

Abstract
Background
Diseases characterized by neurogenic orthostatic hypotension (NOH), such as Parkinson disease (PD) and pure autonomic failure (PAF), are associated with cardiac sympathetic denervation, as reflected by low myocardial concentrations of 6-[18F]fluorodopamine-derived radioactivity. We studied the impact of such denervation on cardiac chronotropic and inotropic function.
Methods
Cardiac inotropic function was assessed by the preejection period index and the systolic time ratio index in response to the directly acting beta-adrenoceptor agonist, isoproterenol, and to the indirectly acting sympathomimetic amine, tyramine, in patients with PD+NOH or PAF (PD+NOH/PAF group, N=13). We compared the results to those in patients with multiple system atrophy, which usually entails NOH with normal cardiac sympathetic innervation (MSA, N=15), and in normal control subjects (N=5).
Results
The innervated and denervated groups did not differ in baseline mean preejection period index or systolic time ratio index. Tyramine increased cardiac contractility in the MSA patients and controls but not in the PD+NOH/PAF group. For similar heart rate responses, the PD+NOH/PAF group required less isoproterenol (p<0.01) and had lower plasma isoproterenol levels (p<0.01) than did the MSA group.
Conclusions
Among patients with NOH those with cardiac sympathetic denervation have an impaired inotropic response to tyramine and exaggerated responses to isoproterenol. This pattern suggests that cardiac denervation is associated with decreased ability to release endogenous norepinephrine from sympathetic nerves and with supersensitivity of cardiac beta-adrenoreceptors.
Keywords: Parkinson disease, pure autonomic failure, sympathetic denervation, pre-ejection period
Sympathetic noradrenergic innervation of the heart constitutes a major effector for central neural control of cardiovascular function. Norepinephrine released from sympathetic nerves occupies adrenoceptors on myocardial cells, exerting a positive inotropic effect. The same neurotransmitter released from sympathetic nerves in the adventitia and media of arteriolar walls evokes vasoconstriction in most vascular beds. Generalized increases in sympathetic noradrenergic outflows augment cardiac output and increase peripheral vascular resistance, thereby increasing blood pressure.
Parkinson disease, pure autonomic failure, and multiple system atrophy are in a class of neurodegenerative disorders called synucleinopathies.[1] All three diseases feature autonomic dysfunction manifested by neurogenic orthostatic hypotension, but with clear differences in the status of the sympathetic innervation of the heart. Over the past decade compelling evidence has accrued for profound cardiac sympathetic denervation in both pure autonomic failure and in Parkinson disease with neurogenic orthostatic hypotension.[2–4] In contrast, most patients with multiple system atrophy have intact cardiac and overall sympathetic innervation.[3]
The functional effects of cardiac sympathetic denervation in synucleinopathies have been unknown.
In the present study we compared patients with neurogenic orthostatic hypotension and denervated hearts (Parkinson disease and pure autonomic failure) to patients with neurogenic orthostatic hypotension and innervated hearts and to normal controls in terms of hemodynamic and neurochemical responses to tyramine and isoproterenol. We hypothesized that patients with neurogenic orthostatic hypotension and cardiac sympathetic denervation (Parkinson disease with neurogenic orthostatic hypotension or pure autonomic failure) would have attenuated inotropic responses to i.v. administration of the indirectly acting sympathomimetic amine tyramine and intact inotropic and chronotropic responses to the directly acting beta-adrenoceptor agonist, isoproterenol. In contrast, patients with neurogenic orthostatic hypotension and intact cardiac sympathetic innervation (multiple system atrophy) would have normal responses.
METHODS
Subjects
The study population consisted of a total of 28 patients with chronic autonomic failure, stratified in two groups. The first group consisted of 13 patients (9 Parkinson disease with neurogenic orthostatic hypotension, 4 pure autonomic failure, aged 67± (standard error of mean) 2 years old, 7 males) who had neuroimaging evidence for cardiac sympathetic denervation (DEN group). The Hoehn and Yahr stage of the Parkinson disease subjects ranged from 1 to 5. The second group consisted of 15 multiple system atrophy with neurogenic orthostatic hypotension patients who had neuroimaging evidence of intact sympathetic innervation (INN group, 56±3 years old, 12 males). Five additional healthy volunteers (aged 48±9 years old, 4 males) served as a normal control group.
