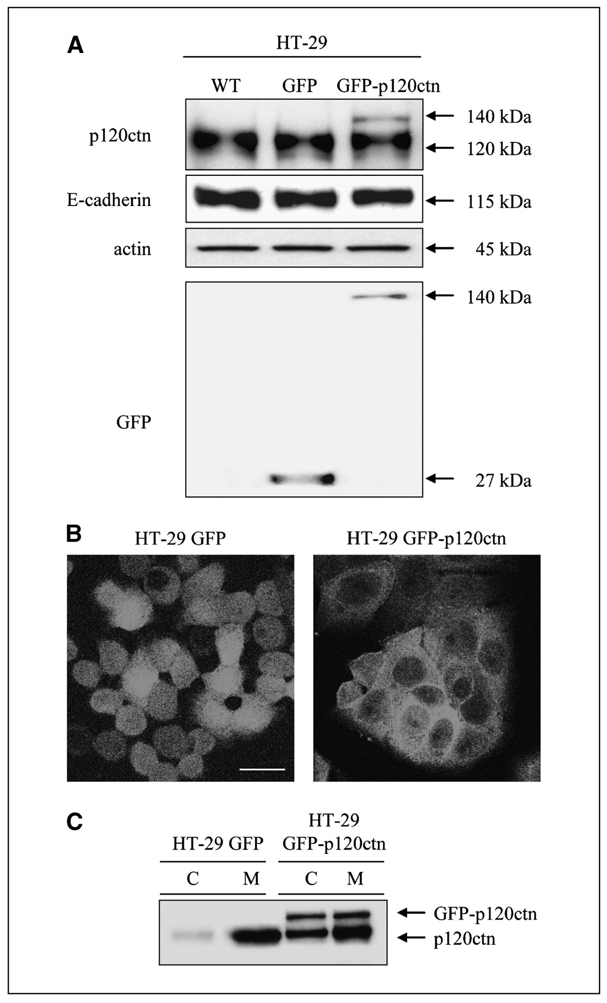
Overexpressed p120ctn accumulates in the cytoplasm. A, WT HT-29 cells (WT) were transfected with pEF6-GFP (GFP) or pEF6-GFP-p120ctn (GFP-p120ctn). Expression levels of p120ctn, GFP, and E-cadherin were analyzed by Western blotting of whole-cell lysates. Actin blotting was used as a loading control. B, GFP or GFP-p120ctn HT-29 cells cultured for 48 h on coverslips were fixed in 3% paraformaldehyde before confocal microscopy analysis. Bar, 50 μm. C, total GFP or GFP-p120ctn cell lysates were separated into cytoplasmic (C) and membrane-associated (M) fractions and detection of endogenous and exogenous p120ctn was achieved by Western blotting.
These results suggest that in HT-29 cells, a moderate overexpression of p120ctn is sufficient to induce cadherin saturation without altering their expression and favors an increase in the cytoplasmic pool of p120ctn.
Overexpression of p120ctn slows down the progression of HT-29 cells through the cell cycle
One cellular function that can be regulated by cell adhesion molecules is progression through the cell cycle (20). In the course of our experiments, we observed a growth delay in HT-29 GFP-p120ctn cells compared with HT-29 GFP cells. Whereas HT-29 GFP cells typically reached confluency 120 h after plating and formed a cell monolayer, GFP-p120ctn cells remained sparsely clustered without reaching full confluency (Fig. 2A). To quantify this growth delay, we seeded an equal number of HT-29 GFP or HT-29 GFP-p120ctn cells and established growth curves by counting live cells every 24 h (Fig. 2B). During the first 48 h of culture, the number of living cells increased slowly in HT-29 GFP cells, with no change in HT-29 GFP-p120ctn cells. After this lag period, the number of living cells increased in both populations although at a slower rate in HT-29 GFP-p120ctn cells. Indeed, at 96 h, we observed 43% less living HT-29 GFP-p120ctn cells compared with HT-29 GFP cells. These data suggest a delayed progression in the cell cycle. To investigate the effects of p120ctn overexpression on DNA replication, we did flow cytometry analysis of cells subjected to a short pulse of BrdUrd incorporation. Compared with control cells (HT-29 GFP), HT-29 GFP-p120ctn showed significantly decreased BrdUrd incorporation into their nuclei (>33% less), indicating a reduced rate of DNA replication, perhaps partly accounting for their delayed growth (Fig. 2C). Impaired DNA replication slows down S-phase progression. We therefore investigated the effects of p120ctn overexpression on cell cycle progression (Fig. 2D). HT-29 GFP and GFP-p120ctn cells were synchronized at G1-S transition by aphidicolin treatment and then released by replacing the medium with complete medium supplemented with fresh serum. Flow cytometry analysis showed that HT-29 GFP cells progressed correctly through the cell cycle: the DNA content was totally duplicated when cells reached G2-M 8 to 10 h after the release (75% of cells were in G2-M) and a complete cycle was observed after 24 h (Fig. 2D, left). In contrast, HT-29 GFP-p120ctn cells remained arrested in G1-S up to 8 h after aphidicolin release (90% of cells in G1-S; Fig. 2D, right). Subsequently, these cells progressed into S phase but only 16% reached G2-M within 24 h.
Figure 2.
