

Abstract
Reactive gliosis is characterized by enhanced glial fibrillary acidic protein (GFAP) expression, cellular hypertrophy, and astrocyte proliferation. The cellular and molecular mechanisms underlying this process are still largely undefined. We investigated the role of endothelin-1 (ET-1) in reactive gliosis in corpus callosum after lysolecithin (LPC)-induced focal demyelination and in cultured astrocytes. We show that ET-1 levels are upregulated in demyelinated lesions within 5 d after LPC injection, together with enhanced astrocyte proliferation, GFAP expression, and JNK phosphorylation. Infusion of the pan-ET-receptor (ET-R) antagonist Bosentan or the selective ETB-R antagonist BQ788 into the corpus callosum prevented postlesion astrocyte proliferation and JNK phosphorylation. In cultured astrocytes, ET-1-induced activation of ETB-Rs promotes a reactive phenotype by enhancing both GFAP expression and astrocyte proliferation. In the same cells, ET-1 activates both JNK and p38MAPK pathways, and induces c-Jun expression at the mRNA and protein levels. By using selective pharmacological inhibitors, we also provide evidence that ET-1 induces astrocyte proliferation and GFAP expression through activation of ERK- and JNK-dependent pathways, consistent with the previous observation of ET-1-induced activation of ERK (Schinelli et al., 2001). Finally, we show by gain and loss of function that increased c-Jun expression enhances the proliferative response of astrocytes to ET-1, whereas c-jun siRNA prevents ET-1-induced cell proliferation. Our results indicate that the effects of ET-1 on astrocyte proliferation depend on c-Jun induction and activation through ERK- and JNK-dependent pathways, and suggest that ET-R-associated pathways might represent important targets to control reactive gliosis.
Keywords: demyelination, cell cycle, endothelin receptors, glial fibrillary acidic protein, JNK kinase, ERK kinase
Introduction
Reactive astrogliosis and microgliosis is a common occurrence in CNS trauma, and reducing the presence of excess reactive glial cells is important for normal cellular function (Miyake et al., 1989; Ridet et al., 1997; McGraw et al., 2001; Buffo et al., 2005; Sofroniew, 2005). After activation by injury, astrocytes and microglia release factors that recruit other astrocytes and microglia to the injury site. This recruitment can lead to glial scar formation, which has the potential to block the growth and maturation of neural progenitors and also impede neovascularization, thus inhibiting recovery after injury (Fawcett and Asher, 1999). A previous study demonstrated the harmful effect of astrogliosis on oligodendrocyte progenitor development and remyelination after demyelination (Back et al., 2005).
Endothelin-1 (ET-1) is implicated in many CNS pathologies that involve reactive gliosis (Jiang et al., 1993; Yamashita et al., 1994; Zhang et al., 1994; Nie and Olsson, 1996; Stiles et al., 1997; Baba, 1998; Hasselblatt et al., 2001, 2003; Egnaczyk et al., 2003; Sofroniew, 2005). Expression of ET-1 and its receptors (ET-Rs) is strongly upregulated in astrocytes after brain injury (Jiang et al., 1993; Zhang et al., 1994; Hino et al., 1996; Nie and Olsson, 1996; Rogers et al., 1997; Sakurai-Yamashita et al., 1997; Baba, 1998). Astrocytes are autocrine targets of ET-1 action, which results in increased cell proliferation (Lazarini et al., 1996; Hasselblatt et al., 2001, 2003; Rogers et al., 2003; Desai et al., 2004).
A direct role for ET-1 in reactive gliosis was demonstrated by in vivo infusion of exogenous ETs or ET-R agonists, which caused astrocyte hypertrophy and proliferation (Koyama et al., 1999). Opposite effects were obtained after injection of ET-R antagonists (Uesugi et al., 1996). In cultured astrocytes, ET-1 promotes cell proliferation (MacCumber et al., 1990; Couraud et al., 1991; Stanimirovic et al., 1995; Teixeira et al., 2000) and causes marked cytoskeletal changes (Cazaubon et al., 1997; Koyama and Baba, 1999), but the specific signal transduction pathways underlying the effects of ET-1 are still poorly characterized. Previous studies demonstrated activation of several intracellular pathways by ET-1 (Simonson and Herman, 1993; Kasuya et al., 1994; Stanimirovic et al., 1995; Lazarini et al., 1996; Koyama et al., 2003, 2004), including PKC/ERK and p38MAPK (Schinelli et al., 2001).
In this study, we used the myelin toxin lysolecithin (LPC) to test the hypothesis that ET-mediated signaling events promote glial activation initiated by white matter lesion in the mouse corpus callosum (CC) (Woodruff and Franklin, 1999). We sought to identify signal transduction pathways that are causally associated with the effects of ET-1 on astrocyte proliferation and reactive gliosis. We investigated the effects of ET-1 on astrocytes in vivo and in vitro, and demonstrate that both JNK and ERK pathways contribute additively to mediate the cellular effects of ET-1 on these astrocytes. Our findings indicate that molecular targeting of ET-1/ET-Rs and the c-Jun/JNK pathway could be used as a strategy aimed at attenuating reactive gliosis after brain injury.
Materials and Methods
Materials.
ET-1 and anti-ET-1 antibodies were both from EMD Biosciences (San Diego, CA). The following antibodies were purchased from Cell Signaling Technology (Beverly, MA): Thr183- and Tyr185-phosphorylated JNK-1,2 and -3, dually phosphorylated (Thr180/Tyr 182) p38MAPK, phosphorylation state-independent p38MAPK, dually phosphorylated (Thr183/Tyr185) JNK, phosphorylation state-independent JNK, phospho (Ser63) c-jun, phosphorylation state-independent c-Jun, and phospho (Ser) ATF-2. Polyclonal anti-hemagglutinin and anti-ATF-2 (s.c.-243) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal antibodies directed against dually phosphorylated ERK5 and against phosphorylation state-independent ERK5 were from Biosource (Camarillo, CA). Bosentan was a kind gift from Actelion Pharmaceuticals (Allschwil, Switzerland). Fetal bovine serum was from Hyclone (Logan, UT). Trizol and all other reagents for cell cultures were from Invitrogen (Carlsbad, CA). The protein kinase inhibitors SB202190, SB203580, SP600125, U-0132, and PD98019 were from Alexis (Lausen, Switzerland). All the other reagents were purchased from Sigma (St. Louis, MO), including the mouse monoclonal anti-glial fibrillary acidic protein (GFAP) antibody.
DNA plasmids.
DNA fragments were excised from agarose gels and purified with kits from Qiagen (Chatsworth, CA). Plasmids were purified with the Maxiprep kit. TAM67 was excised from pCMV-TAM67 as a RsaI-BamHI fragment and cloned into the EcoRV-BamHI site of the pIRES vector (Clontech, Palo Alto, CA). To construct a GFP fusion, a BamHI-HindIII fragment was cut from the pIRES-TAM67 plasmid and then ligated into the BamHI-HindIII sites of pEGFP-C3. To shift the TAM67 insert into frame with the GFP, this plasmid was linearized with HindIII, and the recessed ends were filled with T4 polymerase (Invitrogen) and religated to generate an in-frame fusion between GFP and TAM67, designated pGFP-TAM67. To express GFP-TAM67 from a bicistronic vector, the chimeric DNA encoding GFP-TAM67 was excised from pGFP-TAM67 with Eco47III and BamHI and cloned into the EcoRV and BamHI sites of pIRES to generate pGFP-TAM67.
Animals and cell cultures.
The generation of the CNP-EGFP mouse used throughout this study has been described previously (Yuan et al., 2002). Two separate CNP-EGFP transgenic lines were used (C1 and D1), and data were pooled because no differences were noted between the two. All animal procedures complied with guidelines of the National Institutes of Health and Children's Research Institute. Purified astrocyte cultures were prepared from 20-d-old Sprague Dawley rat embryos as previously described (Gallo and Armstrong, 1995; Schinelli et al., 2001). Animals were killed, and cortices were dissected and then mechanically dissociated by means of a fire-polished Pasteur pipette. Cells were then plated in 75 cm2 tissue culture flasks in DMEM high-glucose medium, containing 2 mm glutamine and 10% fetal bovine serum (Schinelli et al., 2001). Approximately 24 h after plating, the medium was completely replaced, and the cells were grown for 8–10 d in vitro with a complete medium change every 48 h to 60–70% confluency. The cultures comprised >95% GFAP-positive cells (Gallo and Armstrong, 1995; Schinelli et al., 2001).
Cell proliferation assays.
Rat cortical astrocytes grown for ∼10 d in vitro were trypsinized and seeded on 60 mm tissue culture dishes at a density of 50,000 cells/cm2. After 24 h, subconfluent astrocytes (60–70% confluency) were rinsed twice with DMEM and transfected with lipofectamine reagent (Invitrogen). Cortical astrocytes were transfected with pGFP-IRES vectors. To rule out nonspecific or toxic effects of vector–liposome complexes, control cells were transfected with pGFP-IRES empty vector. For each dish, 8 μg of vector were added to 92 μl of DMEM, while, in a separate tube, 25 μl of lipofectamine were added to 75 μl of DMEM; the two suspensions were mixed and incubated for 30 min at room temperature to allow the formation of vector-liposome complexes. The transfection mixture (200 μl) was added to cells in tissue culture dishes containing 800 μl of DMEM. After 8 h, 1 ml of fresh DMEM medium containing 20% FBS was added to the cells, without removing the transfection mixture, for an additional 12 h. The medium was then completely replaced with fresh DMEM containing 10% FBS, and cells were allowed to recover for 48 h. Cells were then starved overnight (∼16 h) in serum-free DMEM before treatment with the indicated agents and lysis for Western blot analysis. The average efficiency of the transfection procedure (evaluated at 48 h after transfection) in primary cortical astrocytes (% of GFP+ cells) was between 30 and 40% for all the constructs. Bromodeoxyuridine (BrdU) and anti-Ki67 stainings were performed as previously described (Sohn et al., 2006). For BrdU immunostaining, because the necessary alkaline denaturation of cell DNA (0.07N NaOH) was partially quenching GFP fluorescence, anti-GFP immunostaining (1:100; polyclonal anti-GFP; Santa Cruz Biotechnology) was performed before BrdU staining, to allow accurate detection of transfected cells. The effect of kinase inhibitors on ET-1-induced proliferation of cortical astrocytes was assessed using the Cell Titer 96 aqueous One Solution Cell proliferation assay kit (Promega, Madison, WI). This single colorimetric assay is based on the ability of living cells to form a formazan product after incubation with the compound 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS), in the presence of the electron coupling reagent phenazine ethosulfate. The amount of formazan product was assessed by the 490 nm absorbance and was directly proportional to the number of viable cells in the culture. Cortical astrocytes were plated at 5000 cells/well in 96-well plates, pretreated with protein kinase inhibitors for 1 h, and then stimulated with ET-1 100 nm for 48 h. At the end of the stimulation period, 20 μl of MTS stock solution was added to each well and the mix incubated for 3 h at 37°C in 5% CO2 atmosphere. Absorbance was then measured in a microplate reader (M-550; Bio-Rad, Hercules, CA) at 490 nm. Cell proliferation was expressed as percentage decrease compared with cells untreated with kinases inhibitors.
Immunocytochemistry in cultured cells.
Astrocytes were plated at a density of 35,000 cells/cm2 and grown for 48 h in the presence or in the absence of ET-1 (100–200 nm). ET-R antagonists were added to the cultures 2 h before ET-1. Cells were fixed in 4% paraformaldehyde/PBS and stained with rabbit anti-Ki67 (1:500; Novocastra Laboratories, Newcastle, UK) and chicken anti-GFAP (1:500; Abcam, Cambridge, MA) and counterstained with 4′,6′-diamidino-2-phenylindole (DAPI). The percentage of Ki-67+ cells was calculated as a percentage of total DAPI+ cells in 10 microscopic fields per coverslip. A total of three coverslips was counted for each experiment from three experiments (total, 9 coverslips per culture condition). For P-c-Jun immunostaining, subconfluent cultures were starved for 18 h before ET-1 stimulation (100 nm, 20 min) and fixed in paraformaldehyde. Astrocytes were then stained with chicken anti-GFAP and rabbit anti-P-c-Jun (1:500; Cell Signaling Technology).
siRNAs.
