

Abstract
Background
Developmental alcohol (EtOH) exposure produces long-term changes in the photic regulation of rat circadian behavior. Because entrainment of circadian rhythms to 24-hr light/dark cycles is mediated by phase shifting or resetting the clock mechanism, we examined whether developmental EtOH exposure also alters the phase-shifting effects of light pulses on the rat activity rhythm.
Methods
Artificially reared Sprague-Dawley rat pups were exposed to EtOH (4.5 g/kg/day) or an isocaloric milk formula (gastrostomy control; GC) on postnatal days 4 to 9. At 2 months of age, rats from the EtOH, GC, and suckle control groups were housed individually, and wheel-running behavior was continuously recorded first in a 12-hr light/12-hr dark photoperiod for 10 to 14 days and thereafter in constant darkness (DD). Once the activity rhythm was observed to stably free-run in DD for at least 14 days, animals were exposed to a 15-min light pulse at either 2 or 10 hr after the onset of activity [i.e., circadian time (CT) 14 or 22, respectively], because light exposure at these times induces maximal phase delays or advances of the rat activity rhythm.
Results
EtOH-treated rats were distinguished by robust increases in their phase-shifting responses to light. In the suckle control and GC groups, light pulses shifted the activity rhythm as expected, inducing phase delays of approximately 2 hr at CT 14 and advances of similar amplitude at CT 22. In contrast, the same light stimulus produced phase delays at CT 14 and advances at CT 22 of longer than 3 hr in EtOH-treated rats. The mean phase delay at CT 14 and advance at CT 22 in EtOH rats were significantly greater (p < 0.05) than the light-induced shifts observed in control animals.
Conclusions
The data indicate that developmental EtOH exposure alters the phase-shifting responses of the rat activity rhythm to light. This finding, coupled with changes in the circadian period and light/dark entrainment observed in EtOH-treated rats, suggests that developmental EtOH exposure may permanently alter the clock mechanism in the suprachiasmatic nucleus and its regulation of circadian behavior.
Keywords: Circadian Rhythms, Clock, Ethanol, Photoentrainment, Suprachiasmatic Nucleus
IN HUMANS, THE central nervous system is susceptible to structural damage from alcohol (EtOH) exposure during critical stages of brain development (Clarren et al., 1978; Jones and Smith, 1973; Mattson et al., 1996; Sowell et al., 1996). Detailed animal studies have revealed that developmental EtOH exposure similarly disrupts the structural organization of different brain regions. In rats, EtOH exposure during the brain growth spurt (early postnatal period), which corresponds to the equivalent period of human brain development during the third trimester of gestation (Dobbing and Sands, 1979), has been shown to retard brain growth; produce microencephaly; cause neuronal loss in the hippocampus, cerebellum, and olfactory bulb; and alter neuronal circuitry (Bonthius et al., 1992; Bonthius and West, 1990; Chen et al., 1998; Goodlett et al., 1998; Goodlett and Johnson, 1999; Kelly et al., 1988; Maier et al., 1996; West et al., 1981). Furthermore, hippocampal and cerebellar cell loss in rats exposed to EtOH during the early postnatal period have been observed in association with behavioral deficits in learning/memory and motor performance tasks (Goodlett et al., 1987, 1988; Kelly et al., 1988; Thomas et al., 1996, 1998). However, further analysis is necessary to fully determine the scope of the developmental EtOH-induced damage to the brain and whether the resulting neuroanatomical changes have long-term neurobehavioral consequences.
The circadian clock and its regulation of circadian behavior have recently emerged as a significant area of interest in the analysis of specific neurobehavioral disturbances associated with developmental EtOH exposure. The internal biological clock responsible for the generation of mammalian circadian rhythms is located in the suprachiasmatic nuclei (SCN) of the anterior hypothalamus. The endogenous timekeeping function of the SCN is complemented by its role in mediating the entrainment or synchronization of mammalian circadian rhythms to light/dark cycles. Entraining light/dark signals are transduced by the retina and conveyed to the SCN via the retinohypothalamic tract (RHT), a monosynaptic projection from a subpopulation of retinal ganglion cells that terminates bilaterally within the ventrolateral subfield of each nucleus (Moore, 1983; Pickard, 1982). In rodents, complete ablation of the SCN abolishes circadian rhythmicity in various physiologic and behavioral processes (Moore, 1983; Turek, 1985), whereas destruction of the RHT eliminates entrainment of the activity rhythm to light/dark cycles without affecting other visual functions (Johnson et al., 1988). Initial evidence for EtOH-induced disturbances of circadian clock function has emanated from rodent studies indicating that prenatal EtOH exposure alters the light/dark regulation of circadian rhythms (Sei et al., 2003). Consistent with these effects of developmental EtOH on the SCN clock, our preliminary findings demonstrate that postnatal EtOH treatment disrupts endogenous neurochemical rhythmicity in the SCN and alters the free-running period and light/dark entrainment of circadian rhythms (Earnest et al., 1997; Marchette et al., 2003).
