

Abstract
In rats, neonatal alcohol (EtOH) exposure coinciding with the period of rapid brain growth produces structural damage in some brain regions that often persists into adulthood and thus may have long-term consequences in the neural regulation of behavior. Because recent findings indicate that the circadian clock located in the rat suprachiasmatic nucleus is vulnerable to alcohol-induced insults during development, the present study examined the long-term effects of neonatal alcohol exposure on the photic regulation of circadian behavior in adult rats. Rat pups were exposed to alcohol (3.0, 4.5, or 6.0 g·kg-1·day-1) or isocaloric milk formula on postnatal days 4-9 using artificial-rearing methods. At 2 months of age, animals were housed individually and circadian wheel-running behavior was continuously analyzed to determine the effects of neonatal alcohol treatment on the rate of reentrainment to a 6-h advance in the 12-h light:12-h dark photoperiod and the phase-shifting properties of free-running rhythms in response to discrete light pulses on a background of constant darkness. For all doses, neonatal alcohol exposure had a significant effect in reducing the time for reentrainment such that EtOH-treated rats required four to five fewer days than control animals for stable realignment of the activity rhythm to the shifted light-dark cycle. Coupled with the accelerated rate of reentrainment, the amplitude of light-evoked phase delays at circadian time 14 and advances at circadian time 22 in the 4.5 and 6.0 g·kg-1·day-1 EtOH groups was almost twofold greater than that observed in control animals. The present observations indicate that the mechanisms by which photic signals regulate circadian behavior are permanently altered following alcohol exposure during the period of rapid brain development. These long-term alterations in the photic regulation of circadian rhythms may account, at least partially, for some neurobehavioral consequences of prenatal alcohol exposure in humans such as depression.
Keywords: Circadian rhythms, Clock, Ethanol, Wheel-running behavior, Entrainment, Suprachiasmatic nucleus
1. Introduction
In humans, prenatal exposure to alcohol induces neurobehavioral deficits resulting from damage to specific regions of the central nervous system (CNS). These alcohol-induced changes in CNS structure and function are often permanent and thus have long-term consequences for exposed individuals. Reductions in brain size (i.e., microencephaly) and in the volume of various brain regions (e.g., the anterior cerebellum and corpus callosum) and behavioral impairments (e.g., learning difficulties and attention deficits) have been observed in offspring subjected to maternal alcohol consumption during pregnancy (Roebuck et al., 1999). In rats, the brain growth spurt coinciding with the early postnatal period has been identified as a critical temporal window for CNS vulnerability to the neurotoxic effects of alcohol (Chen & West, 1999). Exposure to alcohol during the early postnatal period, which corresponds to the third trimester equivalent in human brain development (Dobbing & Sands, 1979), produces a wide range of damaging effects on the rat including reductions in brain size and weight (Maier et al., 1996); cell loss in discrete brain regions such as the cerebellum, hippocampus, and olfactory bulb (Bonthius & West, 1990; Bonthius et al., 1992; Chen et al., 1998; Goodlett et al., 1998); altered neuronal circuitry (West et al., 1981); and decreased expression of specific neurochemical signals (Manteuffel, 1996). In the hippocampus and cerebellum, this alcohol-induced cellular damage is, respectively, accompanied by chronic behavioral deficits in learning/memory and motor performance tasks (Goodlett et al., 1987, 1988; Kelly et al., 1988; Thomas et al., 1996, 1998). Consequently, studies using this or similar animal models provide a unique opportunity to delineate the spectrum of developmental alcohol-induced brain injury and dysfunction beyond reported clinical observations on deficits found in affected offspring.
In this regard, recent investigations have revealed that the neural clock responsible for the regulation of circadian or 24-h rhythms is a vulnerable target for alcohol-induced insult during CNS development (Earnest et al., 2001). The internal biological clock responsible for the regulation of mammalian circadian rhythms is located within the suprachiasmatic nucleus (SCN) of the anterior hypothalamus (Moore, 1983). In addition to its endogenous timekeeping function, the SCN mediates the synchronization or entrainment of circadian rhythms to light-dark cycles so as to provide for their alignment with appropriate times of day. Photic signals mediating circadian entrainment are transduced by the retina and communicated to the SCN via the retinohypothalamic tract (RHT), a monosynaptic projection from a subpopulation of retinal ganglion cells that terminate bilaterally within the ventrolateral subdivision of the nucleus (Moore, 1983; Moore et al., 1995). Evidence for the vulnerability of the SCN-RHT photoentrainment pathway to alcohol-induced damage during brain development has evolved from studies demonstrating that prenatal or early postnatal exposure to alcohol permanently alters the responses of rat circadian rhythms to light (Allen et al., in press; Farnell et al., 2004a; Sei et al., 2003). In association with these alcohol-induced effects on the photic regulation of circadian rhythms, we have shown that neonatal alcohol treatment disrupts endogenous neurochemical rhythmicity in the SCN (Allen et al., 2004) and affects oscillations in core molecular components of the SCN clockworks (Farnell et al., 2004b).
To determine whether neonatal alcohol exposure alters other aspects of the photic regulation of circadian behavior, we first examined the long-term impact of this treatment on the rate of reentrainment to an abrupt shift in the light-dark cycle. Specifically, rats were exposed to three different doses of alcohol during the neonatal period and the circadian rhythm in wheel-running behavior was assessed in adult rats for evidence of alcohol-induced changes in the time required for reentrainment to a 6-h shift in the light-dark cycle. Entrainment is a fundamental property of circadian clocks that provides for the temporal coordination of internal processes with external environmental cycles (for review, see Johnson et al., 2003). Circadian photoentrainment requires daily adjustments of the clock mechanism so that its endogenous period is equal to the length (usually 24 h) of the entraining light-dark cycle. These adjustments are mediated through time-dependent phase shifts of the SCN clock mechanism. The phase of free-running circadian rhythms is delayed or reset to a later time by a brief light exposure during the early subjective night (i.e., coinciding with previous dark phase or the animal’s active period), but is advanced to an earlier time when the same photic stimulus is administered during the late subjective night. Because the time for reentrainment of circadian rhythms is largely determined by the amplitude of their phase-shifting responses to light (Aschoff et al., 1975; Pittendrigh & Daan, 1976), parallel analysis was conducted to determine whether neonatal alcohol exposure also alters the phase-shifting responses of adult rats to light. Experiments examined the dose-dependent effects of neonatal alcohol exposure on the phase shifts of the activity rhythm in response to light pulses administered at three different times during the circadian cycle. In control and alcohol-exposed rats, the phase-resetting actions of light were analyzed at 6 h before (i.e., during the midsubjective day at circadian time [CT] 6), 2 h after (i.e., during the early subjective night at CT 14), or 10 h after the onset of activity (i.e., during the late subjective night at CT 22) because light exposure at these times, respectively, induces no change, maximal delays, or maximal advances in the phase of the rat activity rhythm. Perturbations in these fundamental properties of circadian rhythms and their photoentrainment have been associated with sleep-wake disturbances, bipolar affective disorder (i.e., manic-depressive illness), and depression (Moore, 1991a; Schwartz, 1993), all of which are behavioral manifestations of prenatal alcohol exposure in humans (Sher, 2004). Thus, information from this study may be valuable in both the identification and treatment of neurobehavioral disturbances observed in human neonates, children, and adolescents following prenatal exposure to alcohol (Rosett et al., 1979; Sher, 2004; Smith & Eckardt, 1991; Steinhausen & Spohr, 1998; Steinhausen et al., 1993).
