

Abstract
Innate immune system activation is a critical step in the initiation of an effective adaptive immune response; therefore, activation of a class of innate pathogen receptors called pattern recognition receptors (PRR) is a central feature of many adjuvant systems. It has recently been shown that one member of an intracellular PRR, the NLRP3 inflammasome, is activated by a number of classical adjuvants including aluminum hydroxide and saponins [1, 2]. Inflammasome activation in vitro requires signaling of both the Toll-like receptor (TLR) and NLRP3 in antigen-presenting cells. Here we present a class of nanomaterials endowed with these two signals for rapid optimization of vaccine design. We constructed this system using a simple approach that incorporates lipopolysaccharides (LPS) onto the surface of nanoparticles constructed from a biocompatible polyester, poly (lactic-co-glycolic acid) (PLGA), loaded with antigen. We demonstrate that LPS-modified particles are preferentially internalized by dendritic cells compared to uncoated nanoparticles and the system, when administered to mice, elicits potent humoral and cellular immunity against a model antigen, ovalbumin. Wild type macrophages pulsed with LPS-modified nanoparticles resulted in production of the proinflammatory cytokine IL-1β consistent with inflammasome activation. In comparison, NLRP3-deficient and caspase-1-deficient macrophages showed negligible production of IL-1β. Furthermore, when endocytosis and lysosomal destabilization were inhibited, inflammasome activity was diminished, supporting the notion that nanoparticles rupture lysosomal compartments and behave as ‘danger signals’[3]. The generality of this vaccination approach is tested by encapsulation of a recombinant West Nile envelope protein and demonstrated by protection against a murine model of West Nile encephalitis. The design of such an antigen delivery mechanism with the ability to stimulate two potent innate immune pathways represents a potent new approach to simultaneous antigen and adjuvant delivery.
Keywords: PLGA, inflammasome, West Nile virus, Nanoparticles
1. Introduction
Pathogens are continually emerging and changing; therefore, there is a need for flexible vaccine delivery systems. Vaccines have eliminated smallpox and nearly eliminated polio, two of the worst global infectious diseases. By contrast, vaccines for many other infectious diseases, such as human immunodeficiency virus (HIV) and malaria, are poorly developed or simply unavailable [4, 5]. There are a number of significant scientific challenges that have limited the development of vaccines for deadly diseases. First, few if any approaches are available that efficiently prime cell-mediated immunity by direct intracellular delivery of an antigen. Second, ‘tunable’ adjuvants, that is, adjuvants that can be engineered to optimize the magnitude and direction of an immune response are not available [6, 7]. Third, most vaccine approaches are parenteral (i.e. subcutaneous or intramuscular injection) which has made it difficult to deploy vaccines in underdeveloped countries where medical support systems, resources, and even refrigeration are limited. Therefore, there is a critical need for safe and stable vaccine systems that would address these factors.
Several key variables are assembled and integrated in the design of current vaccines [8, 9]. The first variable is the form of the antigen itself, which can be whole inactivated or attenuated organisms, purified proteins and peptides, or DNA encoded antigens. Purified antigens often become less immunogenic compared to whole pathogens or crude extracts, necessitating a means to amplify the immune response against the purified subunit antigen. Thus, a second necessary component of a vaccine involves potentiating or stimulating the innate and adaptive arms of the immune system to the antigen subunit [8, 9]. Immune potentiators may include bacterial products, toxins, or other molecules that augment specific immunity. Finally, to effect optimal stimulation to a given antigen, a formulation is needed that delivers the correct amount of antigen in a repetitive or sustained fashion to the appropriate immune cells and to the appropriate compartments within those cells. A vaccine vehicle should facilitate delivery of both antigen and immune potentiator molecules preferably to target cells of the immune system.
Because of the historical emphasis on eliciting humoral immune responses, current adjuvants, represented predominately by aluminum adjuvants (alum), are optimized for effective induction of Th2-biased responses and high antibody serum titers, but are less effective at eliciting a strong T cell-mediated immune response. While soluble antigen is poorly presented on MHC class I to stimulate cytotoxic T lymphocytes, it has been demonstrated that internalized antigen encapsulated in polymer particles can be effectively cross-presented on MHC Class I [10, 11] yielding an effective CD8+ T cell response [12, 13].
Aluminum adjuvants while having a limited capacity to adsorb many antigens [14, 15] have been a mainstay in current vaccine formulations. One reason for this is that alum is generally safe to administer and a good humoral immune potentiator. Its adjuvanicity has recently been attributed to NLRP3 inflammasome activation [1, 2]. The inflammasome is an intracellular multiprotein complex that mediates caspase-1 cleavage of the inactive precursor of the proinflammatory cytokine, IL-1β, resulting in release of mature IL-1β [16]. Inflammasome-mediated cleavage of pro-IL-1B in vitro depends on signals that activate both TLR and NOD-like receptors (NLR), such as NLRP3. Activation of these innate immune system receptors is now recognized to be a prerequisite of effective adaptive immunity through provision of the necessary combination of stimuli for naïve T cells.
We hypothesized that antigen-loaded nanoparticles constructed from poly(lactic-co-glycolic) (PLGA), a biodegradable and biocompatible, FDA-approved polymer and LPS would prove to be an effective vaccine vector via both TLR and inflammasome activation. Unlike traditional adjuvants, PLGA particles allow for targeted delivery, protection, and sustained release of antigen during vaccination. Here we show that LPS-modified nanoparticles effectively enter antigen-presenting cells (APCs) and elicit both humoral and cellular immunity against encapsulated antigens in mice. We demonstrate the modularity of this system for vaccine development by encapsulation of a West Nile virus envelope protein antigen (rWNVE) and the induction of protection in a murine model of West Nile Encephalitis.
2. Materials and Methods
2.1 Materials
50:50 PLGA with an inherent viscosity of 0.59 dL/g, was purchased from Lactel Polymers, Inc. (Pelham, AL, USA). Polyvinyl alcohol (PVA) (Mw average 30–70 kD), LPS (Escherichia coli strain 0111:B4), LPS-FITC, chicken egg ovalbumin (OVA), rhodamine B, and Nile red were all obtained from Sigma-Aldrich. Methylene chloride was of chromatography grade and supplied by Fisher Scientific. All other reagents were of reagent grade and used as received. Recombinant West Nile virus envelope protein antigen (rWNVE) was made in Drosophila S2 cells as described previously [17].