On the basis of the effects of developmental EtOH exposure on circadian entrainment to light/dark cycles, this study was conducted to determine whether early postnatal EtOH exposure permanently alters other aspects of the photic regulation of circadian behavior. Photoentrainment of circadian rhythms requires daily adjustment of the SCN circadian clock by an amount equal to the difference between its circadian period and the length (usually 24 hr) of the entraining light/dark cycle. Light is thought to mediate these adjustments by inducing time-dependent phase shifts of the SCN clock mechanism. Circadian rhythms freerunning in constant darkness (DD) are phase-delayed or reset to a later time in response to a brief light exposure during the early subjective night (i.e., coinciding with a previous dark phase or the animal’s active period) but are phase-advanced or displaced to an earlier time when light pulses are administered during the late subjective night. Because phase-shifting responses to light are directly correlated with circadian period (Daan and Pittendrigh, 1976, Pittendrigh and Daan, 1976), it is anticipated that light-induced phase shifts of the activity rhythm in adult rats will be altered in association with the long-term changes in free-running period caused by early postnatal EtOH exposure. Consequently, the circadian rhythm of wheel-running behavior was assessed in adult rats for evidence of developmental EtOH-related alterations in phase-shifting responses to light pulses administered at either 2 or 10 hr after the onset of activity [i.e., during the early subjective night at circadian time (CT) 14 or during the late subjective night at CT 22, respectively], because light exposure at these times induces maximal phase delays or advances of the rat activity rhythm. Because partial damage to the SCN alters circadian clock properties (e.g., period) that influence phase-shifting responses to light (Davis and Gorski, 1984; Pickard and Turek, 1982, 1985), this study also examined whether long-term changes in the photic regulation of circadian behavior are accompanied by developmental EtOH-induced insults on SCN integrity. Specifically, neuronal number and density in the SCN were analyzed for evidence of cell loss in adult rats exposed to EtOH during the early neonatal period.
METHODS
Animals
The subjects were 69 male Sprague-Dawley rat pups derived from 19 time-mated litters. All animals were born and reared in the vivarium at the Texas A&M University System Health Science Center under a standard 12-hr light/12-hr dark photoperiod (lights on at 0600 hr). On postnatal day (PD) 1 (the date of birth was designated PD 0), the neonates within each litter were culled to 8 to 10 per litter by using cross-fostering procedures as necessary. On PD 4, the pups were randomly subdivided into three groups: a suckle control group (SC; n = 20) and two artificial-rearing treatment groups. The artificial-rearing groups received either EtOH (n = 26) or maltose-dextrin (n = 23) from PD 4 through 9 and were respectively designated as EtOH and gastrostomy control (GC; EtOH 0 g/kg/day). The EtOH group in this experiment was treated with 4.5 g/kg/day of EtOH (10.2%; v/v) provided in 2 of the daily 12 feedings because this dose and exposure regimen have been shown to consistently produce long-term neuroanatomical changes in several different brain regions (Bonthius et al., 1992; Bonthius and West, 1990; Chen et al., 1998, 1999). The procedures used in this study were approved by the University Laboratory Animal Care Committee at Texas A&M University.
Artificial Rearing
The process of artificial rearing has been described previously in detail (Diaz, 1991; West, 1993). Briefly, on PD 4, pups were anesthetized with isoflurane (VEDCO Inc., St. Joseph, MO), and gastrostomy tubes were inserted down the esophagus and surgically implanted into the stomach. From PD 4 to 9, pups were maintained in the artificial apparatus and received their daily nutritional requirements through formula (diet)-filled syringes that were controlled by timer-activated infusion pumps (Harvard Apparatus, Holliston, MA). The formula was provided every 2 hr for 20 min. Gastrostomized pups received EtOH treatment or isocaloric maltose-dextrin solution from PD 4 to 9. On PD 10, artificially reared pups were fostered to lactating dams. All pups were coded by subcutaneous implantation of identification microchips (Avid, Los Angeles, CA). Animals were weaned on PD 21 and housed two to three per cage. After weaning, food and water were provided ad libitum for the remainder of the experiment.
Blood Alcohol Concentration
Blood alcohol concentration (BAC) was measured in all EtOH-treated pups by using a gas chromatograph (model 3900, Varian, Palo Alto, CA). Blood samples (20 μl) were collected from the pup tails 1.5 hr after the second EtOH feeding on PD 9 and stored in glass vials containing a 200-μl cocktail composed of 0.6 M perchloric acid and 4 mM n-propanol in double-distilled water until BAC analysis.
Experimental Protocol
At 2 months of age, all animals were housed individually in cages equipped with running wheels so that the circadian rhythm of wheel-running activity could be continuously recorded. Animals were allowed to acclimate to the running-wheel apparatus, and baseline activity behavior was recorded for 10 to 14 days during entrainment to a standard 12-hr light/12-hr dark photoperiod. Beginning at the offset of the light/dark cycle (1800 hr), animals were exposed to DD. Upon establishment of a stable free-running period (after ≈2 weeks in DD), animals were exposed to 15-min light pulses at CT 14 or 22 (i.e., 2 or 10 hr after the onset of activity), because illumination at these CTs induces maximal delays or advances in the phase of the mouse activity rhythm (Summer et al., 1984). Light pulses (150-200 lux) were delivered by transferring the home cages of individual animals to a ventilated, light-proofed chamber. After experimental treatment, animals were then allowed to free-run in DD for at least 2 weeks before receiving a second light pulse. Experiments were designed such that each animal received only two light pulses: one at CT 14 and the other at CT 22. This analysis of phase-shifting responses to light was conducted on animals between 3 and 5 months of age. During the period of experimentation, animal care and experimental manipulations in DD were accomplished with an infrared viewer (FJW Optical Systems, Palatine, IL).
Analysis of Circadian Behavior and Light-Induced Phase Shifts
Wheel-running activity was recorded continuously, summed, stored in 10-min bins, graphically depicted in actograms, and analyzed with a computer running ClockLab data-collection and analysis software (Actimetrics, Evanston, IL). Light-induced phase shifts of free-running activity rhythms in DD were measured by using the methods of Daan and Pittendrigh (1976). Briefly, phase shifts were evaluated by using least-squares analyses to establish linear regressions through the activity onsets for 7 to 13 days before the pulse and through those for an equivalent interval after subsequent re-establishment of a steady-state circadian period. Initial activity onsets after a light pulse were not included in this analysis because they usually reflect transient or non-steady-state cycles of the activity rhythm. For each phase shift, amplitude was calculated by measuring the time difference between the expected time of activity onset (as projected with a pretreatment regression line) and the actual time of activity onset at the first steady-state intercept of the posttreatment regression line. Shifts in the circadian phase of the activity rhythm were independently assessed in this manner by two experienced individuals without knowledge of treatment assignment, and reported values reflect the averages of their determinations. SC (n = 1), GC (n = 1), and EtOH (n = 2) animals showing highly irregular onsets of activity or arrhythmicity were excluded from analysis. Statistical analyses were performed on the raw data by using a one-way ANOVA to determine the significance of treatment effects on light-induced phase shifts of the activity rhythm; Newman-Keuls post hoc analyses were applied if necessary.