2. Methods
2.1. Subjects
The subjects were 106 male Sprague-Dawley rat pups derived from 25 time-mated litters. Male subjects were used exclusively in these studies because female rats show considerable day-to-day variability in the circadian regulation of activity behavior (relative to that in males) that would present a confounding factor in the analysis of the effects of neonatal alcohol treatment. In this regard, female rodents are distinguished by daily irregularity in the period of the activity rhythm in constant conditions, the timing of their activity onsets during entrainment to a light-dark cycle, and the total amount of activity due to changing levels of estrogen over the course of the estrous cycle (Albers, 1981; Albers et al., 1981; Morin et al., 1977). All animals were born and reared in the vivarium at the Texas A&M University System Health Science Center under a standard 12-h light:12-h dark photoperiod (LD 12:12; lights-on at 0600 h). On postnatal day (PD) 1 (date of birth designated as PD 0), the newborns within each litter were culled to 8-10 pups per litter, using cross-fostering procedures as necessary. On PD 4, the rat pups were randomly assigned to one of five treatment groups: a suckle-control (SC) group (n = 20) and four artificial-rearing treatment groups. The artificial-rearing groups received either alcohol (EtOH) at 3.0 (EtOH3.0; n = 25), 4.5 (EtOH4.5; n = 19), or 6.0 (EtOH6.0; n = 20) g·kg-1·day-1 or maltose-dextrin (gastrostomy control [GC]; n = 22) from PDs 4 through 9. The University Laboratory Animal Care Committee at Texas A&M University approved the procedures used in this study.
2.2. Artificial rearing
The artificial-rearing process has been described previously in detail (West, 1993). Briefly, on PD 4, pups were anesthetized with isofluorane (VEDCO Inc., St. Joseph, MO) and gastrostomy tubes were inserted down the esophagus and surgically implanted into the stomach (Diaz, 1991; West et al., 1984). From PDs 4 to 9, gastrostomized pups were maintained in the artificial-rearing apparatus and received their daily nutritional requirements through formula (diet)-filled syringes, which were operated by a timer-activated infusion pump (Model# 935 Harvard Apparatus, Holliston, MA). The formula was provided daily in twelve 20-min fractions (i.e., every 2 h). Gastrostomized pups received either alcohol treatment or isocaloric maltose-dextrin solution from PDs 4 to 9. On PD 10, artificially reared pups were fostered to a lactating dam after their gastrostomy tubes had been cut and sealed. SC animals were maintained with lactating dams and were included to control for any effect produced by artificial-rearing methods. To minimize the potential confound of litter effects, no more than two pups from the same litter were assigned to the same treatment condition. Identification of subjects from different treatment groups was accomplished by subcutaneous implantation of each pup with a coded microchip (Avid, Los Angeles, CA) on PD 10. Animals were weaned on PD 21 and housed two to three per cage. After weaning, food and water were provided ad libitum for the remainder of the experiment.
2.3. Drug administration
Beginning around midday of the LD 12:12 (1200 h) from PDs 4 to 9, alcohol solutions of 6.8%, 10.2%, or 13.6% (vol/vol) were administered to the EtOH groups through 2 of the daily 12 feedings to yield alcohol doses of 3.0, 4.5, or 6.0 g·kg-1·day-1, respectively. These doses of alcohol and binge-like treatment regimen have been shown to consistently produce dose-related, long-term neuroanatomical damage (Bonthius & West, 1988, 1990; Bonthius et al., 1992; Chen et al., 1998, 1999; Livy et al., 2003). The GC group (0 g·kg-1·day-1 alcohol) was given the appropriate concentration of isocaloric maltose-dextrin solution in place of alcohol. All animals received regular formula diet during the 10 remaining daily feedings.
2.4. Blood alcohol concentration
Blood alcohol concentration (BAC) for all EtOH-treated pups was measured using a gas chromatograph (Model# 3900, Varian, Palo Alto, CA). Blood samples (20 μl) were collected from pup tails 90 min after the beginning of the second alcohol feeding on PD 6 (peak BAC) (Bonthius et al., 1988; Kelly et al., 1987). Samples were stored in glass vials containing a 200-μl cocktail composed of 0.6 M perchloric acid and 4 mM n-propanol in double distilled water until BAC analysis.
2.5. Experimental protocol
At 2 months of age, animals (n = 106) were housed individually in cages equipped with running wheels so that the circadian rhythm of wheel-running activity could be continuously recorded. All animals were allowed to acclimate to their running-wheel apparatus and the baseline pattern of behavioral activity was recorded for 21 days during entrainment to LD 12:12 (light period from 0600 to 1800 h). For 49 of these animals, the light-dark cycle was abruptly phase advanced by 6 h so that light onset and offset occurred at 2400 and 1200 h, respectively. Rats were maintained in this shifted LD 12:12 cycle (light period from 2400 to 1200 h) for 6 weeks and the time required for reentrainment of the activity rhythm to the new light-dark cycle was determined. For the 57 remaining animals, the baseline analysis of wheel-running activity in LD 12:12 (light period from 0600 to 1800 h) was followed by exposure to constant darkness (DD) beginning at the offset of the light-dark cycle (1800 h). Upon establishment of a stable free-running period (after ≈3 weeks in DD), animals were exposed to 15-min light pulses at three different phases of the circadian cycle. Animals (n = 39) were exposed to light pulses during the subjective night at CT 14 or CT 22 (i.e., 2 or 10 h after the onset activity) because photic stimulation at these times, respectively, induces maximal phase delays or advances of the rat activity rhythm (Summer et al., 1984). After light exposure, animals were allowed to free-run in DD for at least 18 days and reestablish a stable free-running period before receiving a second light pulse. Experiments were designed such that each animal received no more than two light pulses, one at CT 14 and the other at CT 22. In addition, light pulses were administered to a separate group of animals (n = 18) during the subjective day at CT 6 (i.e., 6 h before activity onset) on one to two occasions because illumination during this interval is known to elicit little or no phase-shifting effect on the free-running rhythm of activity (Summer et al., 1984). Light pulses (150-200 lx) were delivered by transferring the home cages of individual animals to a ventilated, light-proof chamber. Phase-shifting analysis was performed on rats between 3 and 5 months of age. Animal care was performed at random times and experimental manipulations in DD were accomplished using an infrared viewer (FJW Optical Systems, Palatine, IL).