Two 21-nt-long siRNAs targeting rat c-jun mRNA (GenBank accession number X1716), marked c-jun1 and c-jun2, were designed according to the standard selection criteria (Bradác et al., 2007; Ui-Tei et al., 2007) and chemically synthesized by Ambion (Woodward, TX). The synthetic siRNAs with 3′ overhang of 2dTs are as follows: c-jun1, GCCGGUUUUCCAUACUCUCtt (175–195); c-jun2, CAAGAAGA-UGCGCC GCAACtt (682–702). In addition to these siRNAs, Silencer Negative Control siRNA having no significant homology to any known gene sequences from mouse, rat, or human was used as a control siRNA. All the designed siRNA sequences were verified by BLAST search, to avoid significant sequence similarity to other genes in the rat genome. Sense and antisense RNA were solved in annealing buffer (100 mm potassium acetate, 30 mm HEPES-KOH, pH 7.4, 2 mm magnesium acetate), incubated for 1 min at 90°C and then for 1 h at 37°C just before transfection. For siRNA transfection, cells were seeded in at a density of 8 × 105 cells/well and cultured for 24 h. Cells were washed with PBS and transfected with 1 ml of Opti-MEM I medium (Invitrogen) containing 50 nm siRNA using oligofectamine (Invitrogen) following the manufacturer's instructions. Total cell lysates were prepared from these cells for Western blot analysis.
Real-time PCR.
Astrocyte cultures were rinsed twice with PBS and treated with the RNA extraction reagent Trizol (Invitrogen). Total RNA was precipitated by centrifugation at 20,000 × g for 15 min at 4°C, washed twice with ethanol, and quantified by UV spectrophotometry at 260 nm. Only RNA with a 260/280 ratio >2.0 was used for the reverse transcription (RT) reaction. Total RNA was treated with 10 U of RNase-free DNase I for 15 min at 37°C to eliminate contaminating genomic DNA. The DNase was then digested with 15 mg of proteinase K at 70°C for 20 min, and the RNA was phenol-chloroform extracted and ethanol precipitated. The target mRNAs were quantified by real-time RT-PCR analysis using SYBR Green. The cDNAs were prepared from 4 μg of total RNA using the Thermoscript RT-PCR system with oligodT18 primers. The cDNAs were used as templates for PCR in 20 μl scale with Fast start SYBR Green kit (Roche, Indianapolis, IN) in the presence of 200 nm of the forward and reverse primers. The PCR was performed on the Light Cycler Instruments (Roche) at 50°C for 2 min and at 95°C for 10 min, followed by running for 45 cycles at 95°C for 15 s and 60°C for 1 min. The primer sets were forward CTGATCATCCAGTCCAGCAA and reverse TGGCTATGCAGTTCAGCTAGG primers for rat c-jun (GenBank accession number X17163, product size 137 bp) and forward TGCACCACCAACTGCTTA and reverse GGATGCAGG-GATGATGTTC primers for rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH; GenBank accession number NM-017008 and product size 177 bp). The primers were designed by using the Primer3 Input software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3.cgi/primer3_www.cgi). The specificity of each primer was verified using the BLAST software. Expression of mRNA was assessed by evaluating threshold cycle (CT) values. The CT values were normalized with the expression level of GAPDH, and the relative amount of mRNA specific to each of the target genes was calculated using the δ-CT method (ref Pfaff). The effect of pharmacological treatments on mRNA c-jun levels was expressed as fold increase over basal values.
Western blot analysis.
For Western blot analysis, cells were starved for 18 h before stimulation with ET-1 and various pharmacological agents. Then, cells were rinsed twice with ice-cold PBS and lysed for 45 min on ice in 500 μl of ice-cold RIPA lysis buffer (50 mm Tris-HCl, pH 7.5, 140 mm NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mm sodium-orthovanadate, 1 mm NaF, 1 mm PMSF, 2 μg/ml aprotinin, 2 μg/ml pepstatin, 2 μg/ml leupeptin, and 1 μm microcystin-LR). The lysates were centrifuged at 40,000 rpm for 10 min at 4°C, and aliquots were taken for protein determination using the Pierce (Rockford, IL) BCA protein assay kit. Western blot analysis was performed either on 60 μg of total cell lysate (for P-p38MAPK, p38MAPK, P-JNK, JNK, P-ERK5, and ERK5) or 100 μg of nuclear protein extracts (for P-c-Jun, c-Jun, P-ATF-2, and ATF-2). Proteins were resolved by 10% SDS-PAGE and transferred to nitrocellulose or Immobilon PVDF membranes by tank blotting (0.8 A constant current) in transfer buffer (25 mm Tris, 192 mm glycine, and 20% v/v methanol, pH 8.3) for 16 h at 4°C. The membranes were rinsed twice in Tween-Tris-buffered saline (TBS-T) (25 mm Tris-HCl, pH 7.5, 140 mm NaCl, and 0.05% Tween 20), incubated for 1 h in TBS-T containing 5% BSA, and then incubated for 16 h at 4°C with primary antibodies in TBS-BSA. After washing three times for 15 min each with TBS-T, the membranes were incubated in TBS plus 5% milk for 1 h at room temperature with either horseradish peroxidase-conjugated goat polyclonal anti-rabbit IgG for polyclonal primary antibodies or horseradish peroxidase-conjugated goat polyclonal anti-mouse for mouse monoclonal primary antibodies. The chemiluminescent signals were detected using the Super signal kit (Pierce) or ECL Advance kit (for P-proteins; GE Healthcare, Piscataway, NJ). X-ray films were then scanned using an Agfa T1200 scanner with Photolook software for quantification.
Lysolecithin injection, demyelination of the corpus callosum, and ET-R antagonist infusion.
Demyelination of the CC was induced in deeply anesthetized 6- to 8-week-old mice by stereotaxic injection of 2 μl of a 1% LPC solution (Sigma) in 0.9% NaCl (Hall, 1972; Nait-Oumesmar et al., 1999). Anesthesia was induced with ketamine (100 mg/kg body weight; Imalgene; Rhone-Merieux, Lyon, France) and xylazine (10 mg/kg body weight; Rompun; Centravet, Plancoet, France) dissolved in 0.9% sterile saline. Lysolecithin was injected into the CC with a 5 μl Hamilton syringe. Stereotaxic coordinates were 5.5 mm anterior to the lambda, 1 mm lateral to the bregma, and 2.5 mm deep from the skull surface. Sham controls were injected with 2 μl of saline only. The injection site was labeled with charcoal dust. Scalp incisions were closed with Vetbond thread. Brain tissue of injected mice was then analyzed by immunohistochemistry at 2, 5, 7, and 11 d after LPC injection (Aguirre et al., 2007).
For Bosentan infusion, the antagonist was dissolved in sterile saline (0.9% NaCl) and placed in a mini-osmotic pump connected to a 30-gauge stainless-steel cannula through polyethylene catheter tubing (Alzet 1002 and brain infusion kit; Durect, Cupertino, CA). The prefilled pumps were placed in 0.9% saline overnight at 37°C to assure the pumping steady state was reached before implantation. Because the flow rate of the pumps was 0.25 ± 0.02 μl/h, as determined by the supplier, the concentration of Bosentan was fixed at 15 μm, to give an application dose of 90 pmol/d, and the concentration of BQ788 was fixed at 50 μm, to give an application dose of 300 pmol/d. The osmotic pump was inserted into a subcutaneous pocket below the neck, leading the catheter to the site for cannula placement. After the skull was exposed, and lysolecithin was injected into the CC with a 5 μl Hamilton syringe as described above, the brain infusion cannula was inserted through the same perforation and attached to the skull using cyanoacrylate adhesive for drug delivery at the site of the lesion (stereotaxic coordinates 5.5 mm anterior to the lambda, 1 mm lateral to the bregma, and 2 mm deep from the skull surface). The length of the connecting catheter tubing was calculated to hold 12 μl, and it was filled up with 0.9% NaCl so that only saline could be infused for the first 2 d after lysolecithin injection, and drug delivery would not interfere with the formation of the lesion. After this period, drug infusion would start and continue for other 5 d. Control animals were infused with 0.9% saline in the same manner. Mice were killed 7 d after lysolecithin injection. Brain tissue was processed for immunohistochemistry as previously described (Aguirre et al., 2007).
To determine ET-1 and GFAP expression levels within the demyelinating lesion, stereotaxic injections of LPC in the CC of the C57BL/6 mice were performed as described above. The procedure for tissue dissection and Western blot analysis was as previously described (Aguirre et al., 2007). Saline and Vestagreen dye (Sigma) were injected ipsilaterally as controls. Five days and 12 d after injection, CC tissue was microdissected from 300-μm-thick coronal sections. The area of LPC-induced lesion was identified and dissected out based on disruption of white matter cytoarchitecture. Corresponding saline-injected CC tissue was dissected based on Vestagreen staining. Proteins were extracted and separated by Western blot as described above. ET-1 protein levels were expressed as arbitrary units after normalization with actin using control levels as reference.
Immunohistochemistry in tissue sections.
Anti-MBP and anti-GFAP immunostaining was performed as previously described (Aguirre et al., 2007). Proliferation of GFAP+ astrocytes after demyelination was assayed by anti-Ki-67 immunostaining, as previously described (Aguirre and Gallo, 2004; Aguirre et al., 2007). Anti-phosphorylated JNK staining was performed as described by Cole-Edwards et al. (2006).
Results
ET-R activation regulates reactive astrogliosis after corpus callosum demyelination
It has been previously proposed that ET-1 might be one of the cellular signals involved in promoting reactive gliosis through activation of ET-Rs in astrocytes (Uesugi et al., 1996; Rogers et al., 2003). Therefore, we used the LPC focal demyelination model to determine whether ET-1 and ET-Rs regulate astrocytic proliferation and reactive gliosis after white matter lesion (Woodruff and Franklin, 1999). We have previously demonstrated that in white matter regions of adult CNP-EGFP mice, EGFP expression is exclusively limited to oligodendrocytes (Yuan et al., 2002; Aguirre et al., 2007); therefore, these mice represent an ideal animal model to investigate the cellular effects of focal demyelination in corpus callosum (see also Aguirre et al., 2007).
Stereotaxic injection of LPC into the rostral CC of CNP-EGFP mice induced focal demyelination within 3–5 d (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material) (Aguirre et al., 2007). Five days after lysolecithin injection [5 d postlesion (dpl)], immunohistochemical analysis revealed a focal depletion of MBP expression and of CC1+EGFP+ oligodendrocytes in the area immediately surrounding the injection site (supplemental Fig. 1A–D, available at www.jneurosci.org as supplemental material). At 2 dpl, no increase in GFAP+ cells was detected in the lesion, compared with the contralateral side (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). By 5 dpl, an increase in the number of GFAP+ astrocytes was observed in the lesion area, compared with sham saline-injected brains (Fig. 1A,B; supplemental Fig. 2C,D, available at www.jneurosci.org as supplemental material) (Woodruff and Franklin, 1999). These astrocytes displayed higher levels of GFAP protein expression (Fig. 1A,B) and enhanced cell proliferation, as demonstrated by anti-Ki67 immunostaining (Fig. 2D,E,K). Western blot analysis of GFAP protein expression in the lesion confirmed the immunocytochemical results. A peak of GFAP expression could be detected at 5 dpl, whereas no significant GFAP upregulation was observed at 2 dpl (Fig. 1C, supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 1.