Histological Analysis
At the conclusion of behavioral analysis, anesthetized animals (sodium pentobarbital; 100 mg/kg) were killed by transcardiac perfusion with 100 ml of 0.1 M phosphate buffer (pH 7.4) containing heparin (2 units/μl) followed by 250 ml of 4% paraformaldehyde. Immediately after perfusion, the brains were removed, postfixed overnight at 4°C, and then stored for at least 24 hr in cryoprotectant solution (30% sucrose in 0.15 M phosphate buffer) before sectioning. The tissue was then frozen and sectioned serially in the coronal plane at 30 μm by using a sliding microtome. Coronal sections containing the SCN were mounted on glass slides, air-dried overnight at room temperature, stained with cresyl violet, and cover-slipped with Permount® (Fisher Scientific, Pittsburgh, PA).
Stereological Methods
The optical dissector was used to estimate the density of SCN neurons, and point counting (applying Cavalieri’s principle) was used to estimate the reference volume (Vref) of the SCN in SC, GC, and EtOH animals (n = 6 for the GC and EtOH groups; n = 7 for the SC group). The C.A.S.T.-GRID software (Olympus Denmark A/S, Albertslund, Denmark) was used for estimating the volume and the neuronal density of the SCN. The microscope was equipped with a computer-controlled stage (x and y axes), and the attached microcator (model ND2818, Heidenhain, Traument, Germany) measured the z axis. The images were transferred to a personal computer (Micron Millennium, Boise, ID) via a Jai color video camera (model 2040, Copenhagen, Denmark). For SCN volume determination, an × 10 objective lens was used, and for the measurement of SCN neuronal density, an × 60 oil-immersion objective lens with a 1.40 aperture was used.
The total number of SCN neurons was estimated from the measurement of Vref and the numerical density of the cells within the Vref. The Vref was determined by point counting and application of Cavalieri’s principle (Gundersen et al., 1988), and SCN neuronal density was determined by using the optical disector method (Gundersen et al., 1988; West and Gundersen, 1990). The general stereological methods used in this study were similar to those described previously (Bonthius et al., 1992), except that frozen sections were used for cell counting in this study. Statistical analysis was performed by using a one-way ANOVA to determine whether the volume, neuronal number, or neuronal density of the SCN was significantly different among the three treatment groups, and Newman-Keuls post hoc analyses were used if necessary. The α value was set at 0.05 for all statistical analyses.
RESULTS
Blood Alcohol Concentration
In the EtOH group, EtOH exposure in 2 consecutive feedings out of the 12 daily feedings on PD 9 produced a mean BAC of 206.5 ± 13.0 mg/dl. The BACs observed in the EtOH group were lower than values established previously with the EtOH dosage of 4.5 g/kg/day (Chen et al., 1998, 2001), although this distinction is probably related to differences in the time at which blood samples were collected. In other studies using this early postnatal regimen, blood samples were collected on PD 6, when the 4.5 g/kg/day dose of EtOH is known to produce peak BACs (Bonthius et al., 1988), whereas BACs were analyzed on PD 9 in this study.
Effects of Developmental EtOH on Light-Induced Phase Shifts of the Rat Activity Rhythm
Because recent studies suggest that prenatal EtOH treatment causes long-term changes in the regulation of rat circadian behavior by light/dark cues (Sei et al., 2003), the rhythm of wheel-running activity in adult rats was analyzed to determine whether developmental EtOH alters circadian responses to the phase-shifting effects of brief light pulses. The relative levels and rhythmic patterns of wheel-running activity in DD (Fig. 1) were comparable among SC, GC, and EtOH rats. Consistent with our preliminary findings (Marchette et al., 2003), the free-running period of the activity rhythm in EtOH-exposed rats was generally shorter than that observed for animals in the SC and GC groups. The phase-shifting responses of control animals to light pulses were similar to those reported previously for normal Sprague-Dawley rats (Summer et al., 1984). In the SC and GC groups, 15-min light pulses consistently induced approximately 2-hr phase shifts of the activity rhythm. Delays occurred at CT 14, and advances occurred at CT 22 (Fig. 1). The amplitude of the phase-shifting responses to light was comparable between these two control groups (Fig. 2). In SC animals, light pulses produced phase delays and advances ranging from -1.2 to 3.0 hr and from +0.9 to 2.9 hr, respectively. GC animals exhibited light-induced phase delays and advances ranging from -1.0 to 3.2 hr and from +0.6 to 3.6 hr, respectively. In contrast, animals in the EtOH group were distinguished by increased phase-shifting responses to the same light stimulus, with phase delays at CT 14 of -2.1 to 6.0 hr and advances at CT 22 of +2.0 to 5.4 hr. The mean phase delay at CT 14 and advance at CT 22 in EtOH rats were significantly greater (both at p < 0.01) than the light-induced shifts observed in the SC and GC groups at these CTs (Fig. 2). In all treatment groups, light-induced phase delays were usually complete within one or two cycles, whereas advancing shifts required approximately four to seven transient cycles before the period of the activity rhythm returned to a new steady state. At the conclusion of these transients, the steady-state period of the activity rhythms in SC, GC, and EtOH rats was similar to that observed before light exposure.
Fig. 1.