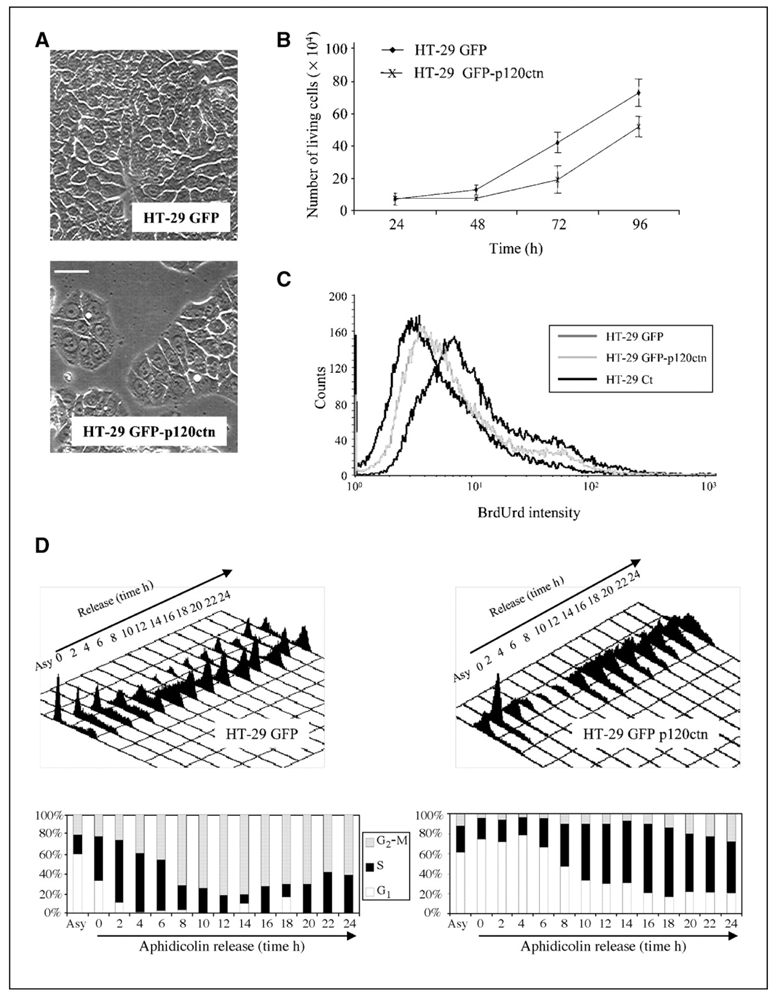
Overexpression of p120ctn slows down HT-29 cell proliferation. A, HT-29 GFP and HT-29 GFP-p120ctn cells were cultured for 120 h and observed under phase-contrast microscopy. Bar, 50 μm. B, HT-29 GFP and HT-29 GFP-p120ctn cells were seeded at 2 × 104 cells per well, grown for the indicated times and living cells, then counted using trypan blue solution. Points, mean of three independent experiments; bars, SE. C, BrdUrd incorporation assay. BrdUrd (5 μmol/L) was added for 15 min to the culture medium of 24-h precultured HT-29 GFP (dark gray line) or HT-29 GFP-p120ctn (gray line) cells. After fixation and BrdUrd immunostaining, positive cells were counted using a Becton Dickinson Facstar flow cytometer. Control cells non–treated with BrdUrd were used as negative control (black line). D, top, HT-29 GFP (left) or HT-29 GFP-p120ctn (right) cells were synchronized at G1-S transition with aphidicolin (5 μg/mL, 24 h) and released in fresh medium for the indicated time. DNA content was then analyzed by FACS after propidium iodide staining. Bottom, histograms indicate the percentage of cells in G1, S, and G2-M phases. Asy, asynchronized.
Taken together, these data indicate that stable overexpression of p120ctn in HT-29 cells induces an overall reduction in cell growth via a negative regulation on cell cycle progression.
Cyclin E is up-regulated in p120ctn-overexpressing cells
Cyclins and their catalytic subunits, the cdks, control cell cycle progression by regulating events that drive the transition between cell cycle phases. Among these, the cdk2/cyclin E complex is involved in G1-S transition and DNA replication (21, 22). The delayed transition observed in HT-29 GFP-p120ctn cells prompted us to examine cyclin E levels and subcellular distributions throughout cell cycle progression (Fig. 3A and B). Western blotting analyses of cyclin E showed dynamic changes according to the cell cycle phases in HT-29 GFP cells: high cyclin E levels in G1-S phase–synchronized cells (0 h) were followed by a sharp decrease after entry into the S and G2-M phases (2–10 h; Fig. 3A, HT-29 GFP). Subsequently, the cyclin concentration increased again, reaching a new steady state following reentry into the G1 phase. In contrast, we observed no such dynamism in HT-29 GFP-p120ctn cells, with a high level of cyclin E maintained in these cells during the first 18 h after aphidicolin release, consistent with the observed persisting G1-S phase in these cells (Fig. 3A, HT-29 GFP-p120ctn). This plateau was followed by a slow decrease in the cyclin E content after S-phase entry (18–24 h). At the same time, the levels of cdk2 and p120ctn remained constant during the cell cycle (Fig. 3A). Confocal microscopy indicated that in asynchronous HT-29 GFP and HT-29 GFP-p120ctn cells, a low amount of cyclin E was localized into cytoplasmic patches and the nucleus (Fig. 3B). After synchronization by aphidicolin, both cell lines displayed a strong nuclear staining for cyclin E. Following entry into the S phase and during the G2-M phase (6–16 h), the labeling of cyclin E became weaker and diffuse in the cytoplasm of HT-29 GFP cells (corresponding to proteolytic degradation of the protein as shown previously by Western blotting). Vesicular cytoplasmic staining of cyclin E reappeared at 24 h when cells entered into a new cell cycle (Fig. 3B, top). In HT-29 GFP-p120ctn cells, a weak nuclear staining was maintained up to 24 h (Fig. 3B, bottom), in agreement with a stabilization of the protein level (Fig. 3A) and the persistence of cells in the G1-S phase (Fig. 2D). The level of cyclin E is strictly regulated throughout the cell cycle by transcriptional and proteolytic mechanisms. It is an unstable protein that is rapidly degraded by two different ubiquitin-dependent proteolysis pathways (23). Indeed, inhibition of the 26S proteasome with MG132 8 h after aphidicolin release resulted in the accumulation of cyclin E protein similar to that observed within HT-29 GFP-p120ctn cells. This suggests that the steady-state level of cyclin E in cells overexpressing p120ctn results from limited degradation of cyclin E (Fig. 3C). Post-translational regulation was confirmed by reverse transcription-PCR experiments indicating similar cyclin E/glyceraldehyde-3-phosphate dehydrogenase mRNA ratios in both cell types (data not shown).