2.6. Analysis of reentrainment and light-induced phase shifts of circadian behavior
Wheel-running activity was continuously recorded, summed, stored in 10-min bins, and graphically depicted in actograms with a computer running ClockLab data-acquisition and analysis software (ActiMetrics, Evanston, IL). The reentrainment of the rhythm in wheel-running behavior was evaluated by determining the time (in days) required to return to the same phase angle (Ψ) between lights-off and the daily onset of activity that was observed prior to the 6-h advance in the light-dark cycle. During entrainment to LD 12:12, the onset of activity for a given cycle was identified as the first bin during which an animal attained 10% of peak running-wheel revolutions (i.e., intensity). To measure Ψ during stable entrainment, least squares analyses were used to establish a regression line through the daily onsets of activity over a 15-day interval and then the number of minutes before (positive) or after (negative) the time of lights-off in the light-dark cycle was determined for each animal. Following the advance in the light-dark cycle, an animal was determined to have reentrained when the new Ψ approximated its preshift value (±30 min) for 15 successive days. The first day of this interval was then used to calculate the time between the advance in the light-dark cycle and stable reentrainment of the activity rhythm. Statistical analyses were performed on the raw data using a one-way analysis of variance to determine significance of treatment effects on reentrainment of the activity rhythm, and Newman-Keuls post hoc analyses were applied if necessary.
Light-induced phase shifts (ΔΦ) of free-running activity rhythms in DD were measured using methods similar to those established by Daan and Pittendrigh (1976). As described previously (Farnell et al., 2004a), phase shifts were evaluated by using least squares analyses to establish linear regressions through the activity onsets for 7-14 days before the pulse and through those for an equivalent interval after subsequent reestablishment of a steady-state circadian period. Initial activity onsets after a light pulse were not included in this analysis because they usually reflect transient or non-steady-state cycles of the activity rhythm. For each phase shift, amplitude was measured by subtracting the actual time of activity onset (at the first steady-state intercept of the posttreatment regression line) from the predicted time of activity onset (as projected by the pretreatment regression line). Thus, negative values denote phase delays (-ΔΦ), whereas positive values indicate phase advances (+ΔΦ). Shifts in the circadian phase of the activity rhythm were independently assessed by two experienced individuals without knowledge of treatment designation, and reported values reflect the averages of their determinations. Analysis of phase-shifting responses to light pulses at CT 6 could not be assessed on three occasions (two GC and one EtOH4.5) because of instability in the free-running activity rhythms of these animals following light treatment. Statistical analyses were performed on the raw data using a one-way analysis of variance to determine significance of treatment effects upon light-induced phase shifts of the activity rhythm, and Newman-Keuls post hoc analyses were applied if necessary.
3. Results
3.1. Blood alcohol concentration
Three groups of EtOH animals were exposed to alcohol at doses of 3.0, 4.5, or 6.0 g·kg-1·day-1. Administration of alcohol in 2 consecutive feedings out of the 12 daily feedings produced dose-dependent differences in peak BAC levels on PD 6 [F(2, 61) = 48.69; p < .01]. For alcohol doses of 3.0, 4.5, and 6.0 g·kg-1·day-1, the peak BACs (mean ± S.E.M.) were 126.0 ± 4.8 (0.126%), 214.1 ± 6.1 (0.214%), and 257.6 ± 16.4 mg/dl (0.258%), respectively. These BAC values are comparable to those established in previous studies using similar doses of alcohol (Bonthius & West, 1988, 1990; Napper & West, 1995). It is noteworthy that the BAC values produced by the low dose (3.0 g·kg-1·day-1) are close to the standard levels for legal intoxication (0.08%) and that even the BACs established with the other doses are often sustained by individuals who abuse alcohol (Urso et al., 1981).
3.2. Effects of neonatal alcohol exposure on reentrainment and light-induced phase shifts of the rat activity rhythm
During the initial 21-day exposure to LD 12:12, entrainment of the activity rhythm was observed in all animals. Actograms for representative animals from all treatment groups are depicted in Fig. 1A. General differences were evident in the activity patterns of control and EtOH rats during entrainment to the light-dark cycle. SC and GC animals entrained to LD 12:12 in a similar fashion such that their daily onsets of activity occurred around 5-15 min after lights-off (1800 h). Similar to previous observations on the general effects of neonatal alcohol exposure on circadian photoentrainment (Allen et al., in press), EtOH-treated rats were distinguished by patterns of entrainment in which the daily wheel-running activity was initiated at more variable and earlier times relative to control animals. In all three EtOH treatment groups, the daily onsets of activity typically preceded the light-dark transition, although these advances in the pattern of entrainment were more pronounced in EtOH4.5 rats. Following an abrupt 6-h advance in the light-dark cycle, all control and EtOH-exposed animals (n = 49) reentrained by gradually advancing the phase of their activity rhythms until the daily onsets of activity were realigned with lights-off in a phase relationship similar to that observed prior to the shift in the light-dark cycle. In SC and GC rats, this resetting of the activity rhythm to earlier times occurred at a rate of approximately 30-50 min/day. The average times to realign the activity onsets and reentrain to the shifted light-dark cycle were 11 ± 0.4 days for the SC group and 9.6 ± 0.6 days for GC animals. In EtOH-treated animals, the phase advances of the activity rhythm in response to new photoperiod were typically much larger and thus the time required to complete reentrainment was significantly less than that observed in control animals [F(4, 44) = 10.00; p < .05]. In the EtOH3.0, EtOH4.5, and EtOH6.0 groups, the average times for reentrainment of the activity rhythm were 7.5 ± 0.9, 6.1 ± 0.6, and 6.7 ± 0.7 days, respectively (Fig. 1B). Similar to the pattern during the preshift interval, the activity onsets of animals in the EtOH groups were generally marked by increased day-to-day variability and an advanced phase angle so as to coincide with the latter portion of the light phase following stable reentrainment to the new photoperiod.
Fig. 1.

Effects of neonatal alcohol exposure on reentrainment of the rat activity rhythm to a shift in the light-dark cycle. (A) Representative activity records of 5 adult rats from the suckle-control (SC), gastrostomy control (GC), and three alcohol-treated groups (EtOH3.0, EtOH4.5, EtOH6.0) maintained under 12-h light:12-h dark photoperiod (LD 12:12) conditions. On the indicated day, the LD 12:12 cycle was abruptly shifted such that the timing of lights-on and lights-off was advanced by 6 h. Actograms are double plotted over a 48-h period. The closed bars at the top and shading on the records signify the timing of the dark phase in the LD 12:12 cycle. (B) Rate of reentrainment in control (SC and GC) and alcohol-exposed (EtOH3.0, EtOH4.5, EtOH6.0) rats to an abrupt shift in the light-dark cycle. Bars denote the mean (+S.E.M.) number of days required for the activity rhythm to reentrain after the LD 12:12 cycle was advanced by 6 h. The time for reentrainment to the shifted light-dark cycle in all three EtOH-treated groups was significantly less (*p < .05) than that observed in SC and GC animals.