Effects of developmental alcohol exposure on light-induced phase shifts of the rat activity rhythm: representative activity records of three adult male rats from the SC, GC, and EtOH groups that were maintained in DD and exposed to a 15-min light pulse at CT 14 (A) or CT 22 (B). Actograms are double-plotted over 48 hr, and shaded circles (with a starred inset) on each record indicate the day and time during which animals were exposed to the light pulse (LP). A line has been fitted through the activity onsets for 7 to 13 days before the light pulse and extended for 25 days after treatment to facilitate visualization of phase shifts in free-running activity rhythm. In EtOH rats, exposure to a 15-min light pulse induced phase delays of the activity rhythm at CT 14 and advances at CT 22 that were larger than those observed in the SC and GC animals.
Fig. 2.
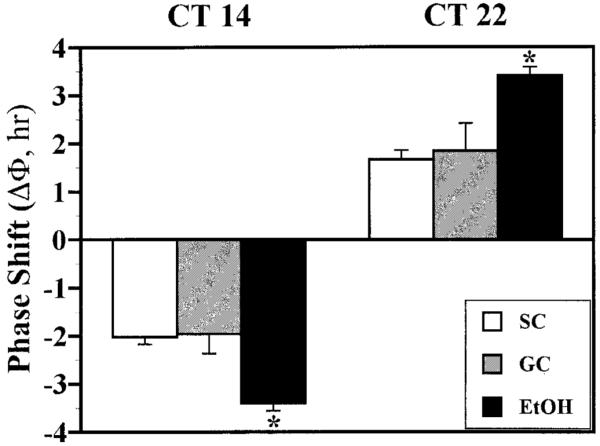
Effects of developmental alcohol exposure on the phase-shifting responses of the rat activity rhythm to light at CT 14 and 22. The mean ± SEM phase shift (ΔΦ) is shown (in hours) of the activity rhythm induced by a 15-min light pulse at CT 14 or 22 in SC, GC, and EtOH rats. Phase delays are indicated by negative values, and advances are denoted by positive values. Light-induced phase delays at CT 14 and phase advances at CT 22 in EtOH rats were significantly greater (*p < 0.05) than those observed in the SC and GC groups.
SCN Cell Counting
In addition to the analysis of phase-shifting responses, we estimated neuronal number and density in the SCN for evidence of cell loss in adult rats exposed to EtOH during the early neonatal period. Table 1 depicts the Vref, estimated neuronal number, and neuronal density for the SCN in SC, GC, and EtOH animals (n = 6 for the GC and EtOH groups; n = 7 for the SC group). The average thickness of the sections was 21.5 μm (measured with a microcator), and this thickness was used to calculate the Vref of the SCN. One-way ANOVAs were conducted to compare each of the dependent measures among all three treatment groups. These comparisons revealed that the SCN Vref and neuronal number were not significantly different among the SC, GC, and EtOH groups. However, analysis of the data for SCN neuronal density revealed a main effect of treatment [F(2,16) = 4.85; p < 0.05]. Further post hoc analyses indicated that SCN neuronal density was significantly reduced (p < 0.05) in EtOH-exposed animals relative to the SC group, but it did not differ between the EtOH and GC groups.
Table 1.
Total Neuronal Number, Reference Volume, and Neuronal Density in the SCN of 4-Month-Old Rats
| Treatment group | Estimated neuronal number (×103) | Reference volume (×10-3 mm3) | Neuronal density (×104 neurons/mm3) |
|---|---|---|---|
| SC | 7.66 ± 0.48 | 8.16 ± 0.41 | 93.8 ± 3.4 |
| GC | 6.97 ± 0.40 | 8.14 ± 0.15 | 87.1 ± 4.1 |
| EtOH | 6.98 ± 0.33 | 8.89 ± 0.41 | 78.7 ± 2.9* |
Significantly different from the SC group (p < 0.05).
DISCUSSION
These findings demonstrate that early postnatal EtOH treatment produces long-term changes in the phase-shifting responses of the rat activity rhythm to light. In rats exposed to EtOH from PD 4 to 9, light-induced phase delays and advances of the activity rhythm were approximately 1.7- to 2-fold greater than those observed in control animals. Because phase-shifting properties and the period of the clock mechanism determine circadian entrainment to light/dark cycles (Daan and Pittendrigh, 1976), this effect is probably linked to the alterations in circadian period and photoentrainment that have been observed previously in rodents exposed to prenatal EtOH (Sei et al., 2003) and in our preliminary studies examining the effects of early postnatal EtOH (Marchette et al., 2003). However, the finding that light-induced phase shifts were increased, rather than decreased, in the EtOH group is somewhat surprising for several reasons. First, most acute treatments with neuropeptides, neurotransmitters, or their agonists inhibit the phase-shifting effects of light on rodent circadian rhythms during the subjective night (Liang et al., 2000; Pickard et al., 1996; Rea et al., 1994; Weber and Rea, 1997; Weber et al., 1998). In addition, it was anticipated that developmental EtOH exposure could permanently affect RHT function in the communication of light signals to the SCN circadian clock because treatment from PD 4 to 9 coincided with the critical period for the synaptogenesis of RHT projections to the SCN (Speh and Moore, 1993). The effect of EtOH on RHT development has been observed previously in studies demonstrating that EtOH administration during the period of RHT synaptogenesis induces apoptosis within the developing visual system (Tenkova et al., 2003). Specifically, a single episode of EtOH exposure [two subcutaneous doses (2.5 g/kg) administered 2 hr apart] during the neonatal period (PD1 to 7) was observed to cause cell death in retinal ganglion cells and neurons in brain regions receiving visual system projections, such as the lateral geniculate nucleus. Although the effects of early postnatal EtOH exposure on components of the visual system were not examined in this study, EtOH-induced destruction of retinal ganglion cells or RHT innervation of the SCN would presumably produce a decrease, rather than the observed increase, in the phase-shifting responses to light in the EtOH group. Thus, the observed effects of developmental EtOH exposure on light-induced phase shifts of the activity rhythm are probably not related to damage to the RHT, but instead may reflect other changes, perhaps in RHT transmission of photic signals to the SCN.