2.2 Preparation of LPS-modified biodegradable nanoparticles
We used a modified water-in-oil-in-water (W/O/W) emulsion method for preparation of LPS-modified PLGA particles. In the first emulsion (W/O), super-concentrated OVA (100 mg/ml ) or rWNVE (20 mg/ml) in phosphate-buffered saline (PBS) was added drop-wise to a vortexing PLGA solution (2 ml) dissolved in methylene chloride. To facilitate nanoparticle detection by fluorescence, rhodamine B or Nile red (0.5 mg) was added to the polymer solution. Polymer and encapsulant were added drop-wise to 5% PVA in the second emulsion (W/O/W). After each emulsion, the samples were sonicated for 30 s on ice using a Tekmar Sonic Distributor fitted with a model CV26 sonicator – amplitude set at 38%. The second emulsion was rapidly added to 0.3% PVA. This external phase underwent vigorous stirring for 3 h at constant room temperature to evaporate methylene chloride. LPS-modified particles were prepared with LPS (20 mg/ml in de-ionized (DI) water) added to the second emulsion containing 5% PVA. Particles were collected at 12,000 rpm for 15 min and washed with DI water three times. The particles were freeze dried and stored at −20°C for later use.
2.3 Characterization of LPS modified-biodegradable nanoparticles
OVA-loaded nanoparticles were visualized by scanning electron microscopy (SEM) and size was assessed by ImageJ software as previously described [18]. Protein encapsulation was quantified using a Micro BCA Protein Assay (Pierce) after dissolving the particles in 0.05 N NaOH with 1% SDS. Release from nanoparticles was measured by performing the Micro BCA Protein Assay on supernatant from nanoparticles incubating in PBS at 37°C over 3 weeks. To quantify the amount of LPS incorporated into the biodegradable particles were prepared LPS-modified nanoparticles with 5% fluorescently labeled LPS (LPS-FITC). The amount of LPS-FITC on particles was calculated from a standard obtained with LPS-FITC and blank particles. Endotoxin content of dissolved LPS-modified and unmodified nanoparticles was confirmed by the Limulus Amebocyte Lysate endotoxin test (Cambrex).
2.4 Uptake studies of fluorescent LPS-modified nanoparticles by dendritic cells
Uptake of particles by dendritic cells (DCs) was assessed by confocal microscopy and flow cytometry. DC2.4, a cell line that presents exogenous antigen on MHC Class I and II molecules, were incubated at 37°C until adherent on glass coverslips in RPMI with 10% fetal bovine serum (FBS) and 2% penicillin-streptomycin (P/S). Cells were primed with rhodamine B-loaded, LPS-modified (LPS/RhodB) and unmodified (-/RhodB) nanoparticles at desired concentrations for 2 h, and were fixed in 4% paraformaldehyde. Cells were permeablized with 1X PBS with 0.1% Triton-X-100 and counterstained with DAPI (nuclear stain) and Phalloidin-FITC (cytoskeleton stain) to visualize cellular viability and morphology. After washing, cells were imaged with a spinning-disk fluorescent confocal microscope (Zeiss).
For flow cytometry experiments DC2.4 were grown to confluence at 37°C in RPMI media with 10% FBS and 2% P/S [11]. Cells were incubated for 2 h with various concentrations of LPS-modified (LPS/Nile red) and unmodified (−/Nile red) nanoparticles loaded with Nile red dye. Subsequently, cells were washed in phosphate buffered saline, fixed at 4°C in 1% paraformaldehyde, and analyzed by flow cytometry using a FACSCalibur system (BD). Nile red fluorescence was detected in the FL2 channel. Significance was analyzed by the student t-test using GraphPad, Prism, version 4.0b.
2.5 Assessment of inflammasome activation
Wild type (WT) (C57Bl/6), NLRP3-deficient, and caspase-1-deficient mice were administered 3% thioglycollate via intraperitoneal injection. After 5 days, macrophages were isolated and transferred to 24-well plates at 5 × 105 cells/well in Dulbecco’s Modified Eagle Media with 10% fetal calf serum, 1% penicillin/streptomycin, and 1% L-glutamine. Cells in triplicate were incubated overnight with 50 ng/ml LPS (Invivogen) or media alone followed by 24 hour of priming with 250 μg of either LPS-modified, OVA-loaded particles; unmodified OVA-loaded particles; or pulsed with ATP for 20 minutes, as a positive control. Cells were incubated with 10 μM for 1 h prior to particle incubation with cytochalasin D (Sigma-Aldrich) or CA-074 Me (Peptide Institute, Osaka, Japan), to inhibit actin polymerization and the lysosomal protease, cathepsin B, respectively. A similar experiment was performed with bone marrow-derived dendritic cells (BMDCs), that were isolated from C57Bl/6 mice by a method previously described [19]. BMDCs were given fresh RPMI media, every two days, supplemented with 10% FBS, L-glutamine, MEM non-essential amino acids, HEPES buffer, gentamicin, β-mercaptoethanol, and 12.5 ng/ml rGM-CSF for 7 days post-isolation. Cells were then pre-stimulated overnight with LPS, as described above, followed by an incubation period with 250 μg particles or Alhydrogel (1.3%) (Accurate Chemicals), preceeded by a pulse with inhibitors, when noted, at the concentrations listed previously. In all groups, supernatant was collected at 24 hours post-particle incubation and analyzed for IL-1β by ELISA (R&D Systems). All experiments were repeated at least twice with reproducible results.
2.6 Antigen-specific CD8+ T cell stimulation
DC2.4 were incubated at 37°C at 1×105/well in 96-well plates until adherent. Cells were pulsed with LPS-modified, OVA-loaded particles (LPS/OVA); unmodified, OVA-loaded particles (−/OVA); blank particles (−/−) with soluble OVA and soluble LPS; soluble OVA and soluble LPS; or soluble OVA for 2 h at 37°C in triplicate. The amount of soluble OVA and LPS was equivalent to the level of these molecules in the particles. Cells were washed and co-cultured with 2×105 splenocytes/well from an OTI mouse – a strain that expresses a transgenic T cell receptor specific for the OVA peptide SIINFEKL in MHC Class I. After 48 h, the supernatant was analyzed for T cell stimulation by IFN-γ secretion using an ELISA kit (BD Pharmingen). This experiment was performed twice on separate occasions with the same findings.