Because the amplitude of the phase shifts induced by light is an important factor in the reentrainment of circadian rhythms (Aschoff et al., 1975; Pittendrigh & Daan, 1976), we examined the activity rhythms of adult rats subjected to neonatal alcohol treatment in separate experiments for evidence of long-term alterations in their phase-shifting responses to light. Based on our previous finding that light-induced phase advances and delays of the activity rhythm are increased in EtOH-treated rats (Farnell et al., 2004a), these experiments were conducted to compare the phase-shifting responses in control groups with rats that were exposed to neonatal alcohol at the same doses used in our reentrainment study. During exposure to DD, all animals (n = 57) showed stable free-running rhythms of wheel-running activity. Although precise determination was not feasible because of the light pulsing paradigm, the free-running period of the activity rhythm tended to be shorter in EtOH-exposed rats than in control animals. This general observation is consistent with our results demonstrating that neonatal alcohol treatment produces decreases in the circadian period of the activity rhythm in adult rats (Allen et al., in press). In both control and EtOH-exposed rats, 15-min light pulses consistently induced phase delays at CT 14 and phase advances at CT 22, but failed to elicit any appreciable change in the phase of the activity rhythm when administered during the subjective day at CT 6. The direction and amplitude of the light-induced phase shifts in SC and GC animals were comparable to those observed previously in Sprague-Dawley rats (Farnell et al., 2004a; Summer et al., 1984), with average delays and advances of about 2 h in response to light pulses administered during the subjective night (Fig. 2). SC rats exhibited light-induced phase delays of -1.4 to -3.4 h at CT 14 (mean ΔΦ = -2.2 ± 0.2 h) and advances of +1.4 to 2.7 h at CT 22 (mean ΔΦ = +2.0 ± 0.2 h). In GC animals, light pulses produced phase delays and advances ranging from -1.3 to 23.2 h (mean ΔΦ = -2.3 ± 0.2 h) and +1.5 to 2.5 h (mean ΔΦ = +2.0 ± 0.2 h), respectively. Although neonatal exposure to the lowest dose of alcohol (EtOH3.0) only had a minor impact on the amplitude of the light-induced phase shifts during the subjective night (mean ΔΦ = -2.6 ± 0.2 h at CT 14 and + 2.3 ± 0.2 h at CT 22) relative to that found in controls, animals in the EtOH4.5 and EtOH6.0 groups were distinguished by increased phase-shifting responses to the same light stimulus. The mean phase delays at CT 14 in EtOH4.5 and EtOH6.0 rats (mean ΔΦ = -3.8 ± 0.4 and -3.6 ± 0.3 h, respectively) were significantly greater [F(4, 34) = 7.49; p < .05] than the light-induced shifts observed in the SC, GC, and EtOH3.0 groups at this CT. Likewise, EtOH4.5 and EtOH6.0 animals showed significant increases in the amplitude of lightinduced phase advances at CT 22 (mean ΔΦ = +3.4 ± 0.2 and +3.2 ± 0.3 h, respectively) relative to both control groups and EtOH3.0 rats [F(4, 34) = 11.72; p < .01]. There were no treatment-related differences in the phase-shifting responses to light at CT 6 with all animals exhibiting negligible shifts that ranged from delays of -3 min to advances of +12 min. In all control and EtOH-treated groups, two to three transient cycles were necessary to complete light-induced phase delays whereas advancing shifts typically required four to seven cycles before a new steady state was established in which the period of the activity rhythm was stable and similar to that observed prior to the light pulse.
Fig. 2.
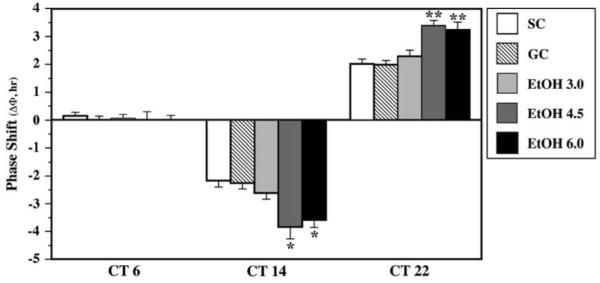
Effects of neonatal alcohol exposure on the phase-shifting responses of the rat activity rhythm to light at circadian time (CT) 6, 14, and 22. Mean (+S.E.M.) phase shift (ΔΦ) in hours of the free-running rhythm of wheel-running behavior in suckle-control (SC), gastrostomy control (GC), and EtOH-treated (EtOH3.0, EtOH4.5, EtOH6.0) rats maintained in constant darkness and exposed to a 15-min light pulse at CT 6, 14, or 22. Phase delays are denoted by negative values and advances are signified by positive values. Control and EtOH-treated groups exhibited no significant differences in their phase-shifting responses to light at CT 6. Light-induced phase delays at CT 14 and phase advances at CT 22 in the EtOH4.5 and EtOH6.0 groups were significantly greater (*p < .05; **p < .01) than those observed in the SC and GC animals.
4. Discussion
The results of this study demonstrate that alcohol exposure during a critical period of CNS development produces interconnected changes in the photic regulation of circadian behavior in adult rats. Adult rats exposed to alcohol from PDs 4 to 9 required four to five fewer days than control animals for reentrainment of the activity rhythm to an abrupt 6-h advance of the light-dark cycle. The long-term impact of neonatal alcohol exposure on the rate of reentrainment was coupled with its effects on the phase-shifting responses of the rat activity rhythm to light. In rats exposed to alcohol at doses of 4.5 or 6.0 g·kg-1·day-1, the amplitude of light-induced phase delays and advances during the subjective night was greatly increased relative to that observed in controls and animals treated with a lower dose of alcohol. The enhanced phase-shifting responses of alcohol-treated rats were comparable to those observed previously when alcohol was administered during the same neonatal period at 4.5 g·kg-1·day-1 (Farnell et al., 2004a), suggesting that this alcohol dose consistently produces BACs necessary for altering the mechanism by which light regulates the phase of circadian rhythms. The observed dissociation between the impact of alcohol on the rate of reentrainment to a shifted light-dark cycle, but not on light-induced phase shifts, in the 3.0 g·kg-1·day-1 EtOH group suggests that neonatal alcohol exposure at a low dose may differentially affect the regulation of these two circadian processes by light. Importantly, neonatal alcohol exposure had no effect on phase-shifting responses to light during the midsubjective day; all control and EtOH-treated groups showed no appreciable change in the phase of their activity rhythms when exposed to a light pulse at CT 6, which corresponds to the “dead zone” of the phase response curve to light (Daan & Pittendrigh, 1976; Summer et al., 1984). This observation indicates that neonatal alcohol exposure alters the amplitude, but not the time-dependent gating, of phase shifts in the clock mechanism induced by nocturnal illumination. In addition to the link with increased phase-shifting responses to light, the observed acceleration of the reentrainment process may be related to the effects of alcohol on circadian period because alterations in this fundamental property of the clock mechanism influence the time required for reentrainment of the activity rhythm (Aschoff et al., 1975; Pittendrigh & Daan, 1976). Although stable free-running period could not be determined in the present study because of experimental design, recent findings indicate that neonatal alcohol exposure alters the free-running period of the activity rhythm in adult rats (Allen et al., in press). In EtOH-exposed rats, the free-running period of the activity rhythm was shorter relative to that found in control animals and consistent with the present results, and accelerated reentrainment in response to an advance in the light-dark cycle would be expected in association with this effect of neonatal alcohol exposure. Thus, neonatal alcohol exposure has complementary effects on circadian period and phase-shifting responses to light that may collectively serve to expedite the rate of reentrainment when the light-dark cycle is advanced.