Although it is unlikely that the increased phase-shifting responses to light in EtOH animals are caused by structural damage to RHT fibers, other changes in neurotransmission of photic signals to the SCN may be responsible for this effect of developmental EtOH exposure. Anatomical and physiologic evidence indicates that a presumptive photopigment, melanopsin, and the neurochemical signals glutamate and pituitary adenylate cyclase-activating polypeptide (PACAP) are involved in RHT transmission of light input to the SCN. Melanopsin is selectively found in retinal ganglion cells that respond intrinsically to light (Hattar et al., 2002), and melanopsin-deficient mice show diminished phase-shifting responses to light (Ruby et al., 2002). Glutamate and PACAP have been localized in RHT fibers and terminals (De Vries et al., 1993; Hannibal, 2002; van den Pol, 1993), and when they are applied to the SCN, both phase-shift circadian rhythms in a manner similar to light (Ding et al., 1994; Harrington et al., 1999; Meijer et al., 1988). Thus, developmental EtOH exposure could permanently alter the phase-shifting effects of light on the SCN clock by enhancing melanopsin-mediated signal transduction or by potentiating light-evoked glutamate or PACAP input to the SCN. Although the effect of developmental EtOH on PACAP-mediated neurotransmission has not been examined, there is some evidence to support its possible modulatory action on glutamatergic function. For example, chronic EtOH exposure during pregnancy has been shown to increase serum glutamate levels in fetal rats (Karl et al., 1995). In addition, evidence for the up-regulation of NMDA-type glutamate receptors in some brain regions after postnatal EtOH treatment (Nixon et al., 2002) may have implications for any effects of developmental EtOH on glutamatergic regulation of phase-shifting responses to light. However, it should be noted that these studies examined only the acute effects of developmental EtOH exposure on glutamate levels and receptor expression, so their persistence in adult rats is uncertain.
Another possible explanation for the changes in the phase-shifting responses of EtOH animals to light is that developmental EtOH exposure may permanently damage the circadian timekeeping mechanism or alter the configuration of its core molecular components. In mammals, the molecular clockwork consists of an interlocked transcriptional/translational feedback loop in which the expression of core elements is periodically suppressed by their protein products (Reppert and Weaver, 2002). Clock, Bmal1 (Mop3), period-1 (Per1), Per2, cryptochrome-1, and cryptochrome-2 have been implicated as core elements of the clock mechanism because mutation or knock-out of these genes in mice alters or abolishes circadian properties of activity rhythm. Consistent with their function in the timekeeping mechanism, these “clock” genes are differentially regulated by light and are involved in mediating light-induced phase shifts of circadian rhythms. Studies examining the effects of decreased clock gene expression in transgenic mice or in response to treatment with antisense oligonucleotides suggest that the Per1 and Per2 genes may contribute to the molecular responses underlying lightinduced phase advances and delays of the activity rhythm, respectively (Albrecht et al., 2001; Wakamatsu et al., 2001). On the basis of these implications for the Per1 and Per2 genes in the photic regulation of SCN circadian function, it is possible that the altered phase-shifting responses to light in EtOH animals may be associated with EtOH-induced changes in the rhythmic regulation of Per gene expression within the SCN. Although the long-term effects of postnatal EtOH exposure on expression of the Per genes and other molecular components of the clock mechanism have not been directly addressed, this possibility warrants further consideration because recent findings indicate that chronic EtOH consumption during adulthood impairs the circadian rhythm of Per2 messenger RNA levels in both the rat SCN and arcuate nucleus (Chen et al., 2003).
This analysis of SCN neuroanatomy for evidence of developmental EtOH-induced cell loss is noteworthy for several reasons. The rat SCN contains approximately 8,000 to 10,000 parvicellular neurons (Guldner, 1976; van den Pol, 1980). Because SCN neurogenesis occurs in utero predominantly between embryonic days 14 and 17 (Altman and Bayer, 1978; Ifft, 1972), it was unclear whether SCN neurons would be vulnerable to EtOH insults during the postnatal period. Previous findings indicate that chronic EtOH consumption for 6 to 12 months does not cause cell death in the SCN of adult rats but instead produces decreases in vasopressin, vasoactive-intestinal polypeptide, and somatostatin messenger RNA levels and the total number of SCN neurons expressing these neuropeptides (Madeira et al., 1997; Madeira and Paula-Barbosa, 1999). In this study, neonatal EtOH exposure induced a small, but significant, decrease in SCN neuronal density relative to that observed in SC animals. However, there were no differences in total neuronal number or the volume of the SCN between the EtOH and both control groups. Although SCN neuropeptide expression was not analyzed, preliminary evidence for neonatal EtOH-induced disturbances in other SCN circadian outputs, such as brain-derived neurotrophic factor (Earnest et al., 1997), suggests that the rhythmic regulation of these output signals may be similarly affected in EtOH animals. This decrease in SCN neuronal density may also have some relation to the increased phase-shifting responses to light in rats exposed to neonatal EtOH. Most, if not all, SCN neurons are autonomous clocks that are endogenously capable of oscillating with their own period (Welsh et al., 1995). Hence, coupling between this ensemble of suboscillators is a critical factor in determining the circadian properties of the SCN clock mechanism. The observed developmental EtOH-induced decrease in SCN neuronal density may reflect changes in intranuclear synaptic connections involved in the coupling of multiple oscillators and thereby alter circadian properties of the SCN clock, such as its phase-shifting responses to light.