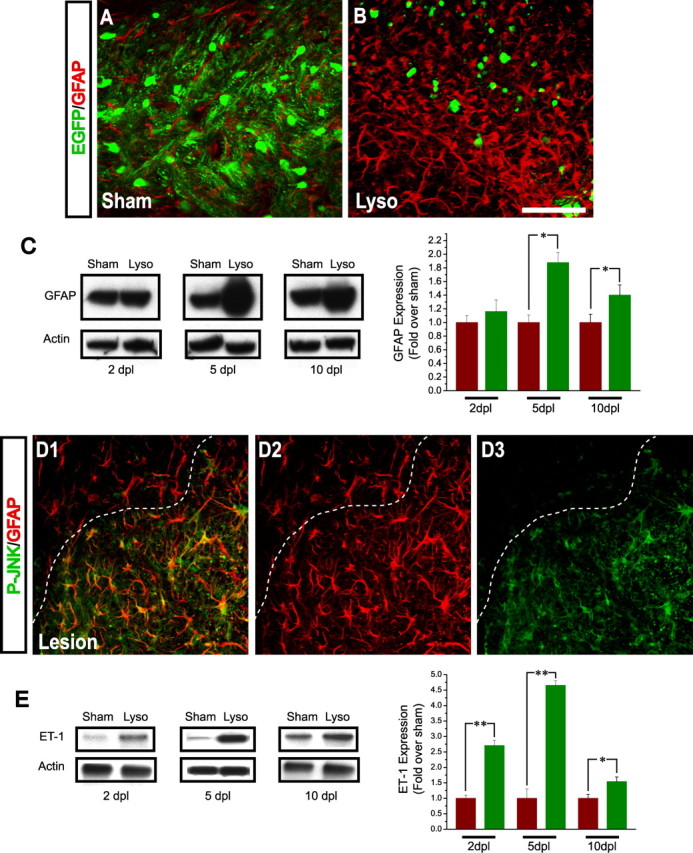
Focal demyelination of the mouse corpus callosum causes ET-1 upregulation in the demyelinated lesion, reactive gliosis, and JNK phosphorylation in reactive astrocytes. CNP-EGFP mouse brains were injected with lysolecithin and immunostained with anti-GFAP at 5 dpl. A, B, Anti-GFAP immunostaining shows significant upregulation of GFAP expression and increase in astrocyte number in the lesion area (B), compared with the contralateral side (A). C, Western blot analysis of GFAP expression in the lesion shows significant upregulation at 5 and 10 dpl but not at 2 dpl. Averages ± SEM are shown (n = 3 independent experiments). *p < 0.05 (Student's t test). D1–D3, Anti-GFAP and anti-P-JNK immunostaining reveals the presence of GFAP+P-JNK+ astrocytes limited to the area of reactive gliosis in the lesion. Scale bar: (in B) A, B, D1–D3, 50 μm. E, Western blot analysis of ET-1 expression in the lesion at 2–10 dpl. Band intensity measurements showed a peak 4.5-fold upregulation of ET-1 levels in the lysolecithin-injected side, compared with sham at 5 dpl. Averages ± SEM are shown (n = 3 independent experiments). *p < 0.05; **p < 0.01 (Student's t test).
Figure 2.
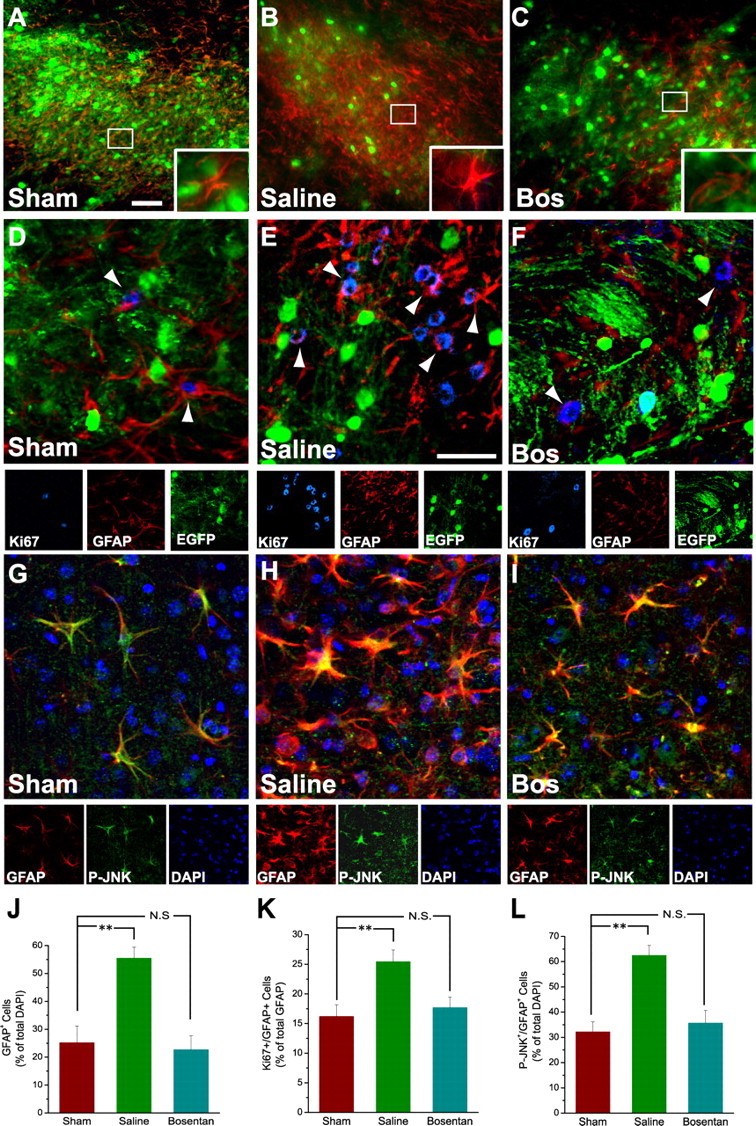
In vivo infusion of a pan-ET-R antagonist reduces JNK phosphorylation and reactive gliosis after corpus callosum demyelination. CNP-EGFP mouse brains were injected with lysolecithin, infused with saline or Bosentan starting at 2 dpl for 5 d, and immunostained with anti-GFAP, anti-Ki67, and anti-P-JNK at 7 dpl. A–C, Focal demyelination after lysolecithin injection enhances GFAP+ astrocyte number and GFAP expression in the demyelinated lesion area infused with saline (B), compared with contralateral sham (A). Infusion with the pan-ET-R antagonist Bosentan (C; 15 μm) for 5 d reduces GFAP+ astrocyte cell number and GFAP upregulation in the demyelinated lesion area, compared with saline infusion (B). Insets, High magnification of typical GFAP+ astrocytes under the different conditions. Scale bar, 50 μm. D–F, Anti-Ki67 immunostaining demonstrates that Bosentan infusion (F) also reduces GFAP+ cell proliferation, compared with saline infusion (E). G–I, Anti-P-JNK immunostaining demonstrates that focal demyelination after lysolecithin injection enhances P-JNK+GFAP+ astrocyte number in the demyelinated lesion area infused with saline (H), compared with contralateral sham (G). Bosentan infusion (I) reduces the number of P-JNK+GFAP+ cells, compared with saline infusion (H). Scale bar: (in E) D–I, 50 μm. J–L, Inhibitory effects of Bosentan on the increase in total GFAP+ cells (J), GFAP+ cell proliferation (K), and total P-JNK+GFAP+ cells (L). Averages ± SEM are shown. Total number of cells counted for each condition ranged between 1280 and 1605 (n = 5 independent experiments). **p < 0.01 (Student's t test). N.S., Not significant.
It has been demonstrated that JNK activation causes GFAP accumulation in astrocytes (Tang et al., 2006) and that JNK phosphorylation is induced in reactive astrocytes after partial sciatic nerve ligation or after kindling (Ma and Quirion, 2002; Cole-Edwards et al., 2006). Therefore, we examined whether increased JNK phosphorylation was detectable in reactive astrocytes in demyelinated lesions. In corpus callosum at 5 dpl, high levels of anti-P-JNK immunostaining were revealed in the majority of GFAP+ reactive astrocytes (Fig. 1D1–D3). Importantly, P-JNK expression was clearly limited only to the lesion area corresponding to the region of higher GFAP+ cell density (Fig. 1D1–D3). JNK phosphorylation was only at background levels in regions of the brain outside of the lesion and was not associated with GFAP+ astrocytes (Fig. 1D1–D3; supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
First, we examined ET-1 expression in the lesion and found that levels of this peptide were strongly upregulated, compared with sham (Fig. 1E). At 2 dpl, a significant increase in ET-1 expression was detected in the lesion, compared with the contralateral, saline-injected side. This upregulation of ET-1 expression was maximal at 5 dpl and decreased to approximately sham levels at 10 dpl (Fig. 1E). At this time, significant remyelination had occurred, whereas reactive gliosis had considerably decreased (Aguirre et al., 2007; A. Gadea and V. Gallo, unpublished data).
Second, we blocked ET-R activation in the lesion with the pan-ET-R antagonist Bosentan, which prevents activation of both ETA-Rs and ETB-Rs (Clozel et al., 1994). Beginning at 2 dpl to avoid interference with the demyelination process, brains were infused for 5 d with a cannula positioned in the CC itself. Bosentan prevented reactive gliosis and significantly decreased the total number of GFAP+ astrocytes around the lesion area (Fig. 2A–C,J). This effect was caused by a significant inhibition of cell proliferation, as demonstrated by a strong decrease in the percentage of GFAP+Ki67+ astrocytes in lysolecithin-injected brains infused with Bosentan, compared with saline-infused brains (Fig. 2D–F,K). The ET-R antagonist also prevented astrocyte hypertrophy, as demonstrated by attenuated GFAP expression (Fig. 2B,C,E,F). Bosentan did not affect GFAP+ astrocyte viability, as determined by caspase-3 immunostaining (<1% of caspase-3+GFAP+ astrocytes were detected in either saline- or Bosentan-infused lesions). Bosentan also partially restored oligodendrocyte numbers in the lesion by ∼40% (Fig. 2C,F), either by a direct effect of the antagonist on ET-R-expressing oligodendrocyte progenitor migration (Gadea and Gallo, 2005) or through indirect effects as a result of reduced astrogliosis (Back et al., 2005).
Finally, Bosentan infusion also reduced the number of P-JNK+GFAP+ astrocytes in the lesion. In sham saline-injected corpus callosum at 7 dpl, only a few GFAP+ astrocytes were immunostained by the anti-P-JNK antibodies, most likely as a consequence of the injection itself (Fig. 2G). Conversely, at the same time point, high levels of anti-P-JNK immunostaining were revealed in all GFAP+ reactive astrocytes in lysolecithin-injected saline-infused brains (Fig. 2H). The percentage of GFAP+P-JNK+ cells in the lesion of saline-infused brains was twofold higher than in sham (Fig. 2L). Finally, consistent with the results obtained on Ki67 immunostaining, Bosentan infusion reduced the number of GFAP+P-JNK+ astrocytes at 7 dpl (Fig. 2I,L).
Based on our previous study (Schinelli et al., 2001) and on the results obtained in cultured astrocytes in the present study (see Fig. 4), we also investigated the effects of the selective ETB-R antagonist BQ788 on astrogliosis in vivo. Consistent with the data obtained with Bosentan and with the analysis in cultured cells (see Fig. 4), infusion of BQ788 reduced the number of GFAP+ astrocytes in the lesion and their proliferation (Fig. 3A,B). Furthermore, the ETB-R antagonist also decreased the percentage of GFAP+P-JNK+ cells in the lesion (Fig. 3C). Similar to Bosentan, also BQ7888 did not affect GFAP+ astrocyte viability, as determined by caspase-3 immunostaining (<1% of caspase-3+GFAP+ astrocytes were detected in either saline- or BQ788-infused lesions).
Figure 4.

ET-1 induces a reactive astrocyte phenotype in culture. A–C, ET-1 promotes GFAP expression and cell proliferation in cultured astrocytes. Cortical astrocytes were incubated with saline (control; A), ET-1 (100 nm; B), or ET-1 (100 nm) plus Bosentan (2 μm; C) for 48 h. In C, cells were preincubated with Bosentan for 30 min, before exposure to ET-1. Cells were then stained with anti-GFAP, anti-Ki67, and DAPI. Note the increase in GFAP and Ki67 immunostaining in cells treated with ET-1 (B), compared with controls (A), and the decrease in GFAP and Ki67 expression in cells cultured with ET-1 plus Bosentan (C), compared with ET-1 alone (B). Small panels show immunostaining for individual antigens in the same microscopic field. Scale bar, 50 μm. D, E, The effects of ET-1 on astrocyte proliferation were quantified by counting the percentage of total DAPI+ cells that were Ki67+ (D) or the total number of cells (E). Incubation with ET-1 increased the percentage of Ki67+ cells and the total astrocyte cell number. The pan-antagonist Bosentan (2 μm) and the ETB-R antagonist BQ788 (2 μm) prevented the effects of ET-1, whereas the ETA-R antagonist BQ123 (2 μm) was ineffective. In both D and E, data are shown as averages ± SEM (n = 3 independent experiments). The total number of cells counted in D ranged between 1560 and 1800. D, E, *p < 0.01. F, Incubation of astrocytes with ET-1 (100–200 nm) for 48 h enhanced GFAP protein expression (top), and the effect of ET-1 was reduced by Bosentan (2 μm; bottom). Actin is also shown from the same blots as internal control for equal protein loading.