The specific mechanisms by which neonatal alcohol exposure permanently modulates photoentrainment of circadian rhythms and their phase-shifting responses to light are unknown, but presumably involve alcohol-induced changes in the development of the SCN and/or the neural substrates responsible for conveying light information to the SCN. Because the brain growth spurt in rats is marked by a series of critical neurodevelopmental events, including cellular proliferation, accelerated synaptogenesis and myelination, the long-term functional effects of neonatal alcohol exposure on the photic regulation of SCN circadian clock may result from the disruption or abnormal progression of these processes. One possibility is that neonatal alcohol exposure may alter the regulation of circadian rhythms by light-dark signals by inducing cell loss in the developing SCN similar to the neurotoxic effects of EtOH on other brain regions (Bonthius & West, 1990; Bonthius et al., 1992; Chen et al., 1998; Goodlett et al., 1998; Ikonomidou et al., 2000). Indeed, neonatal exposure to alcohol at 4.5 g·kg-1·day-1 causes a small, but significant, reduction in SCN neuronal density that may reflect changes in intranuclear synaptic connections (Farnell et al., 2004a). Because the SCN clock consists of an ensemble of cell-autonomous oscillators that are independently capable of generating endogenous rhythmicity (Welsh et al., 1995), alcohol-induced alterations in synaptic communication may perturb the coupling and synchronization of multiple oscillators and thereby affect fundamental clock properties such as its regulation by light.
Another potential explanation for the altered rate of reentrainment and phase-shifting responses to light in EtOH animals is that neonatal alcohol exposure may permanently impact upon development of the visual system. Evidence for the effects of alcohol on the developing visual system has emerged from studies demonstrating that perinatal alcohol exposure causes hypoplasia and decreased myelination of optic nerve axons (Harris et al., 2000; Pinazo-Duran et al., 1997). In addition, recent findings indicate that neonatal alcohol administration induces apoptotic neurodegeneration of both retinal ganglion cells and neurons in brain regions receiving visual system projections such as the lateral geniculate nucleus (Tenkova et al., 2003). It is noteworthy that the alcohol treatment interval from PDs 4 to 9 was coincident with critical period for the synaptogenesis of RHT projections to the SCN (Speh and Moore, 1993). However, it is unlikely that the observed effects of neonatal alcohol exposure on the photic regulation of circadian behavior are caused by damage to retinal ganglion cells or optic nerve fibers that form the RHT because these structural defects would be expected to decrease, rather than increase, both the rate of reentrainment and phase-shifting responses to light in EtOH animals. Instead, other neurodevelopmental changes in the RHT or its communication of photic signals to the SCN may be responsible for these effects of neonatal alcohol exposure. For example, alcohol exposure during the early postnatal period may permanently enhance clock responsiveness to photic signals by disrupting the latter stages of RHT development during which retinal fiber projections to the SCN and surrounding hypothalamic regions are normally pruned back between PD 6 and PD 10 (Moore, 1991b). If neonatal alcohol treatment interferes with this remodeling process, then the resultant excess in RHT connections with SCN neurons would presumably augment the communication of photic signals that mediate the reentrainment and phase-shifting responses of circadian rhythms. Although retinofugal projections to the SCN were not examined in this study, the possible influence of alcohol exposure on RHT development and remodeling warrants further consideration because alcohol exposure, albeit during the prenatal period, has been shown to inhibit the loss of unmyelinated axons in the developing optic nerve and thereby increase their proportion relative to myelinated fibers (Samorajski et al., 1986).
Alternatively or perhaps in conjunction with neuroanatomical changes in RHT development, neonatal alcohol exposure may affect the rate of reentrainment and phase-shifting responses to light by modulating neurochemical substrates responsible for the communication of entraining light signals to the SCN (for review, see Hannibal, 2002; Meijer & Schwartz, 2003). The putative photopigment, melanopsin, and the neurotransmitters, glutamate and pituitary adenylate cyclase-activating polypeptide (PACAP), are thought to mediate the communication of light information to the SCN because (1) melanopsin is distinctly localized in retinal ganglion cells that are endogenously responsive to light (Hattar et al., 2002); (2) glutamate and PACAP have been identified as the principal neurotransmitters of the RHT (De Vries et al., 1993; Hannibal et al., 2000; van den Pol, 1993); (3) transgenic deletion of genes encoding melanopsin, PACAP, and PACAP receptors influences the phase-shifting responses of the murine circadian clock to light (Colwell et al., 2004; Hannibal et al., 2001; Harmar et al., 2002; Ruby et al., 2002); and (4) infusion of glutamate or PACAP into the SCN phase shifts circadian rhythms in a manner comparable to light (Harrington et al., 1999; Meijer et al., 1988). Thus, neonatal EtOH exposure may chronically increase both the entraining and phase-shifting effects of light on the SCN clock by enhancing melanopsin-mediated phototransduction or by potentiating glutamate and/or PACAP function in the communication of photic signals to the SCN. Although the effects of alcohol on melanopsin expression and on PACAP neurotransmission have not been assessed, glutamatergic input to the SCN is a plausible target for alcohol-induced modulation because previous studies indicate that perinatal alcohol exposure increases serum glutamate levels (Karl et al., 1995) and upregulates NMDA-type glutamate receptors in some brain regions (Naassila & Daoust, 2002; Nixon et al., 2002). Similarly, changes in the regulation of serotonin or gamma-aminobutyric acid (GABA) neurotransmission with the SCN may play some role in the long-term effects of neonatal alcohol exposure on the reentrainment and phase-resetting properties of the clock mechanism because these neurotransmitter systems are involved in the photic regulation of circadian rhythms (Cardinali & Golombek, 1998; Pickard & Rea, 1997; Rea et al., 1995; Smale et al., 1990) and their functional regulation is vulnerable to EtOH-induced insult during development (Ledig et al., 1988; Sari & Zhou, 2004).
The present data, coupled with previous findings (Allen et al., 2004, in press; Farnell et al., 2004a, 2004b; Sei et al., 2003), provide clear evidence for the effects of developmental alcohol exposure on circadian rhythms and their synchronization to light-dark signals. It remains to be determined whether these abnormalities in the regulation of circadian rhythms contribute to long-term neurobehavioral manifestations of fetal alcohol syndrome (FAS) and fetal alcohol spectrum disorders (FASD) (Chudley et al., 2005; Hoyme et al., 2005). The enhanced rate of reentrainment and increased phase-shifting responses to light in EtOH-treated subjects might be first perceived as useful or at least inconsequential changes in circadian clock function. However, these findings may have some detrimental implications in the behavioral correlates of FAS and FASD especially when considered in relation to the recent finding that the reentrainment and phase-shifting responses of another circadian rhythm, the oscillation in deep body temperature, are conversely affected in adult rats exposed to alcohol during brain development (Sei et al., 2003). In contrast to our finding that the responses of the activity rhythm to photic cues are permanently enhanced in EtOH-treated animals, the rhythm of deep body temperature was marked by decreases in both the rate of reentrainment to an advance in the light-dark cycle and the amplitude of light-induced phase shifts during the subjective night following prenatal exposure to alcohol. This directional disparity in the effects of alcohol on circadian responses to light may be simply related to differences in the timing of alcohol treatment (i.e., postnatal vs. prenatal). Nevertheless, it is possible is that the effects of alcohol exposure on resetting responses to photic signals may vary between different circadian rhythms irrespective of the time of treatment during brain development. If developmental alcohol exposure differentially affects the rhythms of wheel-running activity and deep body temperature with regard to their resetting by photic signals, then internal desynchronization between these and perhaps other circadian rhythms would be expected to temporarily occur in EtOH-treated animals until reentrainment or phase-shifting responses are completed and a new steady state is achieved. Such alterations in the phase relationships between various circadian rhythms (e.g., sleep-wake cycle and oscillations in hormone levels) have been linked to bipolar affective disorder and depression (Moore, 1991a; Wehr et al., 1983). Consequently, disturbances in the photic regulation of circadian rhythms similar to those observed in this study may represent important neurobehavioral consequences of human alcohol exposure in utero that contribute to the incidence of affective disorders in FAS and FASD (Famy et al., 1998).