Further analysis is necessary to fully identify the long-term effects of developmental EtOH exposure on circadian timekeeping and to elucidate the basic mechanisms underlying this EtOH-induced brain injury. Taken together with our preliminary findings (Earnest et al., 2001; Marchette et al., 2003), these data suggest that the SCN clock mechanism, its circadian outputs, or both are potential targets for the effects of developmental EtOH exposure on the photic regulation of the rat activity rhythm. Perturbations in the normal periodicity of circadian rhythms and their regulation by light/dark signals, whether caused by developmental EtOH exposure or other factors, would be expected to exacerbate sleep disturbances associated with normal aging and affective disorders, affect chronotherapeutic phenomena in the pharmacological treatment of disease, and impair performance in shift workers. [[Chen and West, 1999; Pittendrigh and Daan, 1976; Rollag et al., 2003]]
ACKNOWLEDGMENTS
The authors thank Rodney Walline for excellent technical assistance.
Supported by NIH Grants AA13242 (DJE) and AA05523 (JRW).
REFERENCES
- Albrecht U, Zheng B, Larkin D, Sun ZS, Lee CC. mPer1 and mper2 are essential for normal resetting of the circadian clock. J Biol Rhythms. 2001;16:100–104. doi: 10.1177/074873001129001791. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Development of the diencephalon in the rat. II. Correlation of the embryonic development of the hypothalamus with the time of origin of its neurons. J Comp Neurol. 1978;182:972–994. doi: 10.1002/cne.901820512. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, Bonthius NE, Napper RM, West JR. Early postnatal alcohol exposure acutely and permanently reduces the number of granule cells and mitral cells in the rat olfactory bulb: a stereological study. J Comp Neurol. 1992;324:557–566. doi: 10.1002/cne.903240408. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, Goodlett CR, West JR. Blood alcohol concentration and severity of microencephaly in neonatal rats depend on the pattern of alcohol administration. Alcohol. 1988;5:209–214. doi: 10.1016/0741-8329(88)90054-7. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, West JR. Alcohol-induced neuronal loss in developing rats: increased brain damage with binge exposure. Alcohol Clin Exp Res. 1990;14:107–118. doi: 10.1111/j.1530-0277.1990.tb00455.x. [DOI] [PubMed] [Google Scholar]
- Chen C, Kuhn P, Poplawski M, Advis JP, Sarkar DK. Ethanol consumption impairs the circadian rhythm of proopiomelanocortin (POMC) and Per gene expression in the hypothalamus of male rats (abstract) Alcohol Clin Exp Res. 2003;27(Suppl 5):58A. [Google Scholar]
- Chen W-JA, Berryhill EC, West JR. Zinc supplementation does not attenuate alcohol-induced cerebellar Purkinje cell loss during the brain growth spurt period. Alcohol Clin Exp Res. 2001;25:600–605. [PubMed] [Google Scholar]
- Chen W-JA, Parnell SE, West JR. Neonatal alcohol and nicotine exposure limits brain growth and depletes cerebellar Purkinje cells. Alcohol. 1998;15:33–41. doi: 10.1016/s0741-8329(97)00084-0. [DOI] [PubMed] [Google Scholar]
- Chen W-JA, Parnell SE, West JR. Effects of alcohol and nicotine on developing olfactory bulb: loss of mitral cells and alterations in neurotransmitter levels. Alcohol Clin Exp Res. 1999;23:18–25. [PubMed] [Google Scholar]
- Clarren SK, Alvord EC, Jr, Sumi SM, Streissguth AP, Smith DW. Brain malformations related to prenatal exposure to ethanol. J Pediatr. 1978;92:64–67. doi: 10.1016/s0022-3476(78)80072-9. [DOI] [PubMed] [Google Scholar]
- Daan S, Pittendrigh CS. A functional analysis of circadian pacemakers in nocturnal rodents. II. The variability of phase-response curves. J Comp Physiol. 1976;106:253–266. [Google Scholar]
- Davis FC, Gorski RA. Unilateral lesions of the hamster suprachiasmatic nuclei: evidence for redundant control of circadian rhythms. J Comp Physiol. 1984;154:221–232. [Google Scholar]
- De Vries MJ, Cardazo BN, van der Want J, de Wolf A, Meijer JH. Glutamate immunoreactivity in terminals of the retinohypothalamic tract of the brown Norwegian rat. Brain Res. 1993;612:231–237. doi: 10.1016/0006-8993(93)91665-f. [DOI] [PubMed] [Google Scholar]
- Diaz J. Experimental rearing of rat pups using chronic gastric fistulas. In: Shair HN, Barr GA, Hofer MA, editors. Developmental Psychobiology: New Methods and Changing Concepts. Oxford University Press; New York: 1991. pp. 272–284. [Google Scholar]
- Ding JM, Chen D, Weber ET, Faiman LE, Rea MA, Gillette MU. Resetting the biological clock: mediation of nocturnal circadian shifts by glutamate and NO. Science. 1994;266:1713–1717. doi: 10.1126/science.7527589. [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Earnest DJ, Chen W-JA, West JR. Developmental alcohol and circadian clock function. Alcohol Res Health. 2001;25:136–140. [PMC free article] [PubMed] [Google Scholar]
- Earnest DJ, Mahoney JC, Chen W-JA, Sohrabji F, West JR. Alcohol exposure during rapid brain development mimics the effects of aging on BDNF expression in the SCN and circadian timekeeping (abstract) Soc Neurosci Abstr. 1997;23:20. [Google Scholar]