Figure 3.

In vivo infusion of an ETB-R antagonist reduces JNK phosphorylation and reactive gliosis after corpus callosum demyelination. CNP-EGFP mouse brains were injected with lysolecithin, infused with saline or the ETB-R antagonist BQ788 starting at 2 dpl for 5 d, and immunostained with anti-GFAP, anti-Ki67, and anti-P-JNK at 7 dpl. A, Focal demyelination after lysolecithin injection enhances GFAP+ astrocyte number and GFAP expression in the demyelinated lesion area infused with saline, compared with contralateral sham. Infusion with the ETB-R antagonist BQ788 (50 μm) for 5 d reduces GFAP+ astrocyte cell number and GFAP upregulation in the demyelinated lesion area, compared with saline infusion. B, Anti-Ki67 immunostaining demonstrates that BQ788 infusion also reduces GFAP+ cell proliferation, compared with saline infusion. C, Anti-P-JNK immunostaining demonstrates that focal demyelination after lysolecithin injection enhances P-JNK+GFAP+ astrocyte number in the demyelinated lesion area infused with saline, compared with contralateral sham. BQ788 infusion reduces the number of P-JNK+GFAP+ cells, compared with saline infusion. Averages ± SEM are shown. Total number of cells counted for each condition ranged between 820 and 1641 (n = 3 independent experiments). **p < 0.01 (Student's t test). N.S., Not significant.
In summary, these findings indicate (1) ET-1 levels are strongly upregulated in demyelinated lesions concurrently with reactive gliosis; (2) blockage of ET-Rs with either a pan- or an ETB-R-selective antagonist attenuates reactive gliosis after demyelination in vivo, by preventing astrocyte proliferation and JNK phosphorylation; and (3) blockage of ET-Rs does not affect astrocyte survival in the lesion. These results strongly suggest that ET-1 might be one of the endogenous cellular signals that promote reactive gliosis after demyelination in vivo.
ET-1 induces a reactive phenotype in cultured cortical astrocytes
The changes observed in astrocytes in vivo after Bosentan perfusion could be caused by effects of the antagonist on other cell types present in the lesion expressing ET-Rs, including endothelial cells or oligodendrocyte lineage cells (Gadea and Gallo, 2005). Therefore, we wanted to determine whether ET-1 directly activates ET-Rs in astrocytes to induce phenotypic changes in these cells similar to those observed during the process of reactive gliosis in vivo.
Pure primary astrocyte cultures were incubated with ET-1 (100 nm) for 48 h, and cell proliferation was assessed. Figure 4, A, B, and D, shows that ET-1 strongly promoted astrocyte proliferation, as determined by Ki67 immunostaining of GFAP+ cells. The effects of ET-1 on astrocyte proliferation were prevented by the pan-antagonist Bosentan and by the ETB-R antagonist BQ788, but not by the ETA-R antagonist BQ123 (Fig. 4C,D). ET-1 also increased astrocyte cell number, as determined by total DNA content per dish, and the ETB-R antagonist selectively blocked this effect (Fig. 4E).
It has been previously described that reactive astrocytes express higher GFAP levels than normal astrocytes (Ridet et al., 1997; McGraw et al., 2001; Nolte et al., 2001; Egnaczyk et al., 2003). Incubation of culture astrocytes with ET-1 (100–200 nm) for 48 h significantly enhanced GFAP protein levels (Fig. 4F). Bosentan treatment strongly reduced ET-1-induced GFAP induction (Fig. 4F).
In summary, these data indicate that ET-1 induces a reactive phenotype in cultured astrocytes by enhancing both cell proliferation and GFAP expression.
ET-1 induces JNKs and p38MAPK phosphorylation in cortical astrocytes
Previous studies demonstrated that, in cultured astrocytes, ET-1 mediates its cellular effects via the activation of MEK- and ERK-dependent pathways, which in turn induce the phosphorylation of downstream transcription factor targets (Lazarini et al., 1996; Cazaubon et al., 1997; Schinelli et al., 2001). To investigate the molecular mechanisms by which ET-1 exerts its effects in astrocytes, we first examined whether other MAPK-dependent signal transduction pathways, including JNK and p38MAPK, could be activated by ET-1 in our experimental model.
Time course analysis in confluent rat cortical astrocyte cultures revealed a small increase in ET-1-induced phosphorylation of the p54JNK isoform as early as 5 min after stimulation (Fig. 5A). The effect of ET-1 on p54JNK phosphorylation reached a plateau between 20 and 30 min (Fig. 5A). In contrast, the same stimulation paradigm did not significantly affect the phosphorylation state of the p46JNK isoform (Fig. 5A). The temporal pattern of ET-1-induced p38MAPK phosphorylation also showed a maximal effect at 20 min, but the kinetics of phosphorylation was more rapid, being detectable at 1 min (Fig. 5B). Analysis of the same protein extracts using specific antibodies that recognize JNK isoforms or p38MAPK independently of their phosphorylation state demonstrated that, under our experimental conditions, ET-1 did not modify the total levels of these two kinases (Fig. 5A,B).
Figure 5.
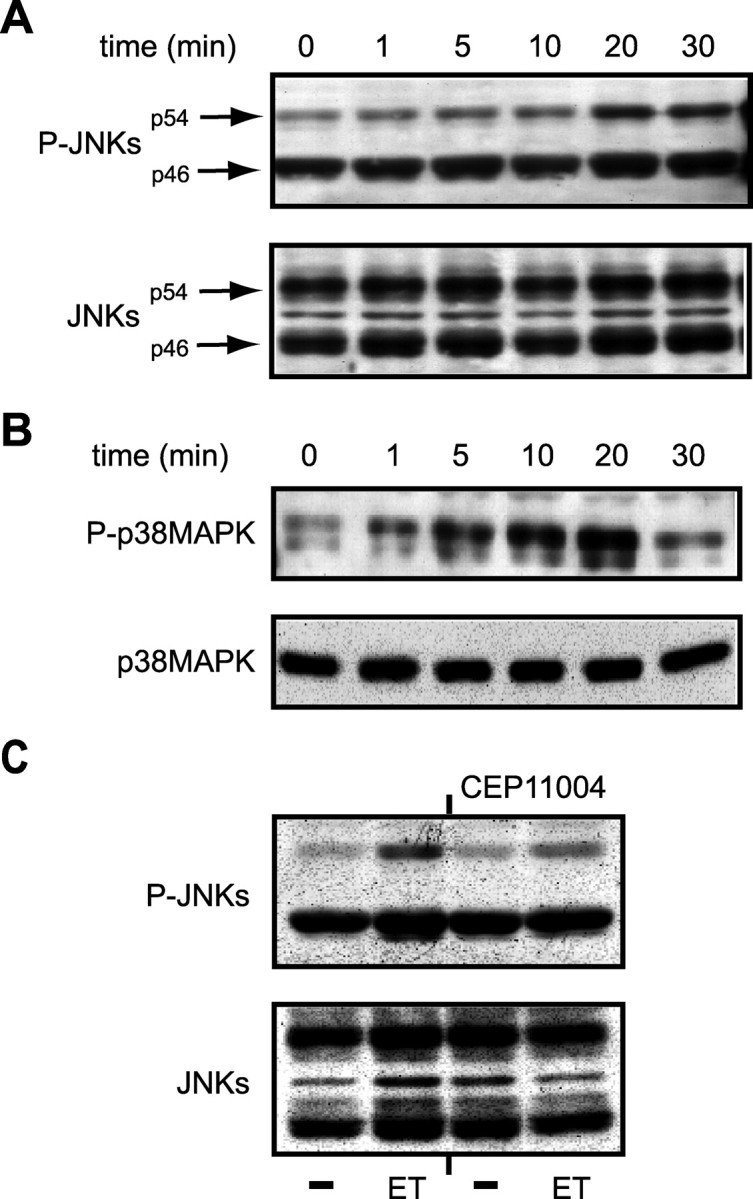
ET-1 induces JNKs and p38MAPK phosphorylation in rat cortical astrocytes. A, B, Time course of ET-1-induced JNK (A) and p38MAPK (B) phosphorylation. Cells were stimulated with 100 nm ET-1 for the indicated times, and total cell lysates were analyzed by Western blot (as described in Materials and Methods) using an anti-P-JNK antibody (A, top) or an anti-P-p38MAPK antibody (B, top). The same blots were then stripped and incubated with phosphorylation-state independent anti-JNK or anti-p38MAPK antibodies to normalize for the total (phosphorylated plus nonphosphorylated) amounts of JNKs (A, bottom) and p38MAPK (B, bottom). C, ET-1 induces JNK phosphorylation via the upstream kinase MLK. Cells were preincubated for 30 min with the MLK inhibitor CEP-11004 (5 mm) and then treated with 100 nm ET-1 for 20 min. Total cell lysates were then analyzed for P-JNK phosphorylation (C, top) or total JNK (C, bottom) as described above. Data are representative of three independent experiments.
Finally, pretreatment of cells for 30 min with CEP-11004 (5 mm), an inhibitor of the mixed lineage kinase (MLK) family acting upstream of JNKs, blocked ET-1-induced p54JNK phosphorylation, without affecting the total levels of JNKs (Fig. 5C). Altogether, these data indicate that ET-1 activates both the JNK and the p38MAPK pathway in astrocytes.
ET-1 induces c-jun phosphorylation through a JNK-dependent pathway and ATF-2 phosphorylation through JNK- and p38MAPK-dependent pathways
In a variety of cell types, activation of the JNK- and p38MAPK-dependent pathways results in the phosphorylation of downstream targets, including the immediate early gene c-jun and the transcription factor ATF-2 (Pearson et al., 2001). Using anti-phospho antibody directed against the Ser-63 residue of c-Jun, we found that ET-1 increased phosphorylation of the immediate early gene within 5 min of exposure, with a maximal effect at 20 min (Fig. 6A, Table 1). This time course of c-Jun phosphorylation correlated with P-JNK levels (Fig. 5A). The overall expression levels of c-Jun were not significantly modified by ET-1 treatment within a 30 min time frame (Fig. 6A). ET-1-induced c-Jun phosphorylation was prevented by the ETB-R antagonist BQ788, but not by the ETA-R antagonist BQ123 (Fig. 6B).
Figure 6.
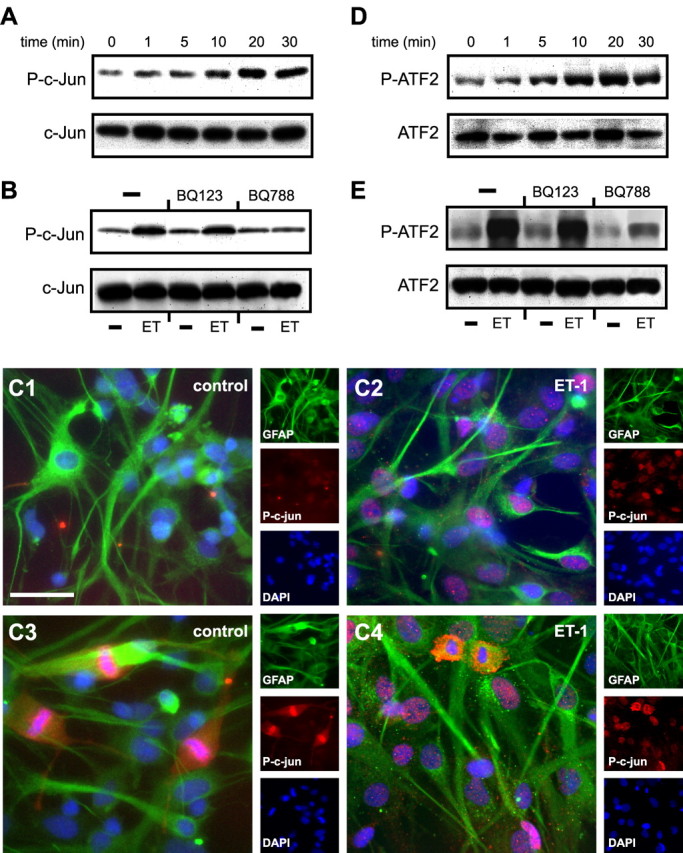
ET-1 induces c-jun and ATF2 phosphorylation in astrocytes. A, D, Time course of ET-1-induced c-Jun (A) and ATF2 (D) phosphorylation. Cells were stimulated with 100 nm ET-1 for the indicated times, and total cell lysates were analyzed by Western blot using an anti-P-c-Jun antibody or an anti-P-ATF2 antibody. The same blots were then stripped and incubated with phosphorylation-state independent anti-c-Jun or anti-ATF2 antibodies to normalize for the total (phosphorylated plus nonphosphorylated) amounts of c-Jun (A, bottom) and ATF2 (D, bottom). C, P-c-Jun immunostaining of cultured astrocytes incubated with saline (C1, C3) or ET-1 (200 nm; C2, C4) for 20 min. In quiescent, unstimulated astrocytes, P-c-Jun was only detected in GFAP+ cells in metaphase (C3), whereas the majority of ET-1-treated astrocytes expressed high levels of nuclear P-c-Jun (C2, C4). Small panels show immunostaining for individual antigens in the same microscopic field. Scale bar, 50 μm. B, E, ET-1-induced c-Jun and ATF2 phosphorylation is selectively blocked by the ETB-R antagonist BQ788, but is not modified by the ETA-R antagonist BQ123. The same blots were then stripped and incubated with phosphorylation-state independent anti-c-jun or anti-ATF2 antibodies to normalize for the total (phosphorylated plus nonphosphorylated) amounts of c-jun (B, bottom) and ATF2 (E, bottom). Data are representative of three independent experiments.