Figure 3.
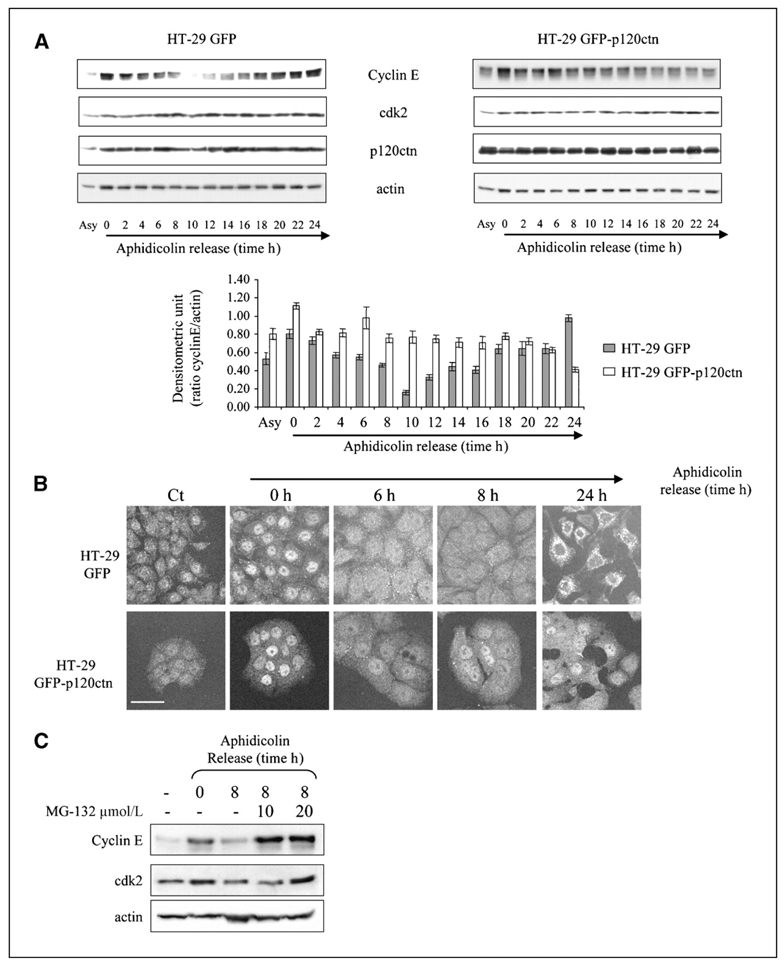
Cyclin E protein is stabilized by p120ctn overexpression. A, top, asynchronized HT-29 GFP or HT-29 GFP-p120ctn cells or cells synchronized at G1-S transition with aphidicolin blockade and released in fresh medium for indicated times were lyzed. Total lysates were analyzed for cyclin E, cdk2, p120ctn, and actin expression by Western blotting. Bottom, histograms represent the densitometric ratio of cyclin E to actin levels. Columns, mean of three independent experiments; bars, SE. B, in parallel, the cells were immunostained for cyclin E and analyzed by confocal microscopy. Bar, 50 μm. C, G2-M–enriched GFP cells were treated or not with the indicated concentrations of proteasome inhibitor MG132. Total cell lysates were then analyzed for cyclin E and cdk2 expression by Western blotting. Actin staining was used as a loading control.
Taken together, these observations strongly suggest that p120ctn overexpression results in impaired proteasome degradation and maintenance within the nucleus, which in turn causes cyclin E level persistence and S-phase lengthening.
Cyclin E and p120ctn accumulate at centrosomes in HT-29 GFP-p120ctn cells
Recently, cyclin E–dependent kinases have been implicated in the control of centrosome duplication, a process associated with their recruitment to this organelle (24). Interestingly, an increase in the cytoplasmic pool of p120ctn, for instance in tumor cells, causes a localization of p120ctn at centrosomes (13, 14). This localization of p120ctn at centrosomes was suggested in our model by confocal microscopy on mitotic cells (Fig. 4A). Double immunofluorescence analysis of GFP-p120ctn and β-tubulin showed the colocalization of both molecules at the mitotic furrow and centrosomes. Centrosomes from HT-29 GFP and GFP-p120ctn cells were then isolated by centrifugation on sucrose gradient as described previously (18). Indeed, immunoblot analysis of this fractionated gradient revealed the presence of p120ctn in fractions reported previously as containing centrosomes that sedimented together with the centrosomal marker Aurora A (Fig. 4B). We then compared the distribution of various endogenous proteins in centrosomal and membrane-enriched fractions prepared from HT-29 GFP and HT-29 GFP-p120ctn cells (Fig. 4C). We detected endogenous cyclin E, cdk2, and p120ctn in the centrosomal fractions isolated from HT-29 GFP cells. Moreover, p120ctn overexpression resulted in increased amounts of cyclin E and cdk2 in these fractions when compared with levels of Aurora A (Fig. 4C, right). Finally, we detected E-cadherin and actin only in the membrane-enriched fractions, indicating that little or no contamination occurred with membranous or cytoplasmic proteins in our centrosome preparation. Altogether, our data confirm that p120ctn localizes to centrosomes and suggest the implication of this catenin in cyclin E and cdk2 regulation at this peculiar location.
Figure 4.