The subjects were studied at the National Institutes of Health Clinical Center after giving informed, written consent to participate in protocols approved by the Intramural Research Board of the National Institute of Neurological Disorders and Stroke.
For each patient, a medical history and physical examination were taken, with emphasis on symptoms and signs of central neurodegeneration and autonomic failure. Drugs known to affect sympathetic neuroeffector function, such as tricyclic antidepressants and sympatholytic agents, were discontinued a week before the study. Other antiparkinsonian drugs including carbidopa/levodopa (9 patients), pramipexole (3 patients), trihexyphenidyl (1 patient), amantadine (2 patients) were discontinued 5 or more days before the study.
Cardiac sympathetic neuroimaging
All subjects had cardiac sympathetic neuroimaging before the study. Each subject was positioned supine, feet-first in a GE Advance™ scanner (General Electric, Milwaukee, WI), with the thorax in the gantry. After transmission scanning for attenuation correction, 6-[18F] fluorodopamine (usual dose 0.037 MBq, specific activity 7.4–37 MBq/mmol, in about 10 mL of normal saline) was infused i.v. at a constant rate for 3 min. Dynamic scanning data were obtained for thoracic radioactivity, with the midpoint of the scanning interval at 7.5 min after injection of the tracer (data collection interval between 5–10 min). Cardiac sympathetic denervation was defined by low concentrations of 6-[18F] fluorodopamine-derived radioactivity in the interventricular septum (less than 5000 Bq/mL per MBq/kg) and left ventricular free wall (less than 4000 Bq/mL per MBq/kg), corresponding to 2 standard deviations below the normal means.
Study protocol
In most subjects, a brachial arterial catheter was inserted for pressure monitoring and drawing blood samples, and an antecubital venous catheter was used to infuse test drugs. Each patient also had electrocardiographic and impedance cardiographic monitoring (BioZ, Cardiodynamics, San Diego, CA, USA). Continuous vital signs data were digitized and recorded using a PowerLab (AD Instruments Ltd., Castle Hill, Australia) data acquisition system.
Tyramine Infusion
On the day of infusion, tyramine was dissolved in 5% dextrose and infused i.v. at a rate of 1 mg/min for 10 min. Subjects were supine, except that in those with severe supine hypertension (systolic pressure more than 200 mm Hg; 4 patients), tyramine was infused during head-up tilting (15–30 degrees), to decrease baseline pressure. Blood samples were drawn after the patient was at rest and then at 10 min during the infusion, transferred to heparinized sample tubes, and placed immediately on ice. The plasma was separated by refrigerated centrifugation. A sample of the tyramine infusate was also taken. The plasma and infusate samples were stored at −70 °C or colder until assayed for catechol contents in our laboratory by batch alumina extraction followed by liquid chromatography with electrochemical detection.[5]
Isoproterenol Infusion
On a separate day from that for tyramine infusion, isoproterenol was infused in 10 patients in the DEN group and 14 in the INN group. The setup and data acquisition for the isoproterenol infusion test were similar to those for tyramine infusion, except that a brachial arterial catheter was not used. A catheter was inserted in a vein in each arm, one catheter for blood sampling and the other for isoproterenol infusion. Beat-to-beat blood pressure was monitored non-invasively using a Finometer™ device (Finapres Medical Systems, Amsterdam, Netherlands). After baseline samples were obtained, isoproterenol freshly diluted in normal saline was infused at four incremental doses (3.5, 7, 14, and 35 ng/kg/min, each step for about 10 min), until heart rate increased by about 25 bpm from the baseline value. Blood samples for plasma catechols and isoproterenol plasma levels were processed together with an aliquot of the isoproterenol and stored and assayed as described above.