Table 1.
Time course of ET-1-induced c-Jun phosphorylation
| Time (min) | Fold increase over control (time 0) |
|---|---|
| 1 | 1.25 ± 0.14 |
| 5 | 1.53 ± 0.17 |
| 10 | 1.78 ± 0.13 |
| 20 | 2.94 ± 0.24** |
| 30 | 1.99 ± 0.19* |
Cultured astrocytes were serum deprived for 18 h and incubated with 100 nm ET-1 for different times (1–30 min). Cells were then harvested and processed for P-c-jun Western blot. Data were obtained from x-ray P-c-jun densitometry and are expressed as fold increase in ET-1-induced c-Jun phosphorylation over unstimulated cells (time 0). Data are averages ± SEM of three independent experiments performed on separate astrocyte cultures. *p < 0.05; **p < 0.001 (Student's t test).
ET-1-induced c-Jun phosphorylation was also revealed by immunostaining of cultured astrocytes with antibodies for P-c-Jun (Fig. 6C). In quiescent, unstimulated astrocytes, P-c-Jun was only detected in GFAP+ cells in metaphase (Fig. 6C1,C3). Conversely, the majority of ET-1-treated astrocytes expressed high levels of nuclear P-c-Jun (Fig. 6C2,C4).
In a second set of experiments, we used an anti-phospho antibody directed against the Ser-73 residue of ATF-2 and demonstrated that ET-1 also increased ATF-2 phosphorylation in astrocytes with a time course very similar to that of c-Jun (Fig. 6D). ET-1 did not modify the total levels of ATF-2 protein (Fig. 6D). Also in this case, the ETB-R antagonist BQ788 blocked ET-1-induced ATF2 phosphorylation, but the ETA-R antagonist BQ123 was ineffective (Fig. 6E).
We then investigated whether different pharmacological inhibitors of the JNK pathway modified the effects of ET-1 on c-Jun and ATF2 phosphorylation. ET-1-induced c-Jun phosphorylation was abolished by preincubating the cells for 30 min with either the MLK inhibitor CEP-11004 (5 mm) or the specific JNK kinase inhibitor SP600125 (10 mm) (Fig. 7A). In contrast, the two specific p38MAPK inhibitors SB203580 (10 mm) and SB202190 (10 mm) had no appreciable effects on ET-1-induced c-Jun phosphorylation (Fig. 7B). Reprobing the membranes with phosphorylation-state independent antibodies showed that ET-1 did not modify the total levels of c-Jun protein (Fig. 7A,B).
Figure 7.

ET-1 induces c-Jun and ATF2 phosphorylation in astrocytes via JNK- and p38MAPK-dependent pathways. A, C, The JNK-dependent pathway mediates ET-1-induced c-Jun (A) and ATF2 (C) phosphorylation. In both sets of experiments, cells were preincubated for 30 min with the MLK inhibitor CEP-11004 (5 mm) or the JNK inhibitor SP600125 (10 mm) and then treated with 100 nm ET-1 for 20 min. Total cell lysates were then analyzed by Western blot using an anti-P-c-jun antibody (A, top) or an anti-P-ATF2 antibody (C, top). B, D, The p38MAPK-dependent pathway mediates ET-1-induced ATF2 (D) but not c-jun (B) phosphorylation. Cells were preincubated for 30 min with the p38MAPK inhibitors SB203580 (10 mm) or SB202190 (10 mm) and then treated with 100 nm ET-1 for 20 min. Total cell lysates were then analyzed by Western blot using an anti-P-ATF2 antibody (D, top) or an anti-P-c-jun antibody (B, top). The blots were then stripped and incubated with phosphorylation-state independent anti-c-jun or anti-ATF2 antibodies to normalize for the total amounts of c-jun (A, B, bottom) and ATF2 (C, D, bottom). Data are representative of three independent experiments.
Inhibition of the JNK-dependent pathway by preincubation of the cells with either CEP-11004 or with SP600125 also decreased ET-1-induced ATF2 phosphorylation (Fig. 7C). Furthermore, the p38MAPK inhibitors SB202580 and SB202190 attenuated ET-1-induced ATF2 phosphorylation to a similar extent without modifying the total levels of ATF2 protein (Fig. 7D).
Together, these data demonstrate that c-Jun phosphorylation is specifically detected in GFAP+ astrocytes induced to proliferate by ET-1. Our results also indicate that ET-1 causes c-Jun phosphorylation in cultured astrocytes primarily through JNK kinases, and that the concomitant activation of JNK- and p38MAPK-dependent pathways causes phosphorylation of the downstream transcription factor ATF2.
ET-1-induces c-jun transcription through activation of ERK- and JNK-dependent pathways
We first assessed the effect of ET-1 on c-jun transcription in astrocytes by measuring c-jun mRNA levels by quantitative real-time RT-PCR. A threefold increase in c-jun mRNA expression was detected within 30 min of exposure to ET-1, and reached a maximum 4.5-fold increase at 60 min (Fig. 8A).
Figure 8.

ET-1 increases c-jun expression via multiple signaling transduction pathways. A, Time course of ET-1-induced c-jun mRNA expression. Cells were stimulated with 100 nm ET-1 for the indicated times, and total RNA was extracted and processed for quantitative real-time RT-PCR analysis. Data are averages ± SEM (n = 3–4 independent experiments). B, ET-1 increases c-jun mRNA expression through activation of ERK/JNK-dependent pathways. Cells were preincubated for 30 min with one of the following inhibitors: p38MAPK inhibitor SB202190 (10 mm), the MEK inhibitor U0126 (10 mm), the JNK inhibitor SP600125 (10 mm), or a combination of U0126 and SP600125. For c-jun mRNA quantification, cells were treated for 60 min with ET-1, and total RNA was processed as described above. Data are averages ± SEM (n = 3–4 independent experiments). *p < 0.001 versus ET-1. **p < 0.05 versus U0126 and SP600125 alone; **p < 0.001 versus ET-1 (Student's t test). C, Time course of ET-1-induced c-jun protein expression. Cells were stimulated with 100 nm ET-1 for the indicated times, and total cell lysates were analyzed by Western blot using an anti-c-jun antibody (C, top) or an anti-tubulin antibody (C, bottom) for normalization of total amount of loaded proteins. D, ET-1 increases c-jun protein expression through activation of ERK/JNK-dependent pathways. Cells were treated with 100 nm ET-1 for 2 h in the presence or absence of inhibitors (see B), and total cell lysates were analyzed by Western blot using an anti-c-jun antibody (D, top) or an anti-tubulin antibody (D, bottom). Note the synergistic effects of SP600125 and U0126 on both ET-1-induced c-jun mRNA and protein expression.
The CRE/AP-1-like transcription factor binding sites present in the c-jun promoter may also bind the transcription factor CREB as homodimer, or as heterodimer in combination with ATF-1. In a previous study, we demonstrated that ET-1 induces CREB and ATF-1 phosphorylation in astrocytes primarily via an ERK-dependent pathway (Schinelli et al., 2001). Consistent with our previous findings and with the results reported in the present study, we observed that ET-1-induced c-jun mRNA expression in astrocytes was not affected by the p38MAPK inhibitor SB202190, but was significantly reduced by the MEK inhibitor U0126 or by the JNK inhibitor SP600125 (Fig. 8B). Combination of the two inhibitors further reduced the effects of ET-1 on c-jun mRNA levels (Fig. 8B).
To rule out a possible effect of ET-1-induced signaling cascades on c-jun translation (Clerk et al., 2002), we also determined the effect of ET-1 on the total amount of c-Jun protein by Western blot. The time course pattern of c-Jun protein expression correlates with that of c-jun mRNA expression except for the typical time shift between transcriptional and translational mechanisms. The expression of c-Jun protein slightly increased within 60 min, reached a peak at ∼2 h, and then declined to almost basal levels within 4 h (Fig. 8C). These findings indicate that in our cell system the main regulatory mechanism of the observed increased c-Jun protein expression induced by ET-1 occurs at transcriptional level.
Similar to c-jun mRNA, both the MEK inhibitor U0126 and the JNK inhibitor SP600125 partially reduced ET-1-induced c-Jun protein upregulation (Fig. 8D), and the combination of the two inhibitors completely prevented c-Jun increase triggered by the peptide (Fig. 8D).
Together, these observations show that the ERK- and the JNK-dependent pathways are the two main signaling cascades involved in the regulation of c-jun transcription by ET-1. Activation of ERK and JNK pathways involves phosphorylation of the downstream transcription factors CREB and ATF-1 (Schinelli et al., 2001), and c-Jun and ATF-2, respectively.
ET-1 induces astrocyte proliferation and GFAP expression through activation of ERK- and JNK-dependent pathways
Phosphorylation of the transcription factors c-Fos and c-Jun increases their heterodimer complex affinity for DNA AP-1 binding site, and triggers gene transcription. Previous reports demonstrated that ET-1 promotes astrocyte proliferation through regulated transcription of a variety of genes, including cyclin kinases, which contain AP-1 binding sites in their promoter regions (Shaulian and Karin, 2001). We have previously shown that ET-1 causes an increase in c-Fos expression in cultured astrocytes through ERK activation, and here we demonstrate that ET-1 induces an increase in c-Jun expression through JNK. Therefore, we wanted to determine whether the kinase pathway inhibitors that prevent ET-1-induced c-Fos and c-Jun expression in astrocytes also affect ET-1-induced cell proliferation.
ET-1 treatment of astrocytes increased cell proliferation by ∼40%, compared with untreated cultures (Fig. 9A). The inhibitors SP600125 (10 mm) and U0126 (10 mm) by themselves did not modify the effects of ET-1 on cell proliferation (Fig. 9A), whereas treatment with the combination of U0126 (10 mm) and SP600125 (10 mm) completely inhibited the effects of ET-1 on cell proliferation (Fig. 9A). Similarly, U0126 and SP600125 by themselves did not modify the effects of ET-1 on GFAP expression in astrocytes, but completely prevented ET-1-induced GFAP expression when used in combination (Fig. 9B).
Figure 9.