2.7 Animal vaccination
C57BL/6 mice at 6–8 weeks of age were purchased from Charles River Laboratories for all vaccination experiments. National Institute of Health guidelines for the care and use of laboratory animals were observed. All animals were maintained under specific pathogen-free conditions and routinely checked by the Yale University Animal Resource Center staff. For subcutaneous (s.c.) vaccination studies with OVA antigen-loaded particles, animals (n=5 per group) were injected with a single dose of 100 μg of OVA at the base of the tail with either OVA-loaded nanoparticles (−/OVA); LPS-modified, OVA-loaded nanoparticles (LPS/OVA); OVA in a 20mM Tris-HCl, 100mM NaCL buffer absorbed to 100 μL Alhydrogel; or OVA emulsified in Complete Freund’s Adjuvant according to the manufacturers diferections (Pierce). LPS-modified, ”empty” nanoparticles were used as a negative control. Blood and spleens were harvested at 2 weeks post-vaccination. Vaccination with the OVA antigen were repeated twice.
For subcutaneous vaccinations with the recombinant West Nile viral antigen, rWNVE, mice (n=10) received a single-dose of antigen-loaded, LPS-modified particles (LPS/rWNVE) (20 μg) in PBS at the base of the tail. Mice vaccinated intranasally (n=10) were administered 25 μg of antigen encapsulated in LPS/rWNVE in 20 μl of PBS in small droplets to the tip of the nose with a micropipette, allowing the unanesthetized mice to inhale the dose. Mice vaccinated orally (n=10) were given 150 μg of encapsulated antigen in LPS/rWNVE by oral gavage in 300 μl of PBS. To demonstrate the protection of these particles during oral vaccination, 150 μg of rWNVE was administered by oral gavage. Intranasally and orally vaccinated animals were given an identical booster dose at 2 weeks post-initial vaccination. LPS/OVA particles were administered s.c. as a control. Mice were injected intraperitoneally with 1000 PFU of WN virus isolate 2741 at 2 weeks or 4 weeks post-initial vaccination for s.c. and oral/nasal vaccinations, respectively. Mice were monitored daily for morbidity and mortality for 21 days post-challenge. Survival results are the combined curves from two experiments (total n per group=20). Statistical analysis was performed by the Logrank (Mantel-Cox) test using GraphPad, Prism, version 4.0b.
2.8 Analysis of serum antigen-specific antibodies
Blood was collected retro-orbitally at week 2 for OVA vaccinations and s.c. rWNVE vaccinations and at week 4 for nasal and oral rWNVE vaccinations. Samples were incubated at 4°C overnight and centrifuged at 3000 rpm for 10 min. Serum was isolated and stored at −80°C for later analysis. Antigen-specific IgG titers were analyzed by ELISA. In brief, plates were coated with 100 ng OVA or rWNVE per well at 4°C overnight and blocked using 1X PBS with 10% goat serum. Serum (unpooled) was diluted in blocking buffer and added to the washed plate. After 2 h incubation at room temperature, plates were again washed and incubated with an excess of horseradish peroxidase-conjugated goat anti-mouse IgG (H+L) (Zymed) for 1 h. Enzymatic activity was assessed using TMB substrate. End-point antibody titer was the average reciprocal dilution that corresponded to an absorbance two standard deviations above the control (LPS/−) absorbance. Data shown are the average titer from a single experiment. Two identical experiments were performed with the same outcome.
2.9 Antigen-specific T cell stimulation in OVA-vaccinated mice
Mice were sacrificed 2 weeks post-vaccination. Splenocytes were isolated and pooled after red blood cell lysis with ACK lysis buffer (Quality Biological). Splenocytes were cultured in 96-well, round bottom plates at 2×105 cells/well with 100 μg/ml OVA in RPMI + 10% FBS supplemented with L-glutamine, MEM non-essential amino acids, HEPES buffer, gentamicin, and β-mercaptoethanol. Supernatant was collect at 48 h and analyzed for IFN-γ content by ELISA (BD Pharmingen). As with serum IgG analysis, data shown are represntative of mulitple experiments.
3. Results
3.1 Characterization of LPS-modified nanoparticles
LPS-modified and unmodified OVA-loaded nanoparticles (Fig. 1A) exhibited a characteristic sustained release of encapsulated antigen at a statistically similar rate (Fig. 1B). Because our strategy for formulating the vaccine particle involved simultaneous encapsulation and surface modification, we examined how the addition of LPS might affect the encapsulation efficiency of the model antigen, ovalbumin (OVA). The amount of encapsulated OVA in modified and unmodified PLGA nanoparticles was 44 ± 0.4 μg and 37 ± 0.1 μg per 1 mg of particles, respectively; therefore, LPS functionalization enhanced OVA encapsulation by about 16 percent. Others have found similar effects on encapsulation efficiency of molecules with the addition of fatty acids or lipids to PLGA during the encapsulation process[20, 21]. A possible mechanism for this effect might involve increased particle stability due to the presence of amphipathic molecules such as LPS, facilitating enhancements in PLGA particle formation and encapsulation efficiency [22]. LPS-modified nanoparticles loaded with rWNVE contained 17 ± 0.2 μg of antigen per 1 mg of particles. LPS-FITC was used as a means to quantify the amount of LPS incorporated on the surface. Total LPS incorporation was found to be roughly 13 μg LPS per mg of particles and activity was confirmed by the Limulus Amebocyte Lysate endotoxin assay. While ovalbumin from Sigma does contain LPS, the endotoxin activity of unmodified, OVA-loaded nanoparticles was far less (12.7 ± 5.2 EU/mg) compared to LPS-modified, OVA-loaded nanoparticles (641 ± 0.4 EU/mg). Particles displayed a size distribution in the range of 100–400 nm in diameter (Fig. 1C,D).
Figure 1.