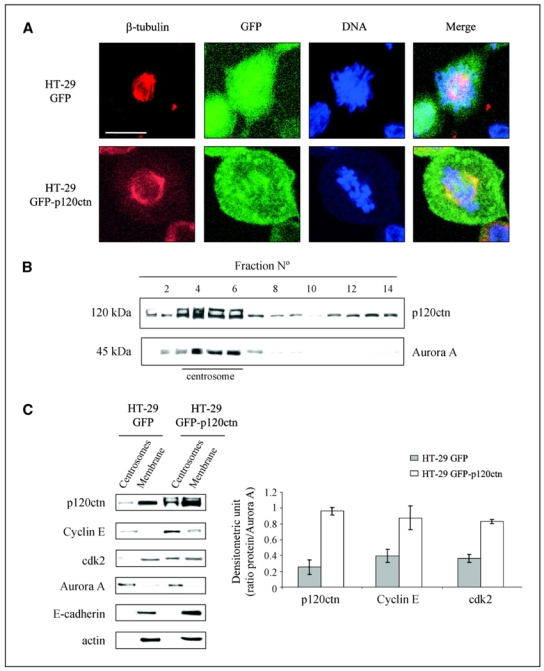
p120ctn localizes to the mitotic furrow and the centrosomes in mitotic HT-29 cells. A, HT-29 GFP (top) and HT-29 GFP-p120ctn (bottom) cells were grown for 48 h on coverslips, fixed with 3% paraformaldehyde, and immunostained for β-tubulin before observation under a confocal microscope DNA was stained blue with 4′,6-diamidino-2-phenylindole (DAPI). Chosen fields are representative of mitotic cells in each condition. Bar, 10 μm. B, centrosome fractions were enriched from mitotic HT-29 GFP-p120ctn cells using a discontinuous sucrose gradient (see Material and Methods). Antibodies against p120ctn and Aurora A were used for Western blotting analyses of each collected fraction. C, left, centrosome- and cell membrane–enriched fractions obtained from the previously described discontinuous sucrose gradient were analyzed by Western blotting using antibodies against p120ctn, cyclin E, cdk2, Aurora A, E-cadherin, and actin. Right, histograms represent the densitometric ratio of p120ctn, cyclin E, and cdk2 to Aurora A levels in centrosomal fractions. Columns, mean of three independent experiments; bars, SE.
The cyclin E/cdk2 complex is associated with p120ctn in centrosomes
From our results, we hypothesized that the increased levels of cyclin E and cdk2 in the centrosome could arise from their association with p120ctn. A RXL motif is required for stable binding of cyclin E/cdk2 complexes to substrates (25, 26). Sequence analysis of p120ctn revealed the presence of six putative RXL motifs in the protein structure. Confocal analysis showed that GFP-p120ctn colocalized with cyclin E and cdk2 in structures corresponding to centrosomes during mitosis (Fig. 5A). To further determine whether p120ctn binds cyclin E/cdk2 complexes during mitosis, we immunoprecipitated cdk2 from synchronized HT-29 GFP and GFP-p120ctn cells and assayed its association with cyclin E and p120ctn by immunoblotting (Fig. 5B). We observed the presence of cyclin E/cdk2/p120ctn complexes in both cell types. As expected, normalized to the amount of immunoprecipitated cdk2, the association of p120ctn and cyclin E with cdk2 was respectively 6.5- and 2.5-fold greater in HT-29 GFP-p120ctn cells than in control. One of the main targets of cdk2/cyclin E at centrosomes is nucleophosmin, a protein allowing centrosome duplication following phosphorylation on Thr199. Staining of HT-29 GFP-p120ctn cells with an antibody recognizing phosphorylated nucleophosmin (Thr199) indicated the presence of the phosphorylated protein in the cytoplasm and the centrosome of dividing cells (Fig. 5C). We then detected levels of phosphorylated nucleophosmin in centrosomal and total fractions of HT-29 GFP or GFP-p120ctn cells by Western blotting (Fig. 5D). We found more phosphorylated nucleophosmin in HT-29 GFP-p120ctn than in control cells, suggesting a functional association between the increased recruitment of the cdk2/cyclin E complex and its increased phosphorylation activity within the centrosomes.
Figure 5.
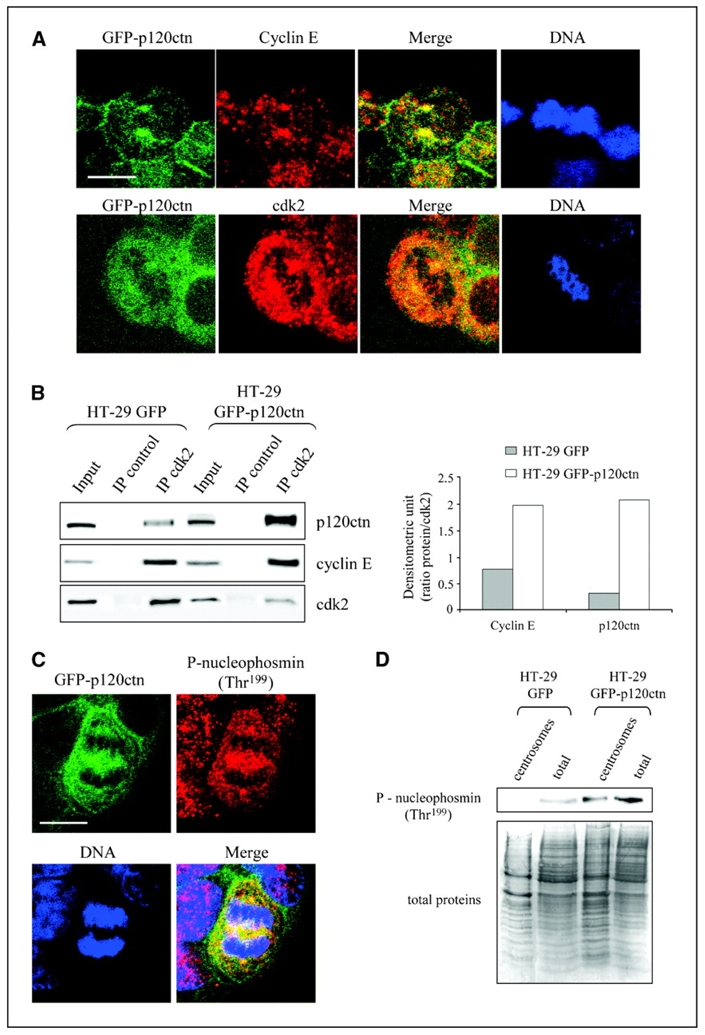
p120ctn interacts with the cdk2/cyclin E complex. HT-29 GFP-p120ctn cells were grown for 48 h on coverslips, fixed with 3% paraformaldehyde, and immunostained for cyclin E and cdk2 (A) or phosphorylated nucleophosmin (P-nucleophosmin; C) before confocal microscopic analysis. DNA was stained blue with DAPI. Bar, 10 μm. B, left, HT-29 GFP or GFP-p120ctn cells were synchronized at G1-S transition with aphidicolin and released in fresh medium for 6 h. cdk2 was immunoprecipitated (IP) from total cell lysates and levels of p120ctn, cyclin E, and cdk2 were detected by Western blotting. Right, histograms represent the densitometric ratio of cyclin E and p120ctn to immunoprecipitated cdk2 levels. This experiment was reproduced at least thrice. D, centrosomeenriched and total fractions from HT-29 GFP and GFP-p120ctn cells were analyzed by Western blotting using antibodies against nucleophosmin phosphorylated on Thr199 (top). Coomassie blue staining of the transferred membrane was used as a loading control (bottom).