Gain and loss of function demonstrate that ET-1 induces astrocyte proliferation via c-jun. A, ET-1 increases cell proliferation through ERK/JNK-dependent pathways. Cells were treated for 24 h with 100 nm ET-1 in the presence or in the absence of the following inhibitors: the p38MAPK inhibitor SB202190 (10 mm), the MEK inhibitor U0126 (10 mm), the JNK inhibitor SP600125 (10 mm), or a combination of U0126 and SP600125. Cell proliferation was then assessed by a colorimetric assay, as described in Materials and Methods. Data are expressed as a percentage of BrdU incorporation in cells cultured without ET-1 (100%). Data are averages ± SEM (n = 3 independent experiments). *p < 0.01 (Student's t test). B, ET-1-induced GFAP expression is prevented by the combination of SP600125 and U0126 but is not modified by the inhibitors by themselves. All data are representative of three independent experiments. C, Overexpression of c-jun protein increases astrocyte proliferation and enhances ET-1-induced cell proliferation. Conversely, expression of dominant-negative c-jun dn (c-jun-IRES) prevents the effects of ET-1. Cells were transfected with the indicated c-jun-IRES bicistronic vectors encoding GFP and then treated for 24 h with 100 nm ET-1. Data are expressed as a percentage of GFP+ cells that were also BrdU+. Results are means ± SEM (n = 3 independent experiments), where >1000 GFP+ cells were counted in triplicate coverslips for each condition. *p < 0.01 compared with cells transfected with empty vector; **p < 0.01 compared with cells transfected with empty vector (Student's t test). D, Downregulation of c-jun protein after c-jun siRNA transfection. Cells were untreated (lane 1), transfected for 2 d with oligofectamine alone (lane 2), transfected with scrambled siRNA (lane 3), or transfected with a mix of two c-jun siRNAs (lane 4). Total cell lysates were analyzed by Western blot using an anti-c-jun antibody (D, top) or an anti-actin antibody for normalization (D, bottom). E, c-Jun knock-down prevents ET-1-induced astrocyte proliferation. In parallel experiments, c-jun siRNA-treated cells (D) were stimulated with ET-1 for 24 h, and total cell number was measured as in Figure 4. Data are averages ± SEM (n = 3 independent experiments). *p < 0.05 (Student's t test). F, The same conditions as in E were tested in a separate set of experiments, and cells were stained with anti-Ki-67 antibodies. A total of 600–800 cells per condition were counted. Data are expressed as averages ± SEM (n = 3 independent experiments). *p < 0.01 (Student's t test).
These findings are consistent with a combined and synergistic contribution of signaling cascades converging on c-Fos and c-Jun activation in the modulation of rat cortical astrocyte proliferation and GFAP expression elicited by ET-1.
Modulation of c-Jun expression by gain and loss of function modifies astrocyte proliferation
The finding that ET-1 promotes c-Jun expression and astrocyte proliferation through convergent signal transduction pathways prompted us to investigate by a gain- and loss-of-function approach whether direct modulation of c-Jun levels modified the effects of ET-1 on cell proliferation.
In a first set of experiments, we transfected astrocytes with an IRES bicistronic vector encoding GFP, together with wild-type c-jun (wt c-jun-IRES) or with the truncated c-jun form TAM67 (dn c-jun-IRES), which lacks the AP-1 binding site domain (Freemerman et al., 1996). The effects of overexpressing these two exogenous c-Jun protein isoforms were then assessed by BrdU incorporation (Fig. 9C). Expression of wt c-jun-IRES increased both basal and ET-1-induced astrocyte proliferation, compared with cells transfected with the empty vector (Fig. 9C). Conversely, overexpression of dn c-jun-IRES did not significantly modify basal astrocyte proliferation, but abrogated the effects of ET-1 (Fig. 9C).
In a second set of experiments, we used the complementary loss-of-function approach of small interference siRNA transfection to modify c-Jun expression levels. Astrocytes were transfected with a combination of two synthetic double-stranded siRNAs targeted to c-jun mRNA. In siRNA-treated cells, basal c-Jun levels were reduced by ∼95% (Fig. 9D), whereas in untreated astrocytes or in astrocytes transfected with scrambled siRNA, c-jun mRNA levels were not significantly modified (Fig. 9D). Consistent with these findings, total cell number estimates (Fig. 9E) and anti-Ki-67 immunostaining (Fig. 9F) showed that ET-1-induced astrocyte proliferation was prevented only in c-jun siRNA-treated cells, compared with untreated astrocytes, but not in cells transfected with scrambled siRNA. Astrocyte viability was not affected by c-jun overexpression or siRNA transfection, as assessed by caspase-3 immunostaining of the cultures. Under all conditions shown in Figure 9C–F, <1% of the cells were caspase-3+ (data not shown).
In conclusion, these pharmacological and gain- and loss-of-function studies demonstrate that ET-1-induced astrocyte proliferation functionally requires c-Jun.
Discussion
The endothelinergic system is involved in a wide variety of functions and pathological processes in the CNS (MacCumber et al., 1990; Rubanyi and Polokoff, 1994; Nie and Olsson, 1996; Schinelli, 2006), and astrocytes are a major target cell population of ET-1 in the brain (MacCumber et al., 1990; Cazaubon et al., 1997; Rogers et al., 1997, 2003; Baba, 1998; Schinelli et al., 2001; Hasselblatt et al., 2003; Koyama et al., 2003; Blomstrand et al., 2004; Blomstrand and Giaume, 2006; Schinelli, 2006). Our report defines the involvement of a specific intracellular signal transduction pathway, JNK/c-Jun, in the cellular effects of ET-1 on astrocytes, particularly on cell proliferation (Fig. 10). Therefore, our study is relevant to both astrocyte pathophysiology and to our understanding of the role of endothelins in the CNS.
Figure 10.
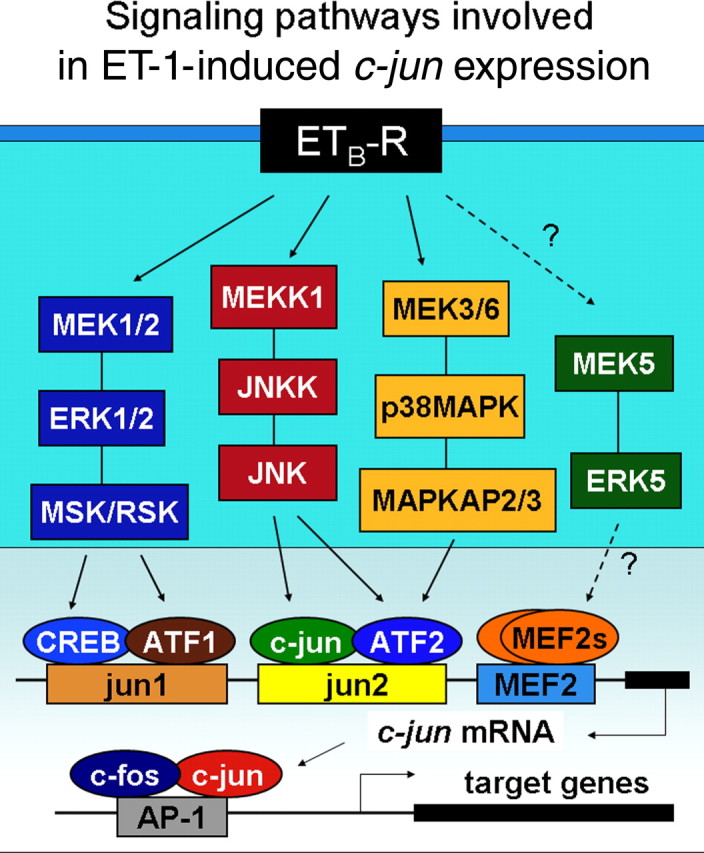
Signaling pathways involved in ET-1-induced c-jun expression. Shown is a representative scheme of the main signaling pathways involved in ET-1-induced/ETB-R-mediated phosphorylation of downstream transcription factors in the c-jun promoter. Increased expression of the immediate early genes c-fos and c-jun, which bind the AP-1 site in promoters of target genes, modulate astrocyte proliferation. Dashed lines and question marks indicate unknown links between elements of signaling pathways.
The detrimental effects of astrogliosis on brain cell repair are well established in different injury models, including white matter demyelination (Woodruff and Franklin, 1999; Back et al., 2005). A recent study demonstrated a functional link between JNK activation and GFAP accumulation in astrocytes (Tang et al., 2006). Furthermore, c-Jun activation and JNK phosphorylation have been demonstrated in reactive astrocytes in different animal models of pathology (Lee et al., 2004; Nakagawa and Schwartz, 2004; Hashimoto et al., 2005; Cole-Edwards et al., 2006). However, the extracellular signals that induce c-Jun expression in reactive astrocytes have not been identified, and a direct causal relationship between c-Jun activation and reactive gliosis has not been established. In the present study, we show that ET-1 induces both c-Jun activation and a reactive phenotype in astrocytes, and that ET-1-induced astrocyte proliferation requires functional activation of the c-Jun/JNK pathway.
We show that the ETB-R subtype mediates the effects of ET-1 on astrocyte proliferation. Both ETA- and ETB-Rs are expressed by astrocytes in vivo and in culture (MacCumber et al., 1990; Rubanyi and Polokoff, 1994; Lazarini et al., 1996; Cazaubon et al., 1997; Rogers et al., 1997, 2003; Sakurai-Yamashita et al., 1997; Baba, 1998; Koyama et al., 1999; Teixeira et al., 2000; Schinelli et al., 2001; Schinelli, 2006). Previous studies indicate that ET-1-induced astrocyte proliferation and hypertrophy are mainly mediated by ETB-Rs (Koyama et al., 1999; Rogers et al., 2003). ETB-R activation has also been linked to enhanced expression of both BDNF (brain-derived neurotrophic factor) (Koyama et al., 2005b) and neurotrophin-3 (Koyama et al., 2005a) in astrocytes. In the present study, we used selective ET-R antagonists to demonstrate that both ET-1-induced astrocyte proliferation and c-Jun phosphorylation are attributable to activation of ETB-Rs.
Consistent with a role of ET-1 in reactive gliosis after injury, we observed a significant increase in ET-1 levels in demyelinated lesions of the corpus callosum within the same time frame of astrogliosis. The cellular source of ET-1 in these lesions is not yet established, although endothelial cells and astrocytes are likely to play a major role (Tsang et al., 2001). ET-1 is expressed in reactive murine and human astrocytes (MacCumber et al., 1990; Jiang et al., 1993; Ma et al., 1994; Zhang et al., 1994; Tsang et al., 2001; Desai ate al., 2004), and preliminary analysis in our laboratory indicates that cultured astrocytes synthesize ET-1 and release this peptide in the extracellular medium (Gadea and Gallo, unpublished data). Moreover, we also observed increased ET-1 immunoreactivity in GFAP+ astrocytes in vivo within 1 week after lysolecithin injection in corpus callosum (Gadea and Gallo, unpublished data). Further analysis will establish the cell type(s) releasing ET-1 after demyelination and will determine whether this peptide causes physiological changes in other classes of cells (e.g., oligodendrocytes) in white matter lesions.
Our analysis in cultured astrocytes shows that both the ERK- and the JNK-dependent signal transduction pathways are critically involved in the effects of ET-1 on cell proliferation and GFAP expression. These pathways appear to be activated also in reactive astrocytes in vivo, as demonstrated by enhanced JNK phosphorylation, which is prevented by Bosentan infusion. Although we have not yet characterized the downstream genes that are controlled by the ERK- and JNK-dependent pathways and that might be directly involved in promoting astrocyte proliferation, it is likely that many of these genes are activated by transcription factors binding to AP-1 regulatory elements, including dimers of c-Fos, c-Jun, and ATF family members. Recent studies using cultured cells and genetically modified mice (Shaulian and Karin, 2001) demonstrated that AP-1 transcription factors, particularly c-Jun (Raivich and Behrens, 2006), might affect cell proliferation and survival in the developing and adult injured brain through their ability to modulate expression of cyclin D1 (Bakiri et al., 2000; Teixeira et al., 2000) and cyclin D3 (Koyama et al., 2004). Our previous report (Schinelli et al., 2001) and the present study demonstrate that ET-1 regulates both components of the AP-1 complex c-Fos and c-Jun in astrocytes. Therefore, it is likely that cell cycle regulators, such as cyclin D1 and D3, might be downstream targets of the ERK- and JNK-dependent pathways activated by ET-1 in astrocytes.