Cardiac evaluation
Stroke volume, cardiac output, velocity index, acceleration index, pre-ejection period (PEP), left ventricular ejection time (LVET) and electromechanical systole were measured non-invasively using the BioZ impedance cardiographic device, before and during the tyramine and isoproterenol infusions. Total peripheral resistance was calculated as mean arterial pressure divided by cardiac output. Heart rate-corrected indices including LVET index (LVETI) and PEP index (PEPI) were calculated according to Weissler’s equations[6] as follows: LVETI=1.7 heart rate+LVET and PEPI=0.4 heart rate+PEP. Systolic time ratio and systolic time ratio index were calculated as follows: systolic time ratio =PEP/LVET and systolic time ratio index=PEPI/LVETI. PEP the interval between the electrocardiographic Q wave and aortic valve opening, corresponds to the isovolumetric ventricular contraction time. PEP and PEPI have been regarded as inverse measures of cardiac contractility.[6–11] A pattern of prolongation of the PEP and shortening of the LVET characterizes acute myocardial infarction, angina pectoris, and heart failure.[11–13] The ratio of PEP to LVET (PEP/LVET) has also been regarded as an inverse measure of left ventricular systolic performance. PEP reduction reflects a positive inotropic effect of the cardiac sympathetic nerves on the myocardium.[14]
Baroreflex evaluation
The Valsalva maneuver was performed as described previously.[15] Baroreflex-cardiovagal gain was calculated from the slope of the relationship between cardiac interbeat interval (with one beat delay) and systolic blood pressure during Phase II of the maneuver. Baroreflex failure was defined as low baroreflex-cardiovagal gain (less than 3 ms/mm Hg) corresponding to about 2 standard deviations below the normal means.
Data Analysis and Statistics
Statistical analyses were performed using SPSS version 11.5 (SPSS Inc., Chicago, IL, USA). Changes associated with drug infusion were compared using paired t tests. Group differences in mean values at baseline and at the end of the tests and in mean responses were analyzed by means of independent means t tests (or the Mann-Whitney u test, depending on normality of the data distributions). Mean values were expressed ± standard error of mean. A p value less than 0.05 was considered statistically significant.
RESULTS
Baseline
Supine systolic blood pressure, diastolic blood pressure, and mean arterial pressure tended to be higher (p=0.06, p=0.07, and p=0.03) in the DEN than INN group (Figure 1, Table 1). There were no group differences in mean stroke volume, cardiac output, total peripheral resistance, PEP, PEPI, LVET, LVETI, systolic time ratio, or systolic time ratio index (Table 1, Figure 2). Mean electromechanical systole time was longer in DEN patients.
Figure 1.
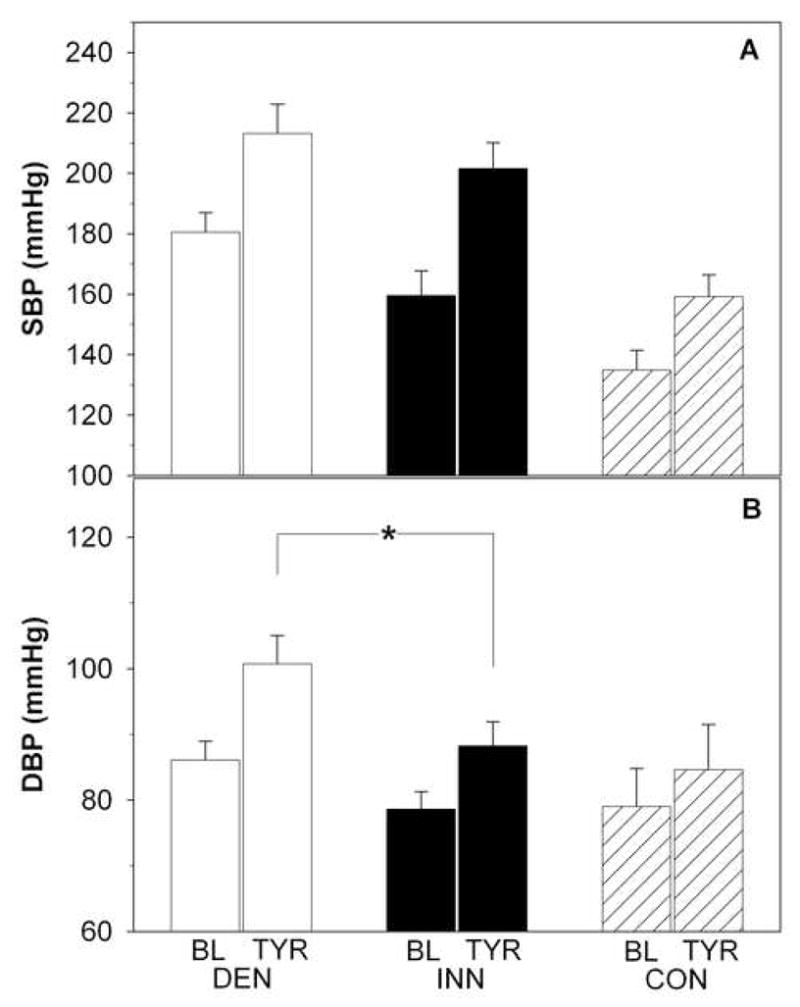
Systolic (SBP; panel A) and diastolic (DBP; panel B) blood pressure (BP) at baseline (BL) and during tyramine (TYR) infusion in patients with cardiac sympathetic denervation (DEN; white bars), in patients with normal cardiac innervation (INN; black bars), and in healthy controls (CON; dashed bars). *p<0.05.