- Goodlett CR, Johnson TB. Temporal windows of vulnerability to alcohol during the third trimester equivalent: why ‘knowing when’ matters. In: Hannigan JH, Spear LP, Spear NE, Goodlett CR, editors. Alcohol and Alcoholism: Effects on Brain and Development. Erlbaum; Mahwah, NJ: 1999. pp. 59–91. [Google Scholar]
- Goodlett CR, Kelly SJ, West JR. Early postnatal alcohol exposure that produces high blood alcohol levels impairs development of spatial navigation learning. Psychobiology. 1987;15:64–74. [Google Scholar]
- Goodlett CR, Nonneman AJ, Valentino ML, West JR. Constraints on water maze spatial learning in rats: implications for behavioral studies of brain damage and recovery of function. Behav Brain Res. 1988;28:275–286. doi: 10.1016/0166-4328(88)90130-1. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Pearlman AD, Lundahl KR. Binge neonatal alcohol intubations induce dose-dependent loss of Purkinje cells. Neurotoxicol Teratol. 1998;20:285–292. doi: 10.1016/s0892-0362(97)00102-5. [DOI] [PubMed] [Google Scholar]
- Guldner F-H. Synaptology of the rat suprachiasmatic nucleus. Cell Tissue Res. 1976;165:509–544. doi: 10.1007/BF00224478. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Bendtsen TF, Korbo L, Marcussen N, Moller A, Nielsen K, et al. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS. 1988;96:379–394. doi: 10.1111/j.1699-0463.1988.tb05320.x. [DOI] [PubMed] [Google Scholar]
- Hannibal J. Neurotransmitters of the retino-hypothalamic tract. Cell Tissue Res. 2002;309:73–88. doi: 10.1007/s00441-002-0574-3. [DOI] [PubMed] [Google Scholar]
- Harrington ME, Hoque S, Hall A, Golombek D, Biello S. Pituitary adenylate cyclase activating peptide phase shifts circadian rhythms in a manner similar to light. J Neurosci. 1999;19:6637–6642. doi: 10.1523/JNEUROSCI.19-15-06637.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattar S, Liao H-W, Takao M, Berson DM, Yau K-W. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. 2002;295:1065–1070. doi: 10.1126/science.1069609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifft JD. An autoradiographic study of the time of final division of neurons in rat hypothalamic nuclei. J Comp Neurol. 1972;144:193–204. doi: 10.1002/cne.901440204. [DOI] [PubMed] [Google Scholar]
- Johnson RF, Moore RY, Morin LP. Loss of entrainment and anatomical plasticity after lesions of the hamster retinohypothalamic tract. Brain Res. 1988;460:297–313. doi: 10.1016/0006-8993(88)90374-5. [DOI] [PubMed] [Google Scholar]
- Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in early infancy. Lancet. 1973;2:999–1001. doi: 10.1016/s0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- Karl PI, Kwun R, Slonim A, Fisher SE. Ethanol elevates fetal serum glutamate levels in the rat. Alcohol Clin Exp Res. 1995;19:177–181. doi: 10.1111/j.1530-0277.1995.tb01488.x. [DOI] [PubMed] [Google Scholar]
- Kelly SJ, Goodlett CR, Hulsether SA, West JR. Impaired spatial navigation in adult female but not adult male rats exposed to alcohol during the brain growth spurt. Behav Brain Res. 1988;27:247–257. doi: 10.1016/0166-4328(88)90121-0. [DOI] [PubMed] [Google Scholar]
- Liang FQ, Allen G, Earnest DJ. Role of brain-derived neurotrophic factor in the circadian regulation of the suprachiasmatic pacemaker by light. J Neurosci. 2000;20:2978–2987. doi: 10.1523/JNEUROSCI.20-08-02978.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira MD, Andrade JP, Lieberman AR, Sousa N, Almeida OF, Paula-Barbosa MM. Chronic alcohol consumption and withdrawal do not induce cell death in the suprachiasmatic nucleus, but lead to irreversible depression of peptide immunoreactivity and mRNA levels. J Neurosci. 1997;17:1302–1319. doi: 10.1523/JNEUROSCI.17-04-01302.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira MD, Paula-Barbosa MM. Effects of alcohol on the synthesis and expression of hypothalamic peptides. Brain Res Bull. 1999;48:3–22. doi: 10.1016/s0361-9230(98)00131-2. [DOI] [PubMed] [Google Scholar]
- Maier SE, Chen W-JA, West JR. The effect of timing and duration of alcohol exposure on development of the fetal brain. In: Abel EL, editor. Fetal Alcohol Syndrome. From Mechanism to Prevention. CRC Press; Boca Raton, FL: 1996. pp. 27–50. [Google Scholar]
- Marchette L, West JR, Chen W-JA, Earnest DJ. Developmental alcohol exposure alters the circadian regulation of rat wheel-running behavior (abstract) Alcohol Clin Exp Res. 2003;27(Suppl 5):44A. [Google Scholar]
- Mattson SN, Riley EP, Sowell ER, Jernigan TL, Sobel DF, Jones KL. A decrease in the size of the basal ganglia in children with fetal alcohol syndrome. Alcohol Clin Exp Res. 1996;20:1088–1093. doi: 10.1111/j.1530-0277.1996.tb01951.x. [DOI] [PubMed] [Google Scholar]
- Meijer JH, van der Zee EA, Dietz M. Glutamate phase shifts circadian activity rhythms in hamsters. Neurosci Lett. 1988;86:177–183. doi: 10.1016/0304-3940(88)90567-8. [DOI] [PubMed] [Google Scholar]
- Moore RY. Organization and function of a central nervous system circadian oscillator: the suprachiasmatic nucleus. Fed Proc. 1983;42:2783–2789. [PubMed] [Google Scholar]
- Nixon K, Hughes PD, Amsel A, Leslie SW. NMDA receptor subunit expression following early postnatal exposure to ethanol. Brain Res Dev Brain Res. 2002;139:295–299. doi: 10.1016/s0165-3806(02)00515-1. [DOI] [PubMed] [Google Scholar]