Acknowledgments
The authors wish to thank Nichole Neuendorff for excellent technical assistance. This study was supported by National Institutes of Health (NIH) grants AA13242 and MH60147 (D.J.E.) and NIH grant AA05523 (J.R.W.).
References
- Albers HE. Gonadal hormones organize and modulate the circadian system of the rat. Am J Physiol. 1981;241:R62–R66. doi: 10.1152/ajpregu.1981.241.1.R62. [DOI] [PubMed] [Google Scholar]
- Albers HE, Gerall AA, Axelson JF. Effect of reproductive state on circadian periodicity in the rat. Physiol Behav. 1981;26:21–25. doi: 10.1016/0031-9384(81)90073-1. [DOI] [PubMed] [Google Scholar]
- Allen GC, West JR, Chen W-JA, Earnest DJ. Developmental alcohol exposure disrupts circadian regulation of BDNF in the rat suprachiasmatic nucleus. Neurotoxicol Teratol. 2004;26:353–358. doi: 10.1016/j.ntt.2004.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GC, West JR, Chen W-J, Earnest DJ. Neonatal alcohol exposure permanently disrupts the circadian properties and photic entrainment of the activity rhythm in adult rats. Alcohol Clin Exp Res. 2005;29:1845–1852. doi: 10.1097/01.alc.0000183014.12359.9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschoff J, Hoffmann K, Pohl H, Wever R. Reentrainment of circadian rhythms after phase-shifts of the zeitgeber. Chronobiologia. 1975;2:23–78. [PubMed] [Google Scholar]
- Bonthius DJ, Bonthius NE, Napper RM, West JR. Early postnatal alcohol exposure acutely and permanently reduces the number of granule cells and mitral cells in the rat olfactory bulb: a stereological study. J Comp Neurol. 1992;324:557–566. doi: 10.1002/cne.903240408. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, Goodlett CR, West JR. Blood alcohol concentration and severity of microencephaly in neonatal rats depend on the pattern of alcohol administration. Alcohol. 1988;5:209–214. doi: 10.1016/0741-8329(88)90054-7. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, West JR. Blood alcohol concentration and microencephaly: a dose-response study in the neonatal rat. Teratology. 1988;37:223–231. doi: 10.1002/tera.1420370307. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, West JR. Alcohol-induced neuronal loss in developing rats: increased brain damage with binge exposure. Alcohol Clin Exp Res. 1990;14:107–118. doi: 10.1111/j.1530-0277.1990.tb00455.x. [DOI] [PubMed] [Google Scholar]
- Cardinali DP, Golombek DA. The rhythmic GABAergic system. Neurochem Res. 1998;23:607–614. doi: 10.1023/a:1022426519297. [DOI] [PubMed] [Google Scholar]
- Chen W-JA, Parnell SE, West JR. Neonatal alcohol and nicotine exposure limits brain growth and depletes cerebellar Purkinje cells. Alcohol. 1998;15:33–41. doi: 10.1016/s0741-8329(97)00084-0. [DOI] [PubMed] [Google Scholar]
- Chen W-JA, West JR. Alcohol-induced brain damage during development: potential risk factors. In: Hannigan JH, Spear LP, Spear NE, Goodlett CR, editors. Alcohol and Alcoholism: Effects on Brain and Development. Lawrence Erlbaum Assoc.; Mahwah: 1999. pp. 17–37. [Google Scholar]
- Chudley AE, Conry J, Cook JL, Loock C, Rosales T, LeBlanc N. Fetal alcohol spectrum disorder: Canadian guide-lines for diagnosis. CMAJ. 2005;172(5 Suppl):S1–S21. doi: 10.1503/cmaj.1040302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS, Michel S, Itri J, Rodriguez W, Tam J, Lelievre V, Hu Z, Waschek JA. Selective deficits in the circadian light response in mice lacking PACAP. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1194–R1201. doi: 10.1152/ajpregu.00268.2004. [DOI] [PubMed] [Google Scholar]
- Daan S, Pittendrigh CS. A functional analysis of circadian pacemakers in nocturnal rodents. II. The variability of phase response curves. J Comp Physiol. 1976;106:253–266. [Google Scholar]
- De Vries MJ, Cardazo BN, van der Want J, de Wolf A, Meijer JH. Glutamate immunoreactivity in terminals of the retinohypothalamic tract of the brown Norwegian rat. Brain Res. 1993;612:231–237. doi: 10.1016/0006-8993(93)91665-f. [DOI] [PubMed] [Google Scholar]
- Diaz J. Experimental rearing of rat pups using chronic gastric fistulas. In: Shair HN, Barr GA, Hofer MA, editors. Developmental Psychobiology: New Methods and Changing Concepts. Oxford University Press; New York: 1991. pp. 272–284. [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Earnest DJ, Chen W-JA, West JR. Developmental alcohol and circadian clock function. Alcohol Res Health. 2001;25:136–140. [PMC free article] [PubMed] [Google Scholar]
- Famy C, Streissguth AP, Unis AS. Mental illness in adults with fetal alcohol syndrome or fetal alcohol effects. Am J Psychiatry. 1998;155:552–554. doi: 10.1176/ajp.155.4.552. [DOI] [PubMed] [Google Scholar]
- Farnell YZ, West JR, Chen W-JA, Allen GC, Earnest DJ. Developmental alcohol exposure alters light-induced phase shifts of the circadian activity rhythm in rats. Alcohol Clin Exp Res. 2004a;28:1020–1027. doi: 10.1097/01.alc.0000130807.21020.1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnell YZ, West JR, Chen W-JA, Allen GC, Marchette LD, Earnest DJ. Developmental alcohol exposure disrupts clock gene rhythms in the rat SCN and cerebellum. (abstract) Alcohol Clin Exp Res. 2004b;28(5 Suppl):164A. [Google Scholar]
- Goodlett CR, Kelly SJ, West JR. Early postnatal alcohol exposure that produces high blood alcohol levels impairs development of spatial navigation learning. Psychobiology. 1987;15:64–74. [Google Scholar]
- Goodlett CR, Nonneman AJ, Valentino ML, West JR. Constraints on water maze spatial learning in rats: implications for behavioral studies of brain damage and recovery of function. Behav Brain Res. 1988;28:275–286. doi: 10.1016/0166-4328(88)90130-1. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Pearlman AD, Lundahl KR. Binge neonatal alcohol intubations induce dose-dependent loss of Purkinje cells. Neurotoxicol Teratol. 1998;20:285–292. doi: 10.1016/s0892-0362(97)00102-5. [DOI] [PubMed] [Google Scholar]
- Hannibal J. Neurotransmitters of the retino-hypothalamic tract. Cell Tissue Res. 2002;309:73–88. doi: 10.1007/s00441-002-0574-3. [DOI] [PubMed] [Google Scholar]