p120ctn-mediated deregulation of cyclin E is associated with centrosome amplification and genomic instability
Because various reports have indicated a common association between up-regulated cyclin E and such centrosome defects and genetic instability (16), p120ctn stabilization of cyclin E might also alter centrosome duplication. To address this question, we immunomarked centrosomes with anti-γ-tubulin and compared centrosome profiles of HT-29 GFP versus HT-29 GFP-p120ctn cells under a confocal microscope. Whereas almost all dividing HT-29 GFP cells contained two centrosomes, we reproducibly detected centrosome amplifications in HT-29 GFP-p120ctn cells (Fig. 6A). We found a low frequency of centrosome amplification in wild-type (WT) HT-29 cells (<2%), which showed a poorly significant increase in HT-29 GFP cells (<4%). Conversely, >12% of HT-29 GFP-p120ctn cells showed a pronounced amplification of centrosomes (Fig. 6B, left). Statistical analysis indicated that these cells displayed three to eight centrosomes with a peak of the histogram distribution at four centrosomes per cell (56%; Fig. 6B, right). Observation by phase-contrast microscopy showed an increased number of multinucleated cells in HT-29 GFP-p120ctn (Fig. 6C), indicating that p120ctn-mediated cyclin E overexpression was translated into dysfunctioning mitosis and genetic instability. According to the time of culture, we noticed a decline in the percentage of these cells, in correlation with an increase in caspase-3 activity (Fig. 6D). These observations suggest the involvement of an apoptotic pathway in the elimination of these multinucleated cells in vitro. Therefore, p120ctn overexpression induces centrosome amplification and polynucleation, events both depending on impaired cyclin E proteolysis.
Figure 6.
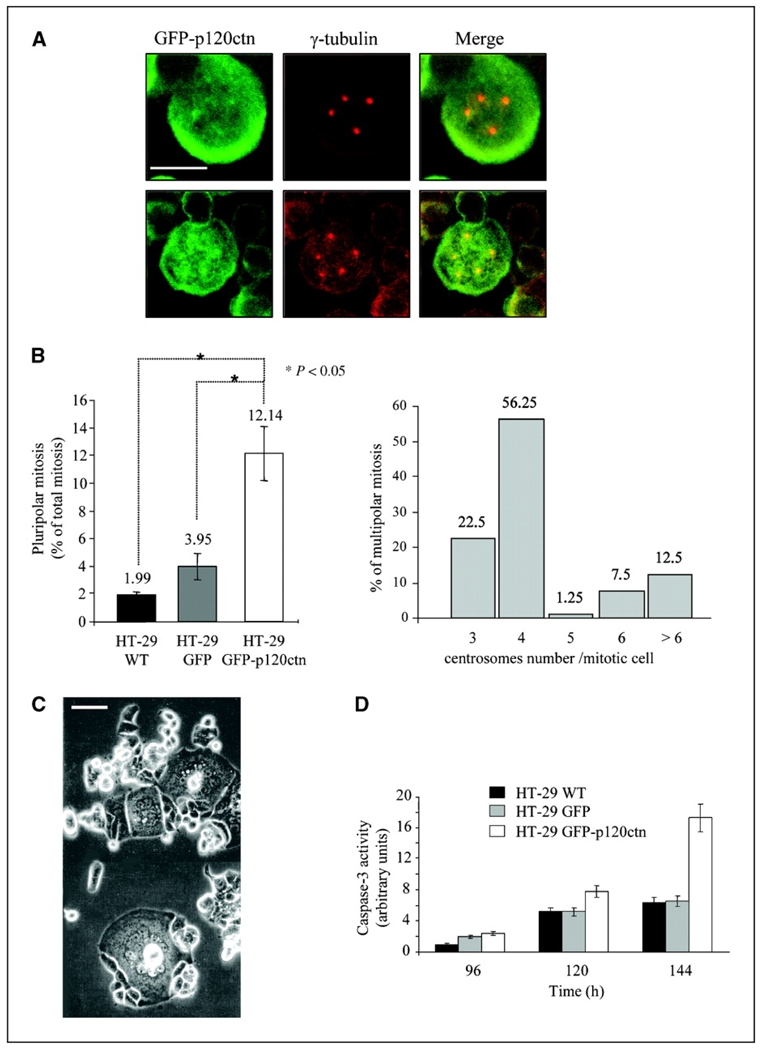
p120ctn overexpression leads to overduplication of centrosomes and polyploidy. A, HT-29 GFP-p120ctn cells were grown for 48 h on coverslips, fixed with 3% paraformaldehyde, and immunostained for γ-tubulin before confocal microscopic observation. Bar, 10 μm. B, the number of centrosomes per mitotic cell was then quantified under confocal microscopy. Multipolar mitotic cells were represented as the percentage of total mitotic cells (n = 200). *, P < 0.05, Student’s t test (left). Histogram showing the repartition of HT-29 GFP-p120ctn cells with supernumerary centrosomes (n = 200; right). C, polynuclear HT-29 GFP-p120 cells were observed under phase-contrast microscopy after 48 h of culture. D, caspase-3/caspase-7 activity was analyzed using Apo-One homogeneous assay in HT-29 GFP or HT-29 GFP-p120ctn cells according to the manufacturer’s instructions. Fluorescence emission at 527 nm was quantified using a Fluoroskan Ascent. All experiments were done in triplicate.