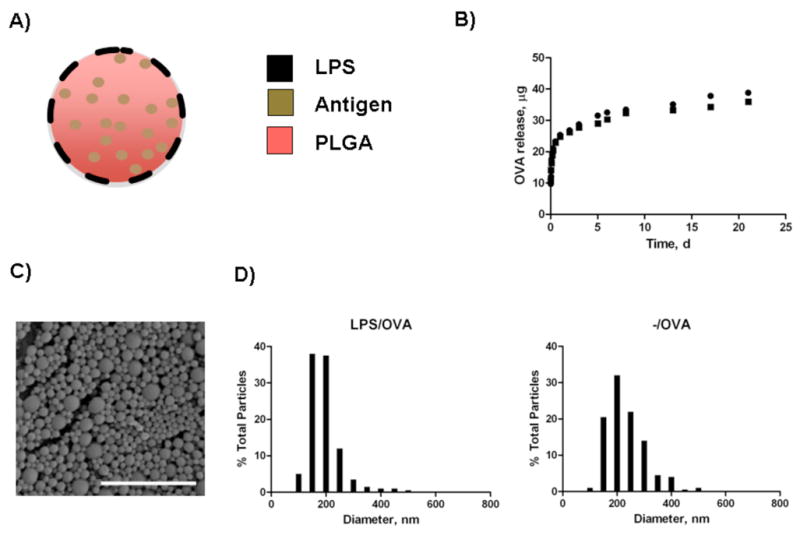
Characterization of nanoparticles. A) Schematic of biodegradable particles with lipopolysaccharide modification. B) Controlled release profile of OVA from LPS/OVA (●) and -/OVA (■) particles. Figure represents release from nanoparticles at 37°C in phosphate-buffered saline in triplicate over 3 weeks. C) Scanning Electron Microscopy (SEM) image of LPS-modified nanoparticles. Scale bar is 2 μm. D) Size distribution profiles for LPS-modified and unmodified OVA-loaded nanoparticles.
3.2 LPS–modified particles are efficiently internalized by dendritic cells compared to unmodified particles
To study the effect of LPS surface modification on the internalization of particles by dendritic cells, particles with and without LPS were incubated with DCs and internalization was assessed by microscopy and flow cytometry. LPS-modified nanoparticles were internalized more effectively than unmodified particles after 2 hours of incubation (Fig. 2A,B). In Fig. 2A, representative images showed DCs internalized a high amount of LPS-modified nanoparticles, whereas unmodified nanoparticles were less likely to be endocytosed. These findings are consistent with previous reports of enhanced macrophage phagocytosis upon TLR stimulation [23]. Flow cytometry analysis of cells pulsed with nanoparticles at various concentrations supported the preferential internalization of LPS-modified versus unmodified particles (P=0.09) (Fig. 2B).
Figure 2.
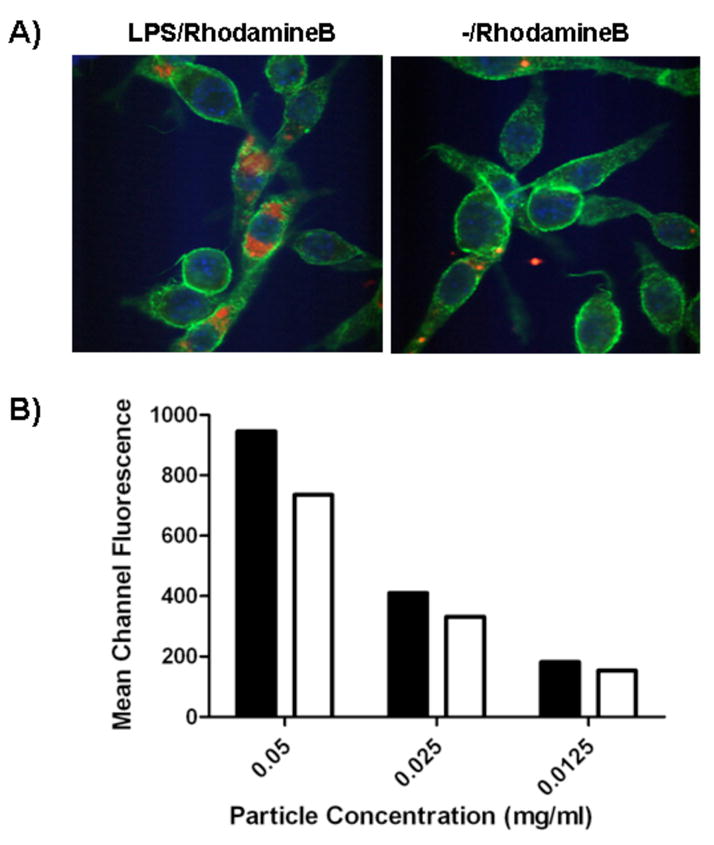
Evaluation of uptake efficiency. A) Dendritic cells were incubated with nanoparticles encapsulating rhodamine B and analyzed by confocal microscopy (green: phalloidin-FITC, blue: DAPI, and red: rhodamine B). A representative image is shown. B) Flow cytometry analysis of dendritic cells pulsed with Nile red-loaded nanoparticles: LPS/Nile red (■) and -/Nile red (□). Mean fluorescence in FL2 is shown.
3.3 LPS-modified particles stimulate the inflammasome
To assess if nanoparticles stimulated the NLRP3 inflammasome pathway (Fig. 3A), macrophages were isolated from WT; NLRP3-deficient; and caspase-1-deficient mice, treated with modified and unmodified nanoparticles, and analyzed for IL-1β secretion (Fig. 3B). Wild-type macrophages, but neither NLRP3-deficient nor caspase-1-deficient macrophages, exhibited a significant level of IL-1β secretion post-incubation with LPS-modified particles with and without prior LPS stimulation. Thus, the LPS-modified particles alone offered both signals necessary to activate the TLR and NLRP3 inflammasome pathways, unlike alum[1], which requires LPS stimulation in vitro and endogenous signals in vivo for inflammasome activation. Unmodified particles did not produce significant levels of the inflammatory cytokine in WT, NLRP3-deficient, or caspase-1-deficient mice even with pre-stimulation with soluble LPS, likely because of poor uptake by APCs as demonstrated in Figure 2. This finding likely explains why previous studies of the inflammatory properties of polystyrene nanoparticles have failed to find a role for the NLRP3 inflammasome [24].
Figure 3.