- Pickard GE. The afferent connections of the suprachiasmatic nucleus of the golden hamster with emphasis on the retinohypothalamic projection. J Comp Neurol. 1982;211:65–83. doi: 10.1002/cne.902110107. [DOI] [PubMed] [Google Scholar]
- Pickard GE, Turek FW. Splitting of the circadian rhythm of activity is abolished by unilateral lesions of the suprachiasmatic nuclei. Science. 1982;215:1119–1121. doi: 10.1126/science.7063843. [DOI] [PubMed] [Google Scholar]
- Pickard GE, Turek FW. Effects of partial destruction of the suprachiasmatic nuclei on two circadian parameters: wheel-running activity and short-day induced testicular regression. J Comp Physiol. 1985;156:803–815. [Google Scholar]
- Pickard GE, Weber ET, Scott PA, Riberdy AF, Rea MA. 5HT1B receptor agonists inhibit light-induced phase shifts of behavioral circadian rhythms and expression of the immediate-early gene c-fos in the suprachiasmatic nucleus. J Neurosci. 1996;16:8208–8220. doi: 10.1523/JNEUROSCI.16-24-08208.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittendrigh CS, Daan S. A functional analysis of circadian pacemakers in nocturnal rodents. V. Pacemaker structure: a clock for all seasons. J Comp Physiol. 1976;106:333–355. [Google Scholar]
- Rea MA, Glass JD, Colwell CS. Serotonin modulates photic responses in the hamster suprachiasmatic nuclei. J Neurosci. 1994;14:3635–3642. doi: 10.1523/JNEUROSCI.14-06-03635.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Ruby NF, Brennan TJ, Xie X, Cao V, Franken P, Heller HC, O’Hara BF. Role of melanopsin in circadian response to light. Science. 2002;298:2211–2213. doi: 10.1126/science.1076701. [DOI] [PubMed] [Google Scholar]
- Sei H, Sakata-Haga H, Ohta K, Sawada K, Morita Y, Fukui Y. Prenatal exposure to alcohol alters the light response in postnatal circadian rhythm. Brain Res. 2003;987:131–134. doi: 10.1016/s0006-8993(03)03329-8. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Jernigan TL, Mattson SN, Riley EP, Sobel DF, Jones KL. Abnormal development of the cerebellar vermis in children prenatally exposed to alcohol: size reduction in lobules I-V. Alcohol Clin Exp Res. 1996;20:31–34. doi: 10.1111/j.1530-0277.1996.tb01039.x. [DOI] [PubMed] [Google Scholar]
- Speh JC, Moore RY. Retinohypothalamic tract development in the hamster and rat. Brain Res Dev Brain Res. 1993;76:171–181. doi: 10.1016/0165-3806(93)90205-o. [DOI] [PubMed] [Google Scholar]
- Summer TL, Ferraro JS, McCormack CE. Phase-response and Aschoff illuminance curves for locomotor activity of the rat. Am J Physiol. 1984;246:R299–R304. doi: 10.1152/ajpregu.1984.246.3.R299. [DOI] [PubMed] [Google Scholar]
- Tenkova T, Young C, Dikranian K, Labruyere J, Olney JW. Ethanol-induced apoptosis in the developing visual system during synaptogenesis. Invest Ophthalmol Vis Sci. 2003;44:2809–2817. doi: 10.1167/iovs.02-0982. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Goodlett CR, West JR. Alcohol-induced Purkinje cell loss depends on the developmental timing of alcohol exposure and correlates with motor performance. Brain Res Dev Brain Res. 1998;105:159–166. doi: 10.1016/s0165-3806(97)00164-8. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Wasserman EA, West JR, Goodlett CR. Behavioral deficits induced by bingelike exposure to alcohol in neonatal rats: importance of developmental timing and number of episodes. Dev Psychobiol. 1996;29:433–452. doi: 10.1002/(SICI)1098-2302(199607)29:5<433::AID-DEV3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Turek FW. Circadian neural rhythms in mammals. Annu Rev Physiol. 1985;47:49–64. doi: 10.1146/annurev.ph.47.030185.000405. [DOI] [PubMed] [Google Scholar]
- van den Pol AN. The hypothalamic suprachiasmatic nucleus of rat: intrinsic anatomy. J Comp Neurol. 1980;191:661–702. doi: 10.1002/cne.901910410. [DOI] [PubMed] [Google Scholar]
- van den Pol AN. Glutamate and GABA presence and action in the suprachiasmatic nucleus. J Biol Rhythms (Suppl) 1993;8:S11–S15. [PubMed] [Google Scholar]
- Wakamatsu H, Takahashi S, Moriya T, Inouye ST, Okamura H, Akiyama M, Shibata S. Additive effect of mPer1 and mPer2 antisense oligonucleotides on light-induced phase shift. Neuroreport. 2001;12:127–131. doi: 10.1097/00001756-200101220-00033. [DOI] [PubMed] [Google Scholar]
- Weber ET, Gannon RL, Rea MA. Local administration of serotonin agonists blocks light-induced phase advances of the circadian activity rhythm in the hamster. J Biol Rhythms. 1998;13:209–218. doi: 10.1177/074873098129000057. [DOI] [PubMed] [Google Scholar]
- Weber ET, Rea MA. Neuropeptide Y blocks light-induced phase advances but not delays of the circadian activity rhythm in hamsters. Neurosci Lett. 1997;231:159–162. doi: 10.1016/s0304-3940(97)00559-4. [DOI] [PubMed] [Google Scholar]
- Welsh DK, Logothetis DE, Meister M, Reppert SM. Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron. 1995;14:697–706. doi: 10.1016/0896-6273(95)90214-7. [DOI] [PubMed] [Google Scholar]
- West MJ. Use of pup in a cup model to study brain development. J Nutr. 1993;123:382–385. doi: 10.1093/jn/123.suppl_2.382. [DOI] [PubMed] [Google Scholar]
- West JR, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- West JR, Hidges CA, Black AC., Jr Prenatal exposure to alcohol alters the organization of hippocampal mossy fibers in rats. Science. 1981;211:957–959. doi: 10.1126/science.7466371. [DOI] [PubMed] [Google Scholar]