Discussion
E-cadherin expression is frequently down-regulated in many tumors, thus leading to the accumulation of cytoplasmic and/or nuclear catenins. Recently, Nong et al. described that overexpression of p120ctn in hepatoma cells can recruit β-catenin to the membrane and the cytoplasm while inhibiting cell proliferation by down-regulating survivin and cyclin D1 expression (27). Wildenberg et al. (28) showed that the lack of p120ctn in NIH3T3 allows these cells to proliferate in the absence of serum and reveals the involvement of p120ctn in the regulation of cell contact inhibition. Moreover, the conditional knockdown of p120ctn in mouse epidermis leads to hyperplasia through nuclear factor-κB activation (29). The generation of stable cell lines overexpressing p120ctn allowed us to modify the E-cadherin/p120ctn balance, thus leading to mislocalization of p120ctn and the unexpected revelation of its role in cell cycle progression. Stabilization of cyclin E leads to its overexpression and accumulation in centrosomes, together with p120ctn and cdk2, forming a functional ternary complex. These data are consistent with a previous report describing p120ctn targeting in centrosomes of breast tumor cells (13). However, the underlying mechanism remains unclear. Park et al. (26) showed that β-catenin could interact directly with cdk2 via RXL motifs present in armadillo repeat domains. In silico analysis of p120ctn sequence revealed that it contains six RXL motifs (data not shown) that may be involved in a p120ctn/cdk2 interaction.
Cyclin E plays an essential role in fundamental biological processes, such as cell cycle control, DNA replication, apoptosis, and DNA repair (30). Therefore, its expression must be finely regulated throughout the cell cycle (31). We found an impaired cyclin E down-regulation and accumulation in centrosomes in cells overexpressing p120ctn, suggesting that the p120ctn offers protection from proteasome degradation. In Chinese hamster ovary cells, the expression of the anchoring protein CG-NAP/D localizing in centrosomes throughout the cell cycle causes centrosome amplification by recruiting an abnormally high level of cyclin E/cdk2 complexes (32). In these cells, we found that endogenous p120ctn also colocalizes with centrosomes (data not shown). Similarly to CG-NAP/D, p120ctn may recruit cyclin E/cdk2 complexes to centrosomes or alternatively modify the association of cyclin E with cdk inhibitors, a process described previously as strictly regulating cyclin E/cdk2 activity (22). Whereas cyclin E overexpression is known to accelerate G1-S transition in different cell lines (33, 34), we observed a delay in S-phase progression associated with the reduced proliferation of HT-29 cells overexpressing p120ctn, in accordance with other reports (33–36). This paradox can be explained by the role(s) played by cdks in regulating DNA replication (37). Indeed, high cyclin E/cdk2 activity was found to interfere with the assembly of prereplication complexes leading to defects in replication initiation and possibly fork movement, thereby slowing down the S phase (36).
Overduplication of centrosomes can occur during prolonged S phases, when DNA replication is impaired and in primary tumors or cancer cell lines (38, 39). In many cases, a strong link between centrosome amplification and cyclin E overexpression has been described (32, 40). Our data reveal an increased number of centrosomes in cells overexpressing p120ctn. Nucleophosmin/B23, a chaperonin abundantly found in the nucleolus, has been identified as the primary target of cdk2/cyclin E in the initiation of centrosome duplication (15, 41). We noticed an increase in nucleophosmin phosphorylation on Thr199, strongly suggesting that the p120ctn-mediated cdk2/cyclin E complex is active in centrosomes and may be directly responsible for centrosome amplification. p120ctn-mediated elevated levels of cyclin E could affect processes involved in the faithful duplication and segregation of chromosomes and may generate CIN, as described by Lengauer (42). This hypothesis fits with the significant levels of aneuploidy observed in both fibroblasts and epithelial cells on cyclin E ectopic expression (35). Interestingly, as for tumoral cells, overexpression of p120ctn in normal rat intestinal cells leads to reduced proliferation and an increased cyclin E expression, which accumulates together with p120ctn in centrosomes. These findings correlate with the apparition of p120ctn-positive polyploid cells (Supplementary Data S1). It thus seems that changes in protein stability might be the major reason for cyclin E–dependent effects of p120ctn overexpression in our biological system (normal and tumoral cells). This may also explain the CIN observed in many human colorectal cancers (43). Indeed, cyclin E has been found overexpressed in several solid tumors (reviewed in ref. 44), proving to be a prognostic marker for poor outcome in breast cancer and associated with increased tumor progression (45). Our observations suggest that the frequency of p120ctn-mediated polyploidy in normal cells may represent an early phenomenon that could contribute to tumor initiation. Ex vivo, HT-29 cells expressing these supernumerary centrosomes represented only 10% to 15% of the total population. However, this population remained constant, reflecting equilibrium between new cells acquiring this phenotype and aged cells that die (after 140 h of culture, 15% of cells died by a caspase-dependent process). In vivo, the situation is quite different because the number of cells presenting an abnormal duplication of chromosomes increases with tumor progression (46).
Recent reports support a significant relationship between deregulation of cyclin E protein levels and the development of human cancers (47). It would be interesting to analyze the expression and distribution of p120ctn in these tumors to determine whether this catenin is the primary cause of cyclin E up-regulation. A study done in the breast cancer cell line MDA-MB-468 indicated a link between cyclin E and adhesive properties shown by changes in adhesion associated gene expression (48). Surprisingly, although cyclin E overexpression is associated with an aggressive phenotype, it significantly decreased the invasive potential of MDA-MB-468 cells compared with control clones in this study. Interaction of p120ctn with cyclin E/cdk2 complex may modulate the capacity of catenin to regulate Rho-GTPase activity, responsible for cytoskeletal reorganization and therefore impaired migration. A recent study by Bellovin et al. (8) done on 557 tumors showed a sustained p120ctn expression in the majority of colorectal carcinomas and a cytoplasmic localization as a statistically significant indicator of advanced disease. Interestingly, relatively few tumors exhibited a loss of p120ctn expression (5.4%).