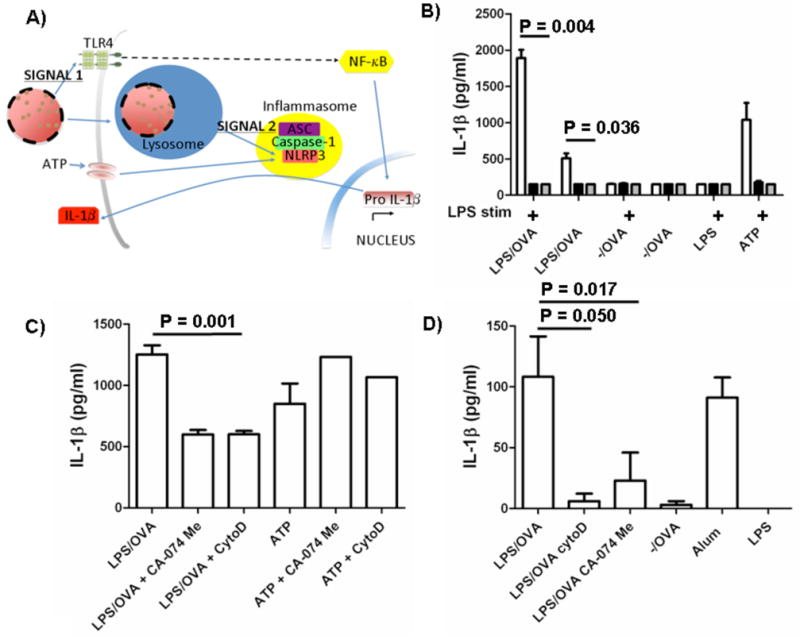
LPS-modified nanoparticles activated the NLRP3 inflammasome. A) Proposed mechanism of activation. Signal 1: LPS triggers Toll-like receptor 4 (TLR4) signaling, which ultimately activates the transcription factor, NF-κB. Pro-IL-1β is upregulated and is cleaved by caspase-1 in an inflammasome complex. Signal 2: Nanoparticles are phagocytosed by cells, disrupt lysosomal compartments, and activate the NLRP3 receptor. NLRP3 recruits ASC, an adapter molecule, which binds caspase-1. B) Macrophages from WT (□), NLRP3-deficient ( ) and caspase-1-deficient (■) mice were isolated and incubated with LPS-modified and unmodified OVA-loaded particles with and without prior stimulation with soluble LPS. ATP with LPS pre-stimulation was used as a positive control. After 24 h, supernatant was analyzed for IL-1β by ELISA. C) WT macrophages and D) BMDCs were pre-stimulated with LPS and then treated with cytochalasin D (cytoD), to inhibit internalization; CA-074 Me, to inhibit cathepsin B; or media alone. After incubation with particles or alum for 24 h or ATP for 20 min, supernatant was analyzed for IL-1β.
) and caspase-1-deficient (■) mice were isolated and incubated with LPS-modified and unmodified OVA-loaded particles with and without prior stimulation with soluble LPS. ATP with LPS pre-stimulation was used as a positive control. After 24 h, supernatant was analyzed for IL-1β by ELISA. C) WT macrophages and D) BMDCs were pre-stimulated with LPS and then treated with cytochalasin D (cytoD), to inhibit internalization; CA-074 Me, to inhibit cathepsin B; or media alone. After incubation with particles or alum for 24 h or ATP for 20 min, supernatant was analyzed for IL-1β.
Recent data have shown that silica crystals and aluminum salts both disrupt lysosomes, propagating an endogenous ‘danger signal’ to the NLRP3 inflammasome [3]. We treated WT macrophages and BMDCs with cytochalasin D, an inhibitor of actin polymerization, or CA-074 Me, an inhibitor of cathepsin B (a lysosomal protease thought to play a role in lysosomal destabilization), to see how inflammasome activity was affected. Both inhibitors abated, but did not eliminate, the IL-1β response to LPS-modified nanoparticles (Fig. 3C,D). This partial response was also observed by Hornung et al [3]. As predicted, inflammasome activity was not hindered in cells treated with both inhibitors followed by ATP, a NLRP3 inflammasome stimulus that is not endocytosed and would, therefore, not be affected by actin polymerization or lysosomal stabilization (Fig. 3C). Lastly, these data support that alum, like LPS/OVA nanoparticles, stimulates the inflammasome (Fig. 3D).
3.4 Dendritic cells primed with LPS-modified particles induce antigen-specific CD8+ T cell responses
Next we evaluated the efficacy of this system in cross-presentation of antigenic epitopes and stimulation of a CD8+ T cell response. For this we studied OVA-specific T cell stimulation by DCs primed with LPS-modified and unmodified nanoparticles loaded with OVA. DC2.4 were incubated with LPS/OVA nanoparticles, −/OVA nanoparticles, soluble OVA and LPS with blank nanoparticles, soluble OVA and LPS, and OVA alone; and then co-cultured with splenocytes from an OTI mouse. While incubation with both modified and unmodified nanoparticles yielded higher T cell responses than soluble antigen as assessed by IFN-γ secretion, it was clear that the LPS/OVA nanoparticles were more effective (Fig. 4A). DCs primed with soluble OVA, or even soluble OVA and free LPS were significantly less effective in stimulating antigen-specific lymphocytes. We note that equivalent amounts of OVA and LPS given with blank PLGA nanoparticles (−/−) were not nearly as effective as the integrated LPS-modified, OVA-loaded particles and that a 60-fold increase in soluble OVA was necessary to generate an equivalent response to the one obtained with LPS-modified nanoparticles with 50 μg/ml encapsulated OVA (Fig. 4B).
Figure 4.
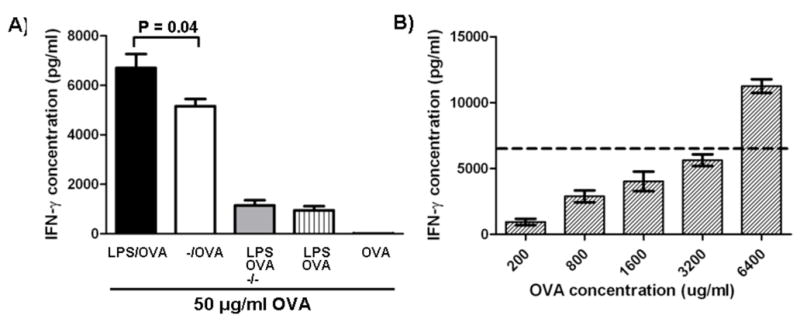
In vitro vaccination with nanoparticles. A) Dendritic cells were pulsed with −/OVA, LPS/OVA, −/− with soluble OVA and LPS, soluble OVA and soluble LPS, or soluble OVA at an OVA concentration of 50 μg/ml. Cells were then co-cultured with OT-1 transgenic splenocytes. Supernatant IFN-γ was quantified by ELISA after 48 hours. B) Dendritic cells were pulsed with soluble OVA at high concentrations and then co-cultured with OT-1 splenocytes in order to achieve similar levels of T cell activity. Dotted line represents IFN-γ levels from cells pulsed with LPS/OVA from (A).