Activation of the JNK pathway demonstrated in the present study is at variance with our previous report describing activation of the ERK and p38 MAPK pathways, but not JNK (Schinelli et al., 2001). We believe that this is attributable to some experimental differences between the present study and the previous report. First, in the present study astrocytes were stimulated with higher concentrations of ET-1 (100 nm) and for longer periods of time (20 min) than in the previous report, in which cells were exposed to 50 nm ET-1 for 10 min. In agreement with our previous report (Schinelli et al., 2001), also in the present study JNK was not significantly activated after a 10 min stimulation with ET-1. Second, in the present study, we used subconfluent astrocyte cultures to correlate the effects of ET-1 on cell proliferation with activation of specific pathways. Conversely, in the previous article, we used confluent cultures (Schinelli et al., 2001). Finally, our previous study was exclusively performed on cultured astrocytes, and we did not examine the effects of ET-1 on cell proliferation and JNK activation in vivo. In the present study, we performed in vivo analysis, and show that the c-jun/JNK pathway is activated in reactive astrocytes in vivo. These differences suggest a context-dependent activation of JNK by ET-1.
Modulation of the expression of immediate early gene (IEG) family members, such as c-fos and c-jun, is mainly regulated by transcription factors acting on specific regulatory sites present in the IEG promoters. The rat c-jun promoter contains several regulatory elements (Han et al., 1992; Harrison et al., 2004); however, it is still unclear which of these sites are relevant for c-jun mRNA transcription. Two AP-1/CRE-like elements, often referred to as the distal jun1 site and the proximal jun2 site, appear to be crucial regulatory DNA sequences. These elements bind c-Jun/ATF2 or CREB/ATF1 transcription factor heterodimers (Harrison et al., 2004), and analysis in different cell types has demonstrated that the transactivating potential of c-Jun and ATF2 is increased after their phosphorylation by the JNKs and p38MAPK protein kinases (Pearson et al., 2001).
Our present study corroborates these findings by showing that stimulation of ETB-Rs in astrocytes induces first an increase in c-Jun phosphorylation and subsequently a rise in c-Jun expression. Moreover, by using pharmacological inhibitors, we distinguished the contribution of the JNK- and p38MAPK-dependent signaling pathways to the phosphorylation of downstream transcription factors that regulate c-jun expression and its phosphorylation. By using specific kinase inhibitors, we demonstrate that, after ET-1 treatment, the JNK-dependent pathway induces the phosphorylation of c-Jun and ATF2, whereas the p38MAPK-dependent pathway phosphorylates only ATF2. Our finding that ET-1-induced c-jun mRNA and protein expression are also partially attenuated by a MEK/ERK-dependent pathway inhibitor points to the involvement of other transcription factors and/or regulatory elements in the modulation of c-jun gene promoter. This hypothesis is consistent with findings in cardiac myocytes, which suggest that ET-1 participates in the regulation of c-jun expression via an ERK/RSK-dependent pathway, and by induction of a CREB/ATF1 dimer complex that binds the proximal c-jun site (Harrison et al., 2004).
The role of the MEK/ERK-dependent pathway and downstream transcription factors in the regulation of c-jun expression has been documented in several experimental models. In HeLa cells, ATF1 phosphorylation via the ERK-dependent pathway is required for EGF (epidermal growth factor)-induced c-jun expression (Gupta and Prywes, 2002). In keratinocytes (Wang et al., 2006) and neoplastic cells (Chen et al., 2000), the same stimulus increases c-jun and downstream gene expression via ERK. These data, together with our previous report showing that ET-1 induces CREB and ATF1 phosphorylation (Schinelli et al., 2001), support the hypothesis that both the ERK-dependent pathway, via the CREB/ATF1 complex, and the JNK/p38MAPK-dependent pathway, through the c-Jun/ATF2 complex, contribute together to enhance c-jun expression in astrocytes.
In conclusion, the main findings of this study point to specific molecular mechanisms that link ET-1, ETB-Rs, and c-Jun activation in astrocytes. We establish a functional link between c-Jun and ET-1-induced astrocyte proliferation and GFAP expression in reactive gliosis. The results reported here and our previous analysis on the effects of ET-1 on astrocytes (Schinelli et al., 2001) emphasize the participation of different signal transduction pathways and the pivotal role of their downstream IEGs, including c-fos and c-jun, in the regulation of delayed response genes responsible for astrocyte proliferation. In view of the ubiquitous response of astrocytes to different types of brain injury (Norenberg, 1994; Ridet et al., 1997; Aschner et al., 2002), our studies have the potential to facilitate the development of selective molecular tools for pharmacological intervention in brain pathologies in which an involvement of the ET system has been documented.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant R21NS048238 (V.G.), NIH Mental Retardation and Development Disabilities Research Center Grant P30HD40677 (V.G.), the Italian Ministero Università e Ricerca (MIUR)–Fondo Investimenti Ricerca di Base (S.S.), and the MIUR–Progetti di Rilevante Interesse Nazionale (S.S.). A.G. was supported by a fellowship from the National Multiple Sclerosis Society. We thank Li-Jin Chew for critically reading this manuscript and for discussion. We thank Dr. Michael J. Birrer (National Cancer Institute, NIH) for providing the pcDNA3.1 wt c-jun and TAM67 c-jun plasmids. We thank Actelion Pharmaceuticals (Allschwil, Switzerland) for the gift of Bosentan.
References
- Aguirre A, Gallo V. Postnatal neurogenesis and gliogenesis in the olfactory bulb from NG2-expressing progenitors of the subventricular zone. J Neurosci. 2004;24:10530–10541. doi: 10.1523/JNEUROSCI.3572-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- Aschner M, Sonnewald U, Tan KH. Astrocyte modulation of neurotoxic injury. Brain Pathol. 2002;12:475–481. doi: 10.1111/j.1750-3639.2002.tb00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba A. Role of Endothelin B receptor in reactive astrocytes. Life Sci. 1998;62:1711–1715. doi: 10.1016/s0024-3205(98)00133-7. [DOI] [PubMed] [Google Scholar]
- Back SA, Tuohy TMF, Chen H, Wallingford N, Craig A, Struve J, Luo NL, Banine F, Liu Y, Chang A, Trapp BD, Bebo BF, Rao MS, Sherman LS. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med. 2005;11:966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 2000;19:2056–2068. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomstrand F, Giaume C. Kinetics of endothelin-induced inhibition and glucose permeability of astrocyte gap junctions. J Neurosci Res. 2006;83:996–1003. doi: 10.1002/jnr.20801. [DOI] [PubMed] [Google Scholar]
- Blomstrand F, Venance L, Sirén AL, Ezan P, Hanse E, Glowinski J, Ehrenreich H, Giaume C. Endothelins regulate astrocyte gap junctions in rat hippocampal slices. Eur J Neurosci. 2004;19:1005–1015. doi: 10.1111/j.0953-816x.2004.03197.x. [DOI] [PubMed] [Google Scholar]
- Bradác I, Svobodova Varekova R, Wacenovsky M, Skrdla M, Plchut M, Polcik M. siRNA selection criteria—statistical analyses of applicability and significance. Biochem Biophys Res Commun. 2007;359:83–87. doi: 10.1016/j.bbrc.2007.05.056. [DOI] [PubMed] [Google Scholar]
- Buffo A, Vosko MR, Erturk D, Hamann GF, Jucker M, Rowitch D, Gotz M. Expression pattern of the transcription factor Olig2 in response to brain injuries: implications for neuronal repair. Proc Natl Acad Sci USA. 2005;102:18183–18188. doi: 10.1073/pnas.0506535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazaubon S, Chaverot N, Romero IA, Girault JA, Adamson P, Strosberg AD, Couraud PO. Growth factor activity of endothelin-1 in primary astrocytes mediated by adhesion-dependent and -independent pathway. J Neurosci. 1997;15:6203–6212. doi: 10.1523/JNEUROSCI.17-16-06203.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BK, Kung HC, Tsai TY, Chang WC. Essential role of mitogen-activated protein kinase pathway and c-Jun induction in epidermal growth factor-induced gene expression of human 12-lipoxygenase. Mol Pharmacol. 2000;57:153–161. [PubMed] [Google Scholar]
- Clerk A, Kemp T, Harrison JG, Mullen AJ, Barton PJR, Sugden PH. Up-regulation of c-jun mRNA in cardiac myocytes requires the extracellular signal-regulated kinase cascade, but c-Jun N-terminal kinases are required for efficient up-regulation of c-jun protein. Biochem J. 2002;368:101–110. doi: 10.1042/BJ20021083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clozel M, Breu V, Gray GA, Kalina B, Loffler BM, Burri K, Cassal JM, Hirth G, Muller M, Neidhart W, Ramuz H. Pharmacological characterization of bosentan, a new potent orally active nonpeptide endothelin receptor antagonist. J Pharmacol Exp Ther. 1994;270:228–235. [PubMed] [Google Scholar]
- Cole-Edwards KK, Musto AE, Bazan N. c-Jun N-terminal kinase activation responses induced by hippocampal kindling are mediated by reactive astrocytes. J Neurosci. 2006;26:8295–8304. doi: 10.1523/JNEUROSCI.1986-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couraud PO, Durieu-Trautmann O, Le Nguyen P, Marin P, Glibert F, Strosberg AD. Functional endothelin-1 receptors in rat astrocytoma C6. Eur J Pharmacol. 1991;206:191–198. doi: 10.1016/s0922-4106(05)80018-5. [DOI] [PubMed] [Google Scholar]
- Desai D, He S, Yorio T, Krishnamoorthy RR, Prasanna G. Hypoxia augments TNF-alpha-mediated endothelin-1 release and cell proliferation in human optic nerve head astrocytes. Biochem Biophys Res Commun. 2004;318:624–628. doi: 10.1016/j.bbrc.2004.04.073. [DOI] [PubMed] [Google Scholar]
- Egnaczyk GF, Pomonis JD, Scmidt JA, Rogers SD, Peters C, Ghilardi JR, Mantyh PW, Maggio JE. Proteomic analysis of the reactive phenotype of astrocytes following endothelin-1 exposure. Proteomics. 2003;3:689–698. doi: 10.1002/pmic.200300407. [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Freemerman AJ, Turner AJ, Birrer MJ, Szabo E, Valerie K, Grant S. Role of c-jun in human myeloid leukemia cell apoptosis induced by pharmacological inhibitors of protein kinase C. Mol Pharmacol. 1996;49:788–795. [PubMed] [Google Scholar]
- Gadea A, Gallo V. ET-1 regulates oligodendrocyte progenitor development through the ETB receptor. Soc Neurosci. 2005 Abstr 144.21. [Google Scholar]
- Gallo V, Armstrong RC. Developmental and growth factor-induced regulation of nestin in oligodendrocyte lineage cells. J Neurosci. 1995;15:394–406. doi: 10.1523/JNEUROSCI.15-01-00394.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Prywes R. ATF1 phosphorylation by the ERK MAPK pathway is required for epidermal growth factor-induced c-jun expression. J Biol Chem. 2002;277:50550–50556. doi: 10.1074/jbc.M209799200. [DOI] [PubMed] [Google Scholar]
- Hall SM. The effect of injections of lysophosphatidyl choline into white matter of the adult mouse spinal cord. J Cell Sci. 1972;10:535–546. doi: 10.1242/jcs.10.2.535. [DOI] [PubMed] [Google Scholar]
- Han TH, Lamph WW, Prywes R. Mapping of epidermal growth factor-, serum-, and phorbol ester-responsive sequence elements in the c-jun promoter. Mol Cell Biol. 1992;12:4472–4477. doi: 10.1128/mcb.12.10.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JG, Sugden PH, Clerk A. Endothelin-1 promotes phosphorylation of CREB transcription factor in primary cultures of neonatal rat cardiac myocytes: implications for the regulation of c-jun expression. Biochim Biophys Acta. 2004;1644:17–25. doi: 10.1016/j.bbamcr.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Parker A, Malone P, Gabelt BT, Rasmussen C, Kaufman PS, Hernandez MR. Long-term activation of c-Fos and c-Jun in optic nerve head astrocytes in experimental ocular hypertension in monkeys and after exposure to elevated pressure in vitro. Brain Res. 2005;1054:103–115. doi: 10.1016/j.brainres.2005.06.050. [DOI] [PubMed] [Google Scholar]