Mechanistically, p120ctn seems to contribute to invasiveness and tumor progression in various independent ways via (a) p120ctn association–dependent regulation of mesenchymal cadherin levels (49); (b) p120ctn-mediated regulation of Rho-GTPases (9); and (c) p120ctn-dependent release of kaiso-mediated repression of gene expression (50). Altogether, our results provide new arguments for the importance of p120ctn in cancer progression: on the one hand, its centrosomal distribution could contribute to aberrant centrosome duplication and cell cycle progression, both leading to genomic instability, and, on the other hand, its cytoplasmic localization could increase invasiveness and migration. Because these modifications are often observed in cancer, p120ctn may represent a new therapeutic target for future therapy.
Acknowledgments
Grant support: Association pour la Recherche sur le Cancer, Ligue Nationale contre le Cancer (Comité de Savoie). N.T. Chartier is the recipient of a fellowship from the Ministère de la Recherche et de l’Enseignement Supérieur.
We thank Géraldine Pawlak and Xavier Renot for their constructive criticism and Alexei Grichine and Brigitte Peyrusse for their excellent technical assistance in confocal microscopy and reprography artwork.
References
- 1.Berx G, Van Roy F. The E-cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res. 2001;3:289–93. doi: 10.1186/bcr309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vleminckx K, Vakaet L, Jr, Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–19. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- 3.Reynolds AB, Carnahan RH. Regulation of cadherin stability and turnover by p120ctn: implications in disease and cancer. Semin Cell Dev Biol. 2004;15:657–63. doi: 10.1016/j.semcdb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Waltzer L, Bienz M. The control of β-catenin and TCF during embryonic development and cancer. Cancer Metastasis Rev. 1999;18:231–46. doi: 10.1023/a:1006321324190. [DOI] [PubMed] [Google Scholar]
- 5.Thoreson MA, Anastasiadis PZ, Daniel JM, et al. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J Cell Biol. 2000;148:189–202. doi: 10.1083/jcb.148.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Hengel J, Vanhoenacker P, Staes K, van Roy F. Nuclear localization of the p120(ctn) armadillo-like catenin is counteracted by a nuclear export signal and by E-cadherin expression. Proc Natl Acad Sci U S A. 1999;96:7980–5. doi: 10.1073/pnas.96.14.7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarrio D, Perez-Mies B, Hardisson D, et al. Cytoplasmic localization of p120ctn and E-cadherin loss characterize lobular breast carcinoma from preinvasive to metastatic lesions. Oncogene. 2004;23:3272–83. doi: 10.1038/sj.onc.1207439. [DOI] [PubMed] [Google Scholar]
- 8.Bellovin DI, Bates RC, Muzikansky A, Rimm DL, Mercurio AM. Altered localization of p120 catenin during epithelial to mesenchymal transition of colon carcinoma is prognostic for aggressive disease. Cancer Res. 2005;65:10938–45. doi: 10.1158/0008-5472.CAN-05-1947. [DOI] [PubMed] [Google Scholar]
- 9.Anastasiadis PZ. p120-ctn: a nexus for contextual signaling via Rho GTPases. Biochim Biophys Acta. 2007;1773:34–46. doi: 10.1016/j.bbamcr.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 10.Daniel JM, Reynolds AB. The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol Cell Biol. 1999;19:3614–23. doi: 10.1128/mcb.19.5.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Roy FM, McCrea PD. A role for Kaiso-p120ctn complexes in cancer? Nat Rev Cancer. 2005;5:956–64. doi: 10.1038/nrc1752. [DOI] [PubMed] [Google Scholar]
- 12.Chen X, Kojima S, Borisy GG, Green KJ. p120 catenin associates with kinesin and facilitates the transport of cadherin-catenin complexes to intercellular junctions. J Cell Biol. 2003;163:547–57. doi: 10.1083/jcb.200305137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franz CM, Ridley AJ. p120 catenin associates with microtubules: inverse relationship between microtubule binding and Rho GTPase regulation. J Biol Chem. 2004;279:6588–94. doi: 10.1074/jbc.M312812200. [DOI] [PubMed] [Google Scholar]
- 14.Yanagisawa M, Kaverina IN, Wang A, Fujita Y, Reynolds AB, Anastasiadis PZ. A novel interaction between kinesin and p120 modulates p120 localization and function. J Biol Chem. 2004;279:9512–21. doi: 10.1074/jbc.M310895200. [DOI] [PubMed] [Google Scholar]
- 15.Okuda M, Horn HF, Tarapore P, et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103:127–40. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 16.Kawamura K, Izumi H, Ma Z, et al. Induction of centrosome amplification and chromosome instability in human bladder cancer cells by p53 mutation and cyclin E overexpression. Cancer Res. 2004;64:4800–9. doi: 10.1158/0008-5472.CAN-03-3908. [DOI] [PubMed] [Google Scholar]
- 17.Chartier NT, Laine M, Gout S, et al. Laminin-5-integrin interaction signals through PI 3-kinase and Rac1b to promote assembly of adherens junctions in HT-29 cells. J Cell Sci. 2006;119:31–46. doi: 10.1242/jcs.02698. [DOI] [PubMed] [Google Scholar]
- 18.Bornens M, Moudjou M. Studying the composition and function of centrosomes in vertebrates. Methods Cell Biol. 1999;61:13–34. doi: 10.1016/s0091-679x(08)61973-1. [DOI] [PubMed] [Google Scholar]
- 19.Reynolds AB, Roczniak-Ferguson A. Emerging roles for p120-catenin in cell adhesion and cancer. Oncogene. 2004;23:7947–56. doi: 10.1038/sj.onc.1208161. [DOI] [PubMed] [Google Scholar]
- 20.Pugacheva EN, Roegiers F, Golemis EA. Interdependence of cell attachment and cell cycle signaling. Curr Opin Cell Biol. 2006;18:507–15. doi: 10.1016/j.ceb.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sauer K, Lehner CF. The role of cyclin E in the regulation of entry into S phase. Prog Cell Cycle Res. 1995;1:125–39. doi: 10.1007/978-1-4615-1809-9_10. [DOI] [PubMed] [Google Scholar]