3.5 Early humoral and cellular immune response induction with LPS-modified nanoparticles
To assess the efficacy of the particle system in the induction of early immune responses, we analyzed serum antigen-specific IgG from mice vaccinated subcutaneously with a single dose of modified and unmodified nanoparticles at 2 weeks post-immunization (Fig. 5A). A significant increase in OVA-specific IgG was observed in mice given LPS-modified particles by subcutaneous injection compared to unmodified particles. Similar titers of IgG were observed after vaccination with LPS-modified particles, OVA absorbed to Alum, and OVA in CFA.
Figure 5.

OVA-specific immune responses 2 weeks after subcutaneous vaccination. Animals received a single vaccination by s.c. injection with particle formulations, alum, or CFA as adjuvants. A) Serum antibody titers were analyzed by ELISA. Data shown are mean OVA-specific IgG titer. B) Splenocytes were stimulated ex vivo with OVA and supernatant was analyzed for IFN-γ secretion at 48 h by ELISA. For all groups: n=5. Experiments were repeated 2 more times (with n=4) with similar results.
Splenocytes from vaccinated animals were stimulated ex vivo with OVA. Antigen-stimulated splenocytes from mice administered LPS/OVA showed greater antigen-specific stimulation in the form of IFN-γ secretion compared to animals vaccinated with -/OVA (Fig. 5B), suggesting that modified particles induced a detectable cellular response in addition to a humoral response. Moreover, while IgG titers were indistinguishable between adjuvantation with LPS-modified particles, alum, and CFA; only LPS-modified particles and CFA displayed a robust cellular response.
3.6 Vaccination with West Nile viral antigen-loaded LPS-modified nanoparticles provided protection to viral challenge
Animals were vaccinated subcutaneously, intranasally, and orally with LPS/rWNVE nanoparticles and compared with a control group of mice injected subcutaneously with LPS/OVA nanoparticles. Animals administered with LPS/rWNVE subcutaneously had IgG titers about 40-fold higher than the control mice (Fig. 6A) and over 20 fold higher than mice given soluble rWNVE subcutaneously (data not shown). Animals intranasally and orally vaccinated with LPS/rWNVE had, in comparison, much lower IgG titers than animals vaccinated subcutaneously, though significantly above the control. In a separate study (Supp. Fig. A) mice administered LPS/rWNVE particles showed an elevated cellular response compared to unmodified particles and rWNVE absorbed to alum. Survival curves (Fig. 6B) show that the nanoparticles when injected subcutaneously confer almost complete protection to a viral challenge after a single dose. Intranasal and oral vaccination with nanoparticles resulted in 80 and 75 percent survival, respectively. Oral vaccination with rWNVE alone resulted in 60 percent survival, indicating that particles may be effective vehicles for transporting antigen across epithelial barriers and may provide antigen protection in the gut. All survival curves from vaccinated mice were significantly different from the control (p=0.0003), by the Logrank (Mantel-Cox) test.
Figure 6.

Antigen-specific IgG and survival in mice after vaccination with encapsulated West Nile virus envelope protein antigen. Mice (n=20) were vaccinated with LPS/rWNVE subcutaneously (s.c.), orally, or intranasally (i.n.). As a negative control, mice were subcutaneously administered LPS/OVA particles. A) Anti-rWNVE IgG titers in the serum pre-challenge were quantified by ELISA. B) Mice were challenged with 1000 PFU of isolate 2741 and monitored for mortality for 21 days. LPS/OVA (●); rWNVE alone ( ); LPS/rWNVE s.c. (
); LPS/rWNVE s.c. ( ); LPS/rWNVE oral (
); LPS/rWNVE oral ( ); and LPS/rWNVE nasal (
); and LPS/rWNVE nasal ( ;). Results are combined data from two experiments (n=10).
;). Results are combined data from two experiments (n=10).
4. Discussion
An approach for constructing vaccine delivery systems in which antigen, immune-potentiator, and delivery vehicle incorporated in a single nanodevice is attractive because it allows control over variables that are important in optimizing an effective vaccine delivery system. As discussed in this study, such systems may be engineered to induce inflammasome activation, much the same as aluminum adjuvants, but can also be designed to activate multiple arms of the innate immune system.
Inflammasome activation leading to the production of the potent inflammatory cytokine IL-1β is a manifestation of activated NOD-like receptor (NLR) signaling mediated by cytosolic ‘danger’ signals. These NLR signals are still being identified but are now known to include alum, silica, and certain microbial products [1, 24]. In vitro, TLR signaling is a prerequisite for IL-1β induction; therefore, we conjectured that integrating TLR and NLR ligands with antigen may provide a highly effective vaccine vector. Results from this study argue for the success of this approach but also highlight a striking effect regarding the importance of combining these signals in a nanoparticulate assembly. LPS-modification effected an elevated rate of nanoparticle internalization by antigen-presenting cells and subsequent T cell activation. Soluble LPS, ovalbumin, and blank PLGA particles (TLR signal, antigen, and NLR activating ligand) did not yield the elevated T cell responses observed with the LPS-modified, OVA-loaded nanoparticle complex. In addition, cellular and humoral responses were markedly higher after subcutaneous vaccination with LPS-modified particles encapsulating ovalbumin compared to unmodified particles. Even in the absence of an antigen boost, the animals displayed antibody titers competitive with mice given OVA in alum or Complete Freund’s Adjuvant. Moreover, immunization with LPS-modified particles resulted in a cellular response comparable to that from CFA, and much higher than alum, the only FDA-approved adjuvant at present.