- Hasselblatt M, Lewczuk P, Loffler BM, Kamrowski-Kruck H, von Ahsen N, Sirén AL, Ehrenreich H. Role of astrocytic ET(B) receptor in the regulation of extracellular endothelin-1 during hypoxia. Glia. 2001;34:18–26. doi: 10.1002/glia.1036. [DOI] [PubMed] [Google Scholar]
- Hasselblatt M, Bunte M, Dringen R, Tabernero A, Medina JM, Giaume C, Sirén AL, Ehrenreich H. Effect of endothelin-1 on astrocytic protein content. Glia. 2003;42:390–397. doi: 10.1002/glia.10224. [DOI] [PubMed] [Google Scholar]
- Hino A, Tokuyama Y, Kobayashi M, Yano M, Weir B, Takeda J, Wang X, Bell GI, Macdonald RL. Increased expression of endothelin B receptor mRNA following subarachnoid hemorrhage in monkeys. J Cereb Blood Flow Metab. 1996;16:688–697. doi: 10.1097/00004647-199607000-00020. [DOI] [PubMed] [Google Scholar]
- Jiang MH, Hoog A, Ma KC, Nie XJ, Olsson Y, Zhang WW. Endothelin-1-like immunoreactivity is expressed in human reactive astrocytes. NeuroReport. 1993;4:935–937. doi: 10.1097/00001756-199307000-00024. [DOI] [PubMed] [Google Scholar]
- Kasuya Y, Abe Y, Hama H, Sakurai T, Asada S, Masaki T, Goto K. Endothelin-1 activates mitogen-activated protein kinases through two independent signaling pathways in rat astrocytes. Biochem Biophys Res Commun. 1994;204:1325–1333. doi: 10.1006/bbrc.1994.2608. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Baba A. Endothelin-induced protein tyrosine phosphorylation of cultured astrocytes: its relationship to cytoskeletal actin organization. Glia. 1999;26:324–332. [PubMed] [Google Scholar]
- Koyama Y, Takemura M, Fujiki K, Ishikawa N, Shigenaga Y, Baba A. BQ788, an endothelin ET(B) receptor antagonist, attenuates stab wound injury-induced reactive astrocytes in rat brain. Glia. 1999;26:268–271. doi: 10.1002/(sici)1098-1136(199905)26:3<268::aid-glia8>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Yoshioka Y, Matsuda T, Baba A. Focal adhesion kinase is required for endothelin-induced cell cycle progression of cultured astrocytes. Glia. 2003;43:185–189. doi: 10.1002/glia.10240. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Yoshioka Y, Shinde M, Matsuda T, Baba A. Focal adhesion kinase mediates endothelin-induced cyclin D3 expression in rat cultured astrocytes. J Neurochem. 2004;90:904–912. doi: 10.1111/j.1471-4159.2004.02546.x. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Baba A, Matsuda T. Endothelins stimulate the expression of neurotrophin-3 in rat brain and rat cultured astrocytes. Neuroscience. 2005a;136:425–433. doi: 10.1016/j.neuroscience.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Tsujikawa K., Matsuda T, Baba A. Endothelin increases expression of exonIII- and exonIV-containing brain-derived neurotrophic factor transcripts in cultured astrocytes and rat brain. J Neurosci Res. 2005b;80:809–816. doi: 10.1002/jnr.20512. [DOI] [PubMed] [Google Scholar]
- Lazarini F, Strosberg AD, Couraud PO, Cauzabon SM. Coupling of ETB endothelin receptor to mitogen-activated protein kinase stimulation and DNA synthesis in primary cultures of rat astrocytes. J Neurochem. 1996;66:459–465. doi: 10.1046/j.1471-4159.1996.66020459.x. [DOI] [PubMed] [Google Scholar]
- Lee S, Hong J, Choi SY, Oh SB, Park K, Kim JS, Karin M, Lee SJ. CpG oligodeoxynucleotides induce expression of proinflammatory cytokines and chemokines in astrocytes: the role of c-Jun N-terminal kinase in CpG ODN-mediated NF-kappaB activation. J Neuroimmunol. 2004;153:50–63. doi: 10.1016/j.jneuroim.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Ma KC, Nie XJ, Hoog A, Olsson Y, Zhang WW. Reactive astrocytes in viral infections of the human brain express endothelin-like immunoreactivity. J Neurol Sci. 1994;126:184–192. doi: 10.1016/0022-510x(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R. partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain. 2002;99:175–184. doi: 10.1016/s0304-3959(02)00097-0. [DOI] [PubMed] [Google Scholar]
- MacCumber MW, Ross CA, Snyder SH. Endothelin in brain: receptors, mitogenesis, and biosynthesis in glial cells. Proc Natl Acad Sci USA. 1990;87:2359–2363. doi: 10.1073/pnas.87.6.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw J, Hiebert GW, Steeves JD. Modulating astrogliosis after trauma. J Neurosci Res. 2001;63:109–115. doi: 10.1002/1097-4547(20010115)63:2<109::AID-JNR1002>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Miyake T, Hattori T, Fukuda M, Kitamura T. Reactions of S-100-positive glia after injury of mouse cerebral cortex. Brain Res. 1989;489:31–40. doi: 10.1016/0006-8993(89)90005-x. [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Decker L, Lachapelle F, Avellana-Adalid V, Bachelin C, Van Evercooren AB. Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur J Neurosci. 1999;11:4357–4366. doi: 10.1046/j.1460-9568.1999.00873.x. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Schwartz JP. Gene expression profiles of reactive astrocytes in dopamine-depleted striatum. Brain Pathol. 2004;14:275–280. doi: 10.1111/j.1750-3639.2004.tb00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie XJ, Olsson Y. Endothelin peptides in brain diseases. Rev Neurosci. 1996;7:177–186. doi: 10.1515/revneuro.1996.7.3.177. [DOI] [PubMed] [Google Scholar]
- Nolte C, Matyash M, Pivneva T, Schipke CG, Ohlemeyer C, Hanisch UK, Kirchhoff F, Kettenmann H. GFAP promoter-controlled EGFP-expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia. 2001;33:72–86. [PubMed] [Google Scholar]
- Norenberg MD. Astrocyte response to CNS injury. J Neuropathol Exp Neurol. 1994;53:213–220. doi: 10.1097/00005072-199405000-00001. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Barman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. End Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Raivich G, Behrens A. Role of AP-1 transcription factor c-Jun in developing, adult and injured brain. Prog Neurobiol. 2006;78:347–363. doi: 10.1016/j.pneurobio.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rogers SD, Demaster E, Catton M, Ghilardi JR, Levin LA, Maggio JE, Mantyh PW. Expression of endothelin-B receptors by glia in vivo is increased after CNS injury in rats, rabbits, and humans. Exp Neurol. 1997;145:180–195. doi: 10.1006/exnr.1997.6468. [DOI] [PubMed] [Google Scholar]
- Rogers SD, Peters CM, Pomonis JD, Hagiwara H, Ghilardi JR, Mantyh PW. Endothelin B receptors are expressed by astrocytes and regulate astrocyte hypertrophy in the normal and injured CNS. Glia. 2003;41:180–190. doi: 10.1002/glia.10173. [DOI] [PubMed] [Google Scholar]
- Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev. 1994;46:325–415. [PubMed] [Google Scholar]
- Sakurai-Yamashita Y, Niwa M, Yamashita K, Kataoka Y, Himeno A, Shigematsu K, Tsutsumi K, Taniyama K. Endothelin receptors in kainic acid-induced neural lesions of rat brain. Neuroscience. 1997;81:565–577. doi: 10.1016/s0306-4522(97)00175-9. [DOI] [PubMed] [Google Scholar]
- Schinelli S. Pharmacology and physiopathology of the brain endothelin system: an overview. Curr Med Chem. 2006;13:627–638. doi: 10.2174/092986706776055652. [DOI] [PubMed] [Google Scholar]
- Schinelli S, Zanassi P, Paolillo M, Wang H, Avvedimento EV, Gallo V. Stimulation of endothelin B receptors in astrocytes induces cAMP response element binding protein (CREB) phosphorylation and c-fos expression via multiple mitogen-activated protein kinase (MAPK) signaling pathways. J Neurosci. 2001;21:8842–8853. doi: 10.1523/JNEUROSCI.21-22-08842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- Simonson MS, Herman WH. Protein kinase C and protein tyrosine kinase activity contribute to mitogenic signaling by endothelin-1. J Biol Chem. 1993;268:9347–9357. [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sohn J, Natale J, Chew LJ, Belachew S, Cheng Y, Aguirre A, Lytle J, Nait-Oumesmar B, Kerninon C, Kanai-Azuma M, Kanai Y, Gallo V. Identification of Sox17 as a transcription factor that regulates oligodendrocyte development. J Neurosci. 2006;26:9722–9735. doi: 10.1523/JNEUROSCI.1716-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanimirovic DB, Ball R, Mealing G, Morley P, Durkin JP. The role of intracellular calcium and protein kinase C in endothelin-stimulated proliferation of rat type I astrocytes. Glia. 1995;15:119–130. doi: 10.1002/glia.440150204. [DOI] [PubMed] [Google Scholar]
- Stiles JD, Ostrow PT, Balos LL, Greenberg SJ, Plunkett R, Grand W, Heffner RR., Jr Correlation of endothelin-1 and transforming growth factor beta 1 with malignancy and vascularity in human gliomas. J Neuropathol Exp Neurol. 1997;56:435–439. doi: 10.1097/00005072-199704000-00012. [DOI] [PubMed] [Google Scholar]
- Tang G, Xu Z, Goldman J. Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem. 2006;281:38634–38643. doi: 10.1074/jbc.M604942200. [DOI] [PubMed] [Google Scholar]
- Teixeira A, Chaverot N, Donny Strosberg A, Cazaubon S. Differential regulation of cyclin D1 and D3 expression in the control of astrocyte proliferation induced by endothelin-1. J Neurochem. 2000;74:1034–1040. doi: 10.1046/j.1471-4159.2000.0741034.x. [DOI] [PubMed] [Google Scholar]
- Tsang MC, Lo AC, Cheung PT, Chung SS, Chung SK. Perinatal hypoxia/ischemia-induced endothelin-1 mRNA in astrocyte-like and endothelial cells. NeuroReport. 2001;20:2265–2270. doi: 10.1097/00001756-200107200-00044. [DOI] [PubMed] [Google Scholar]
- Uesugi M, Kasuya Y, Hama H, Yamamoto M, Hayashi K, Masaki T, Goto K. Endogenous endothelin-1 initiates astrocytic growth after spinal cord injury. Brain Res. 1996;28:255–259. doi: 10.1016/0006-8993(96)00524-0. [DOI] [PubMed] [Google Scholar]
- Ui-Tei K, Naito Y, Saigo K. Guidelines for the selection of effective short-interfering RNA sequences for functional genomics. Methods Mol Biol. 2007;361:201–216. doi: 10.1385/1-59745-208-4:201. [DOI] [PubMed] [Google Scholar]
- Wang YN, Chen YJ, Chang WC. Activation of extracellular signal-regulated kinase signaling by epidermal growth factor mediates c-jun activation and p300 recruitment in keratin 16 gene expression. Mol Pharmacol. 2006;69:85–98. doi: 10.1124/mol.105.016220. [DOI] [PubMed] [Google Scholar]
- Woodruff RH, Franklin RJ. Demyelination and remyelination of the caudal cerebellar peduncle of adult rats following stereotaxic injections of lysolecithin, ethidium bromide, and complement/anti-galactocerebroside: a comparative study. Glia. 1999;25:216–228. doi: 10.1002/(sici)1098-1136(19990201)25:3<216::aid-glia2>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Yamashita K, Niwa M, Kataoka Y, Shigematsu K, Himeno A, Tsutsumi K, Nakano-Nakashima M, Sakurai-Yamashita Y, Shibata S, Taniyama K. Microglia with an endothelin ETB receptor aggregate in rat hippocampus CA1 subfields following transient forebrain ischemia. J Neurochem. 1994;63:1042–1051. doi: 10.1046/j.1471-4159.1994.63031042.x. [DOI] [PubMed] [Google Scholar]
- Yuan X, Chittajallu R, Belachew S, Anderson S, McBain CJ, Gallo V. Expression of the green fluorescent protein in the oligodendrocyte lineage: a transgenic mouse for developmental and physiological studies. J Neurosci Res. 2002;70:529–545. doi: 10.1002/jnr.10368. [DOI] [PubMed] [Google Scholar]
- Zhang WW, Badonic T, Hoog A, Jiang MH, Ma KC, Nie XJ, Olsson Y. Astrocytes in Alzheimer's disease express immunoreactivity to the vaso-constrictor endothelin-1. J Neurol Sci. 1994;122:90–96. doi: 10.1016/0022-510x(94)90057-4. [DOI] [PubMed] [Google Scholar]