- Hannibal J, Jamen F, Nielsen HS, Journot L, Brabet P, Fahrenkrug J. Dissociation between light-induced phase shift of the circadian rhythm and clock gene expression in mice lacking the pituitary adenylate cyclase activating polypeptide type 1 receptor. J Neurosci. 2001;21:4883–4890. doi: 10.1523/JNEUROSCI.21-13-04883.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannibal J, Moller M, Ottersen OP, Fahrenkrug J. PACAP and glutamate are co-stored in the retinohypothalamic tract. J Comp Neurol. 2000;418:147–155. [PubMed] [Google Scholar]
- Harmar AJ, Marston HM, Shen S, Spratt C, West KM, Sheward WJ, Morrison CF, Dorin JR, Piggins HD, Reubi J-C, Kelly JS, Maywood ES, Hastings MH. The VPAC2 receptor is essential for circadian function in the mouse suprachiasmatic nucleus. Cell. 2002;109:497–508. doi: 10.1016/s0092-8674(02)00736-5. [DOI] [PubMed] [Google Scholar]
- Harrington ME, Hoque S, Hall A, Golombek D, Biello S. Pituitary adenylate cyclase activating peptide phase shifts circadian rhythms in a manner similar to light. J Neurosci. 1999;19:6637–6642. doi: 10.1523/JNEUROSCI.19-15-06637.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SJ, Wilce P, Bedi KS. Exposure of rats to high but not low dose of ethanol during early postnatal life increases the rat of loss of optic nerve axons and decreases the rate of myelination. J Anat. 2000;197:477–485. doi: 10.1046/j.1469-7580.2000.19730477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattar S, Liao H-W, Takao M, Berson DM, Yau K-W. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. 2002;295:1065–1070. doi: 10.1126/science.1069609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyme HE, May PA, Kalberg WO, Kodituwakku P, Gossage JP, Trujillo PM, Buckley DG, Miller JH, Aragon AS, Khaole N, Viljoen DL, Jones KL, Robinson LK. A practical clinical approach to diagnosis of fetal alcohol spectrum disorders: clarification of the 1996 Institute of Medicine criteria. Pediatrics. 2005;115:39–47. doi: 10.1542/peds.2004-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:947–948. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Johnson CH, Elliott JA, Foster R. Entrainment of circadian programs. Chronobiol Int. 2003;20:741–774. doi: 10.1081/cbi-120024211. [DOI] [PubMed] [Google Scholar]
- Karl PI, Kwun R, Slonim A, Fisher SE. Ethanol elevates fetal serum glutamate levels in the rat. Alcohol Clin Exp Res. 1995;19:177–181. doi: 10.1111/j.1530-0277.1995.tb01488.x. [DOI] [PubMed] [Google Scholar]
- Kelly SJ, Bonthius DJ, West JR. Developmental changes in alcohol pharmacokinetics in rats. Alcohol Clin Exp Res. 1987;11:281–286. doi: 10.1111/j.1530-0277.1987.tb01308.x. [DOI] [PubMed] [Google Scholar]
- Kelly SJ, Goodlett CR, Hulsether SA, West JR. Impaired spatial navigation in adult female but not adult male rats exposed to alcohol during the brain growth spurt. Behav Brain Res. 1988;27:247–257. doi: 10.1016/0166-4328(88)90121-0. [DOI] [PubMed] [Google Scholar]
- Ledig M, Ciesielski L, Simler S, Lorentz JG, Mandel P. Effect of pre- and postnatal alcohol consumption on GABA levels of various brain regions in the rat offspring. Alcohol Alcohol. 1988;23:63–67. [PubMed] [Google Scholar]
- Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicol Teratol. 2003;25:447–458. doi: 10.1016/s0892-0362(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Maier SE, Chen W-JA, West JR. The effect of timing and duration of alcohol exposure on development of the fetal brain. In: Abel EL, editor. Fetal Alcohol Syndrome. From Mechanism to Prevention. CRC Press; Boca Raton: 1996. pp. 27–50. [Google Scholar]
- Manteuffel MD. Neurotransmitter function: changes associated with in utero alcohol exposure. In: Abel EL, editor. Fetal Alcohol Syndrome. From Mechanism to Prevention. CRC Press; Boca Raton: 1996. pp. 171–189. [Google Scholar]
- Meijer JH, Schwartz WJ. In search of the pathways for light-induced pacemaker resetting in the suprachiasmatic hypothalmic nucleus. J Biol Rhythms. 2003;18:235–249. doi: 10.1177/0748730403018003006. [DOI] [PubMed] [Google Scholar]
- Meijer JH, van der Zee EA, Dietz M. Glutamate phase shifts circadian activity rhythms in hamsters. Neurosci Lett. 1988;86:177–183. doi: 10.1016/0304-3940(88)90567-8. [DOI] [PubMed] [Google Scholar]
- Moore RY. Organization and function of a central nervous system circadian oscillator: the suprachiasmatic nucleus. Fed Proc Fed Am Soc Exp Biol. 1983;42:2783–2789. [PubMed] [Google Scholar]
- Moore RY. Disorders of circadian function and the human circadian timing system. In: Klein DC, Moore RY, Reppert SM, editors. Suprachiasmatic Nucleus: The Mind’s Clock. Oxford University Press; New York: 1991a. pp. 429–441. [Google Scholar]
- Moore RY. Development of the suprachiasmatic nucleus. In: Klein DC, Moore RY, Reppert SM, editors. Suprachiasmatic Nucleus: The Mind’s Clock. Oxford University Press; New York: 1991b. pp. 391–404. [Google Scholar]
- Moore RY, Speh JC, Card JP. The retinohypothalamic tract originates from a distinct subset of retinal ganglion cells. J Comp Neurol. 1995;352:351–366. doi: 10.1002/cne.903520304. [DOI] [PubMed] [Google Scholar]
- Morin LP, Fitzgerald KM, Zucker I. Estradiol shortens the period of hamster circadian rhythms. Science. 1977;196:305–307. doi: 10.1126/science.557840. [DOI] [PubMed] [Google Scholar]
- Naassila M, Daoust M. Effect of prenatal and postnatal ethanol exposure on the developmental profile of mRNAs encoding NMDA receptor subunits in rat hippocampus. J Neurochem. 2002;80:850–860. doi: 10.1046/j.0022-3042.2002.00755.x. [DOI] [PubMed] [Google Scholar]
- Napper RM, West JR. Permanent neuronal cell loss in the cerebellum of rats exposed to continuous low blood alcohol levels during the brain growth spurt: a stereological investigation. J Comp Neurol. 1995;362:283–292. doi: 10.1002/cne.903620210. [DOI] [PubMed] [Google Scholar]
- Nixon K, Hughes PD, Amsel A, Leslie SW. NMDA receptor subunit expression following early postnatal exposure to ethanol. Brain Res Dev Brain Res. 2002;139:295–299. doi: 10.1016/s0165-3806(02)00515-1. [DOI] [PubMed] [Google Scholar]