Our group previously developed a method for functionalizing PLGA particles with amphiphilic protein-fatty acid conjugates [18]. We hypothesized that this method would apply to other amphiphilic molecules, such as LPS, for targeting antigen presenting cells such as macrophages and dendritic cells, creating simple vaccine delivery systems that combine antigen, immunostimulatory agent, and vehicle all in a single nanodevice. Other groups have devised similar systems for incorporating TLR ligands such as CpG oligonucleotides [25–27], a ligand for TLR9 or mono-phosphoryl lipid A (MPL) and RC529, derivatives of LPS [21, 28] in biodegradable particulates. Although CpG is a relatively non-cytotoxic alternative, TLR9 is an intracellular receptor [29] and unlike TLR4, is therefore unable to facilitate entry into the cell. While MPL and RC529 were devised to combat the toxicity of LPS, these molecules are generally considered less potent adjuvants than LPS. A study by Thompson, BS et al [30] concluded that mice given LPS as a vaccine adjuvant had 6 fold higher CD4+ T cell responses compared to mice given MPL and even more of a difference was observed with RC529. Similarly, a number of groups have reported that levels of cytokines relevant to successful vaccination are much lower after treatment with MPL compared to the same amount of LPS [31, 32]. Cytotoxicity analysis showed cellular viability at nanoparticle concentrations used in this study (data not shown). Furthermore, no significant cytotoxicity difference between LPS-modified and unmodified particles was observed. Biodegradable particles surface-modified with LPS largely evade the toxicity associated with the free form of LPS. This feature has been demonstrated previously: incorporation of LPS into liposomes decreased its toxicity 10-fold compared to its free form[33] and by 1000 fold in endotoxin activity [34]. Thus, this system minimizes the negative effects of LPS while maintaining the potency of the whole molecule.
To the best of our knowledge, this is the first report that shows inflammasome activity with the commonly utilized PLGA nanomaterials. This is also the first report to exploit the efficacy of particulate delivery systems as a vaccine vectors against West Nile (WN) virus, a flavivirus, currently without an available vaccine or specific therapy. Examples of flaviviruses include many human pathogens of global epidemiological importance, such as the agents of dengue fever, yellow fever, tick-borne encephalitis, West Nile (WN) meningoencephalitis, Murray Valley encephalitis, Japanese encephalitis, and St. Louis encephalitis. The envelope proteins from these viruses are all related, suggesting that our results may be applicable to a variety of flaviviruses. Surface-modified, delivery vehicles offer the flexibility to generate an immune response against single flaviviral antigens or a combination of antigens – a powerful methodology for the creation of new, effective vaccines against this group of infectious pathogens.
After a single subcutaneous dose with surface-modified particles loaded with recombinant West Nile virus antigen, 95 percent protection to viral infection was conferred to mice. Antigen-specific IgG titers were more than 20 times higher from antigen delivered in particulate form, compared to soluble antigen. Control of flavivirus infection is generally assumed to be primarily mediated by neutralizing antibodies. Interestingly, while titers were relatively low after intranasal and oral vaccination, significant protection was observed. This finding may highlight the importance of the cellular immune response reported here with the model antigen, ovalbumin, which may also play a significant role in survival rates. This is consistent with previous studies that show that CD8+ and CD4+ T cells both participate in the immune response against a challenge with WN virus [35–37]. In those studies it was proposed that CD8+ T cells are essential to fully eliminate the infecting virus and prevent viral persistence.
In summary, this study which demonstrates a technology for construction of vaccines supports an underlying hypothesis: nanoparticles encapsulating antigen can be made to be more effective vaccines by the proper choice and engineering of immune potentiators into the surface. Indeed the proper combination of these potentiators can activate the inflammasome similar to conventional adjuvants such as alum. By coupling the nanoparticles with a simple immune modulator, we were able to enhance the nanoparticles’ ability to elicit both humoral and cellular immune responses as well as provide protection against a viral challenge by multiple routes of administration. Thus, the system described here is ideal for investigating the wide-variety of emerging antigens and immune potentiators, allowing control over variables that are important in optimizing an effective vaccine delivery system.
Supplementary Material
Acknowledgments
This work was supported by NSF NIRT grant #CTS-0609326 as well as NIH grant #AI-070343. Additionally, M.J.C. is supported by NIH grant #AI-066738. E.F. and R.A.F. are investigators of the Howard Hughes Medical Institute. S.C.E was supported by National Institutes of Health T32HL007974 grant and the Bill & Melinda Gates Foundation. We would like to thank Themis Kyriakides, PhD, Eleni Skokos, PhD, Jeremy Blum, PhD, Eric Stern, PhD, Javier Lapeira, Jason Park, and Joachim Hero, MPH for helpful comments and assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008 Jun 19;453(7198):1122–6. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008 Jul 1;181(1):17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008 Aug;9(8):847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh M, O’Hagan DT. Recent advances in vaccine adjuvants. Pharm Res. 2002 Jun;19(6):715–28. doi: 10.1023/a:1016104910582. [DOI] [PubMed] [Google Scholar]
- 5.O’Hagan DT, Valiante NM. Recent advances in the discovery and delivery of vaccine adjuvants. Nat Rev Drug Discov. 2003 Sep;2(9):727–35. doi: 10.1038/nrd1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv Drug Deliv Rev. 2005 Jan 10;57(3):391–410. doi: 10.1016/j.addr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Sesardic D, Dobbelaer R. European union regulatory developments for new vaccine adjuvants and delivery systems. Vaccine. 2004 Jun 23;22(19):2452–6. doi: 10.1016/j.vaccine.2003.11.071. [DOI] [PubMed] [Google Scholar]
- 8.Pashine A, Valiante NM, Ulmer JB. Targeting the innate immune response with improved vaccine adjuvants. Nat Med. 2005 Apr;11(4 Suppl):S63–8. doi: 10.1038/nm1210. [DOI] [PubMed] [Google Scholar]