- 22.Ekholm SV, Reed SI. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol. 2000;12:676–84. doi: 10.1016/s0955-0674(00)00151-4. [DOI] [PubMed] [Google Scholar]
- 23.Won KA, Reed SI. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J. 1996;15:4182–93. [PMC free article] [PubMed] [Google Scholar]
- 24.Lacey KR, Jackson PK, Stearns T. Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci U S A. 1999;96:2817–22. doi: 10.1073/pnas.96.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adams PD, Sellers WR, Sharma SK, Wu AD, Nalin CM, Kaelin WG., Jr Identification of a cyclin-cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol Cell Biol. 1996;16:6623–33. doi: 10.1128/mcb.16.12.6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park CS, Kim SI, Lee MS, et al. Modulation of β-catenin phosphorylation/degradation by cyclin-dependent kinase 2. J Biol Chem. 2004;279:19592–9. doi: 10.1074/jbc.M314208200. [DOI] [PubMed] [Google Scholar]
- 27.Nong CZ, Pan LL, He WS, Zha XL, Ye HH, Huang HY. p120ctn overexpression enhances β-catenin-E-cadherin binding and down regulates expression of survivin and cyclin D1 in BEL-7404 hepatoma cells. World J Gastroenterol. 2006;12:1187–91. doi: 10.3748/wjg.v12.i8.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wildenberg GA, Dohn MR, Carnahan RH, et al. p120-Catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127:1027–39. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 29.Perez-Moreno M, Davis MA, Wong E, Pasolli HA, Reynolds AB, Fuchs E. p120-catenin mediates inflammatory responses in the skin. Cell. 2006;124:631–44. doi: 10.1016/j.cell.2005.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazumder S, DuPree EL, Almasan A. A dual role of cyclin E in cell proliferation and apoptosis may provide a target for cancer therapy. Curr Cancer Drug Targets. 2004;4:65–75. doi: 10.2174/1568009043481669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ekholm SV, Zickert P, Reed SI, Zetterberg A. Accumulation of cyclin E is not a prerequisite for passage through the restriction point. Mol Cell Biol. 2001;21:3256–65. doi: 10.1128/MCB.21.9.3256-3265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishimura T, Takahashi M, Kim HS, Mukai H, Ono Y. Centrosome-targeting region of CG-NAP causes centrosome amplification by recruiting cyclin E-cdk2 complex. Genes Cells. 2005;10:75–86. doi: 10.1111/j.1365-2443.2005.00816.x. [DOI] [PubMed] [Google Scholar]
- 33.Ohtsubo M, Roberts JM. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science. 1993;259:1908–12. doi: 10.1126/science.8384376. [DOI] [PubMed] [Google Scholar]
- 34.Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol. 1994;14:1669–79. doi: 10.1128/mcb.14.3.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 36.Ekholm-Reed S, Mendez J, Tedesco D, Zetterberg A, Stillman B, Reed SI. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J Cell Biol. 2004;165:789–800. doi: 10.1083/jcb.200404092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woo RA, Poon RY. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle. 2003;2:316–24. [PubMed] [Google Scholar]
- 38.Fukasawa K. Centrosome amplification, chromosome instability, and cancer development. Cancer Lett. 2005;230:6–19. doi: 10.1016/j.canlet.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 39.Balczon R, Bao L, Zimmer WE, Brown K, Zinkowski RP, Brinkley BR. Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J Cell Biol. 1995;130:105–15. doi: 10.1083/jcb.130.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G. Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science. 1999;283:851–4. doi: 10.1126/science.283.5403.851. [DOI] [PubMed] [Google Scholar]
- 41.Tarapore P, Okuda M, Fukasawa K. A mammalian in vitro centriole duplication system: evidence for involvement of CDK2/cyclin E and nucleophosmin/B23 in centrosome duplication. Cell Cycle. 2002;1:75–81. [PubMed] [Google Scholar]
- 42.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–7. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 43.Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- 44.Akli S, Keyomarsi K. Cyclin E and its low molecular weight forms in human cancer and as targets for cancer therapy. Cancer Biol Ther. 2003;2:S38–47. [PubMed] [Google Scholar]
- 45.Porter PL, El-Bastawissi AY, Mandelson MT, et al. Breast tumor characteristics as predictors of mammographic detection: comparison of interval- and screen-detected cancers. J Natl Cancer Inst. 1999;91:2020–8. doi: 10.1093/jnci/91.23.2020. [DOI] [PubMed] [Google Scholar]
- 46.Lingle WL, Barrett SL, Negron VC, et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci U S A. 2002;99:1978–83. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24:2776–86. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- 48.Berglund P, Landberg G. Cyclin e overexpression reduces infiltrative growth in breast cancer: yet another link between proliferation control and tumor invasion. Cell Cycle. 2006;5:606–9. doi: 10.4161/cc.5.6.2569. [DOI] [PubMed] [Google Scholar]
- 49.Yanagisawa M, Anastasiadis PZ. p120 catenin is essential for mesenchymal cadherin-mediated regulation of cell motility and invasiveness. J Cell Biol. 2006;174:1087–96. doi: 10.1083/jcb.200605022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kelly KF, Spring CM, Otchere AA, Daniel JM. NLS-dependent nuclear localization of p120ctn is necessary to relieve Kaiso-mediated transcriptional repression. J Cell Sci. 2004;117:2675–86. doi: 10.1242/jcs.01101. [DOI] [PubMed] [Google Scholar]