- Pickard GE, Rea MA. Serotonergic innervation of the hypothalamic suprachiasmatic nucleus and photic regulation of circadian rhythms. Biol Cell. 1997;89:513–523. doi: 10.1016/s0248-4900(98)80007-5. [DOI] [PubMed] [Google Scholar]
- Pinazo-Duran MD, Renau-Piqueras J, Guerri C, Stromland K. Optic nerve hypoplasia in fetal alcohol syndrome: an update. Eur J Ophthalmol. 1997;7:262–270. doi: 10.1177/112067219700700311. [DOI] [PubMed] [Google Scholar]
- Pittendrigh CS, Daan S. A functional analysis of circadian pacemakers in nocturnal rodents. IV. Entrainment: pacemaker as clock. J Comp Physiol. 1976;106:291–331. [Google Scholar]
- Rea MA, Barrera J, Glass JD, Gannon RL. Serotonergic potentiation of photic phase-shifts of the circadian activity rhythm. Neuroreport. 1995;6:1417–1420. doi: 10.1097/00001756-199507100-00014. [DOI] [PubMed] [Google Scholar]
- Roebuck TM, Mattson SN, Riley EP. Prenatal exposure to alcohol: effects on brain structure and neuropsychological functioning. In: Hannigan JH, Spear LP, Spear NE, Goodlett CR, editors. Alcohol and Alcoholism: Effects on Brain and Development. Lawrence Erlbaum Assoc.; Mahwah: 1999. pp. 1–16. [Google Scholar]
- Rosett HL, Snyder P, Sander LW, Lee A, Cook P, Weiner L, Gould J. Effects of maternal drinking on neonatal state regulation. Dev Med Child Neurol. 1979;21:464–473. doi: 10.1111/j.1469-8749.1979.tb01650.x. [DOI] [PubMed] [Google Scholar]
- Ruby NF, Brennan TJ, Xie X, Cao V, Franken P, Heller HC, O’Hara BF. Role of melanopsin in circadian responses to light. Science. 2002;298:2211–2213. doi: 10.1126/science.1076701. [DOI] [PubMed] [Google Scholar]
- Samorajski T, Lancaster F, Wiggins RC. Fetal ethanol exposure: a morphometric analysis of myelination in the optic nerve. Int J Dev Neurosci. 1986;4:369–374. doi: 10.1016/0736-5748(86)90054-7. [DOI] [PubMed] [Google Scholar]
- Sari Y, Zhou FC. Prenatal alcohol exposure causes long-term serotonin neuron deficit in mice. Alcohol Clin Exp Res. 2004;28:941–948. doi: 10.1097/01.alc.0000128228.08472.39. [DOI] [PubMed] [Google Scholar]
- Schwartz WJ. A clinician’s primer on the circadian clock: its localization, function and resetting. Adv Intern Med. 1993;38:81–106. [PubMed] [Google Scholar]
- Sei H, Sakata-Haga H, Ohta K, Sawada K, Morita Y, Fukui Y. Prenatal exposure to alcohol alters the light response in postnatal circadian rhythm. Brain Res. 2003;987:131–134. doi: 10.1016/s0006-8993(03)03329-8. [DOI] [PubMed] [Google Scholar]
- Sher L. Etiology, pathogenesis, and treatment of seasonal and non-seasonal mood disorders: possible role of circadian rhythm abnormalities related to developmental alcohol exposure. Med Hypotheses. 2004;62:797–801. doi: 10.1016/j.mehy.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Smale L, Michels KM, Moore RY, Morin LP. Destruction of the hamster serotonergic system by 5,7-DHT: effects on circadian rhythm phase, entrainment and response to triazolam. Brain Res. 1990;515:9–19. doi: 10.1016/0006-8993(90)90570-2. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Eckardt MJ. The effects of prenatal alcohol on the central nervous system. Recent Dev Alcohol. 1991;9:151–164. [PubMed] [Google Scholar]
- Speh JC, Moore RY. Retinohypothalamic tract development in the hamster and rat. Brain Res Dev Brain Res. 1993;76:171–181. doi: 10.1016/0165-3806(93)90205-o. [DOI] [PubMed] [Google Scholar]
- Steinhausen HC, Spohr H-L. Long-term outcome of children with fetal alcohol syndrome: psychopathology, behavior, and intelligence. Alcohol Clin Exp Res. 1998;22:334–338. doi: 10.1111/j.1530-0277.1998.tb03657.x. [DOI] [PubMed] [Google Scholar]
- Steinhausen HC, Willms J, Spohr H-L. Long-term psychopathological and cognitive outcome of children with fetal alcohol syndrome. J Am Acad Child Adolesc Psychiatry. 1993;32:990–994. doi: 10.1097/00004583-199309000-00016. [DOI] [PubMed] [Google Scholar]
- Summer TL, Ferraro JS, McCormack CE. Phase-response and Aschoff illuminance curves for locomotor activity of the rat. Am J Physiol. 1984;246:R299–R304. doi: 10.1152/ajpregu.1984.246.3.R299. [DOI] [PubMed] [Google Scholar]
- Tenkova T, Young C, Dikranian K, Labruyere J, Olney JW. Ethanol-induced apoptosis in the developing visual system during synaptogenesis. Invest Ophthalmol Vis Sci. 2003;44:2809–2817. doi: 10.1167/iovs.02-0982. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Goodlett CR, West JR. Alcohol-induced Purkinje cell loss depends on developmental timing of alcohol exposure and correlates with motor performance. Brain Res Dev Brain Res. 1998;105:159–166. doi: 10.1016/s0165-3806(97)00164-8. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Wasserman EA, West JR, Goodlett CR. Behavioral deficits induced by bingelike exposure to alcohol in neonatal rats: importance of developmental timing and number of episodes. Dev Psychobiol. 1996;29:433–452. doi: 10.1002/(SICI)1098-2302(199607)29:5<433::AID-DEV3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Urso T, Gavaler JS, van Thiel DH. Blood ethanol levels in sober alcohol users seen in an emergency room. Life Sci. 1981;28:1053–1056. doi: 10.1016/0024-3205(81)90752-9. [DOI] [PubMed] [Google Scholar]
- van den Pol AN. Glutamate and GABA presence and action in the suprachiasmatic nucleus. J Biol Rhythms. 1993;8(Suppl):S11–15. [PubMed] [Google Scholar]
- Wehr TA, Sack D, Rosenthal N, Duncan W, Gillin JC. Circadian rhythm disturbances in manic-depressive illness. Fed Proc. 1983;42:2809–2814. [PubMed] [Google Scholar]
- Welsh DK, Logothetis DE, Meister M, Reppert SM. Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron. 1995;14:697–706. doi: 10.1016/0896-6273(95)90214-7. [DOI] [PubMed] [Google Scholar]
- West JR. Use of pup in a cup model to study brain development. J Nutr. 1993;123:382–385. doi: 10.1093/jn/123.suppl_2.382. [DOI] [PubMed] [Google Scholar]
- West JR, Hamre KM, Pierce DR. Delay in brain growth induced by alcohol in artificially reared rat pups. Alcohol. 1984;1:213–222. doi: 10.1016/0741-8329(84)90101-0. [DOI] [PubMed] [Google Scholar]
- West JR, Hidges CA, Black AC., Jr. Prenatal exposure to alcohol alters the organization of hippocampal mossy fibers in rats. Science. 1981;211:957–959. doi: 10.1126/science.7466371. [DOI] [PubMed] [Google Scholar]