- 9.Bramwell VW, Perrie Y. The rational design of vaccines. Drug Discov Today. 2005 Nov 15;10(22):1527–34. doi: 10.1016/S1359-6446(05)03600-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 1995 Jan 13;267(5195):243–6. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 11.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997 Mar 15;158(6):2723–30. [PubMed] [Google Scholar]
- 12.Men Y, Tamber H, Audran R, Gander B, Corradin G. Induction of a cytotoxic T lymphocyte response by immunization with a malaria specific CTL peptide entrapped in biodegradable polymer microspheres. Vaccine. 1997 Aug-Sep;15(12–13):1405–12. doi: 10.1016/s0264-410x(97)00047-9. [DOI] [PubMed] [Google Scholar]
- 13.Peter K, Men Y, Pantaleo G, Gander B, Corradin G. Induction of a cytotoxic T-cell response to HIV-1 proteins with short synthetic peptides and human compatible adjuvants. Vaccine. 2001 Jul 20;19(30):4121–9. doi: 10.1016/s0264-410x(01)00179-7. [DOI] [PubMed] [Google Scholar]
- 14.Gupta RK, Rost BE, Relyveld E, Siber GR. Adjuvant properties of aluminum and calcium compounds. Pharm Biotechnol. 1995;6:229–48. doi: 10.1007/978-1-4615-1823-5_8. [DOI] [PubMed] [Google Scholar]
- 15.Lindblad EB. Aluminium adjuvants--in retrospect and prospect. Vaccine. 2004 Sep 9;22(27–28):3658–68. doi: 10.1016/j.vaccine.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 16.Sutterwala FS, Ogura Y, Flavell RA. The inflammasome in pathogen recognition and inflammation. J Leukoc Biol. 2007 Aug;82(2):259–64. doi: 10.1189/jlb.1206755. [DOI] [PubMed] [Google Scholar]
- 17.Ledizet M, Kar K, Foellmer HG, Wang T, Bushmich SL, Anderson JF, et al. A recombinant envelope protein vaccine against West Nile virus. Vaccine. 2005 Jun 10;23(30):3915–24. doi: 10.1016/j.vaccine.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Fahmy TM, Samstein RM, Harness CC, Mark Saltzman W. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials. 2005 Oct;26(28):5727–36. doi: 10.1016/j.biomaterials.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 19.Talmor M, Mirza A, Turley S, Mellman I, Hoffman LA, Steinman RM. Generation or large numbers of immature and mature dendritic cells from rat bone marrow cultures. Eur J Immunol. 1998 Mar;28(3):811–7. doi: 10.1002/(SICI)1521-4141(199803)28:03<811::AID-IMMU811>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 20.Wiwattanapatapee R, Lomlim L, Saramunee K. Dendrimers conjugates for colonic delivery of 5-aminosalicylic acid. J Control Release. 2003 Feb 14;88(1):1–9. doi: 10.1016/s0168-3659(02)00461-3. [DOI] [PubMed] [Google Scholar]
- 21.Newman KD, Elamanchili P, Kwon GS, Samuel J. Uptake of poly(D,L-lactic-co-glycolic acid) microspheres by antigen-presenting cells in vivo. J Biomed Mater Res. 2002 Jun 5;60(3):480–6. doi: 10.1002/jbm.10019. [DOI] [PubMed] [Google Scholar]
- 22.Thomasin C, Ho NT, Merkle HP, Gander B. Drug microencapsulation by PLA/PLGA coacervation in the light of thermodynamics. 1. Overview and theoretical considerations. J Pharm Sci. 1998 Mar;87(3):259–68. doi: 10.1021/js970047r. [DOI] [PubMed] [Google Scholar]
- 23.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004 May 14;304(5673):1014–8. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 24.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008 May 2;320(5876):674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jain S, Yap WT, Irvine DJ. Synthesis of protein-loaded hydrogel particles in an aqueous two-phase system for coincident antigen and CpG oligonucleotide delivery to antigen-presenting cells. Biomacromolecules. 2005 Sep–Oct;6(5):2590–600. doi: 10.1021/bm0503221. [DOI] [PubMed] [Google Scholar]
- 26.de Jong S, Chikh G, Sekirov L, Raney S, Semple S, Klimuk S, et al. Encapsulation in liposomal nanoparticles enhances the immunostimulatory, adjuvant and anti-tumor activity of subcutaneously administered CpG ODN. Cancer Immunol Immunother. 2007 Aug;56(8):1251–64. doi: 10.1007/s00262-006-0276-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunter SK, Andracki ME, Krieg AM. Biodegradable microspheres containing group B Streptococcus vaccine: immune response in mice. Am J Obstet Gynecol. 2001 Nov;185(5):1174–9. doi: 10.1067/mob.2001.117658. [DOI] [PubMed] [Google Scholar]
- 28.Kazzaz J, Singh M, Ugozzoli M, Chesko J, Soenawan E, O’Hagan DT. Encapsulation of the immune potentiators MPL and RC529 in PLG microparticles enhances their potency. J Control Release. 2006 Feb 21;110(3):566–73. doi: 10.1016/j.jconrel.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Miyake K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol. 2007 Feb;19(1):3–10. doi: 10.1016/j.smim.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Thompson BS, Chilton PM, Ward JR, Evans JT, Mitchell TC. The low-toxicity versions of LPS, MPL adjuvant and RC529, are efficient adjuvants for CD4+ T cells. J Leukoc Biol. 2005 Dec;78(6):1273–80. doi: 10.1189/jlb.0305172. [DOI] [PubMed] [Google Scholar]
- 31.Tiberio L, Fletcher L, Eldridge JH, Duncan DD. Host factors impacting the innate response in humans to the candidate adjuvants RC529 and monophosphoryl lipid A. Vaccine. 2004 Mar 29;22(11–12):1515–23. doi: 10.1016/j.vaccine.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 32.Salkowski CA, Detore GR, Vogel SN. Lipopolysaccharide and monophosphoryl lipid A differentially regulate interleukin-12, gamma interferon, and interleukin-10 mRNA production in murine macrophages. Infect Immun. 1997 Aug;65(8):3239–47. doi: 10.1128/iai.65.8.3239-3247.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chhibber S, Wadhwa S, Yadav V. Protective role of liposome incorporated lipopolysaccharide antigen of Klebsiella pneumoniae in a rat model of lobar pneumonia. Jpn J Infect Dis. 2004 Aug;57(4):150–5. [PubMed] [Google Scholar]
- 34.Petrov AB, Semenov BF, Vartanyan YP, Zakirov MM, Torchilin VP, Trubetskoy VS, et al. Toxicity and immunogenicity of Neisseria meningitidis lipopolysaccharide incorporated into liposomes. Infect Immun. 1992 Sep;60(9):3897–903. doi: 10.1128/iai.60.9.3897-3903.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Purtha WE, Myers N, Mitaksov V, Sitati E, Connolly J, Fremont DH, et al. Antigen-specific cytotoxic T lymphocytes protect against lethal West Nile virus encephalitis. Eur J Immunol. 2007 Jul;37(7):1845–54. doi: 10.1002/eji.200737192. [DOI] [PubMed] [Google Scholar]
- 36.Shrestha B, Diamond MS. Role of CD8+ T cells in control of West Nile virus infection. J Virol. 2004 Aug;78(15):8312–21. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sitati EM, Diamond MS. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J Virol. 2006 Dec;80(24):12060–9. doi: 10.1128/JVI.01650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.