Table 1.
Hemodynamic parameters in patients with cardiac sympathetic denervation (DEN), normal cardiac innervation (INN) and healthy controls (CON) at baseline and during i.v. infusion of tyramine (TYR).
| DEN (n=13) | INN (n=15) | CON (n=5) | ||||
|---|---|---|---|---|---|---|
| Baseline | TYR | Baseline | TYR | Baseline | TYR | |
| HR (bpm) | 68.8±3.3 | 70±4.1 | 73.5±2.6 | 71.2±3.2 | 61±5 | 55±3.9 |
| SV (mL) | 76.2±5.6 | 82.6±6.0 | 78.3±4.3 | 87.8±3.3 | 82.2±9.2 | 98±11.2 |
| CO (L/min) | 5.14±0.4 | 5.65±0.4 | 5.70±0.3 | 6.23±0.4 | 4.87±0.36 | 5.3±0.55 |
| PEP (ms) | 104±6.9 | 94±7.9* | 99.4±6.9 | 71.8±5.8† | 113±3.5 | 86±6.6 |
| LVET (ms) | 316±7.0 | 324±5.6 | 300±6.4 | 317±7.5 | 314±10 | 349±18 |
| LVETI (ms) | 433±8.7 | 4436.9 | 431±7.3 | 443±8.7 | 417±3 | 442±14 |
| VI (cm/s) | 35.9±3.2 | 40.0±3.5 | 32.9±1.9 | 41.0±3.6 | 35.4±4.8 | 40±5.5 |
| ACI (cm/s) | 61.4±6.4 | 70.4±7.6 | 56.3±5.6 | 68.1±7.6 | 61±11 | 77.2±12 |
| BRG (ms/mm Hg) | 0.86±0.1 | - | 1.51±0.5 | - | 6.1±1.2 | - |
p<0.05, DEN vs. INN
p<0.05, DEN vs. INN for change from baseline
Abbreviations: heart rate (HR), stroke volume (SV), cardiac output (CO), preejection period (PEP), left ventricular ejection time (LVET), left ventricular ejection time index (LVETI), velocity index (VI), acceleration index (ACI), baroreflex gain (BRG)
Figure 2.

Preejection period index (PEPI; panel A) and systolic time ratio index (STRI) defined as ratio of PEPI to left ventricular ejection time index (panel B) at baseline (BL) and during tyramine (TYR) infusion in patients with cardiac sympathetic denervation (DEN; white bars), in patients with normal cardiac innervation (INN; black bars) and in healthy controls (CON; dashed bars). *p<0.05.
Plasma dihydroxyphenylglycol levels at baseline were lower in the DEN than the INN and control groups (p<0.001, Figure 4). Plasma norepinephrine tended to be lower (p=0.06) at baseline in the DEN group. Epinephrine levels were also lower at baseline (p=0.009) in the DEN than in the INN and control groups.
Figure 4.

Plasma levels of dihydroxyphenylglycol (DHPG; panel A) and norepinephrine (NE; panel B) at baseline (BL) and during tyramine (TYR) infusion in patients with cardiac sympathetic denervation (DEN; white bars), in patients with normal cardiac innervation (INN; black bars), and in healthy controls (CON; dashed bars). *p<0.05, ***p<0.001.
Baroreflex-cardiovagal gain was similarly low in the DEN and the INN groups (Table 1).
Tyramine Infusion
Tyramine infusion increased systolic (p<0.001) and diastolic (p<0.05) blood pressure in all patients, without a change in mean heart rate or total peripheral resistance (Figure 1, Table 1).
Tyramine augmented systolic function in the INN group, as reflected by decreases in PEP, PEPI, systolic time ratio, and systolic time ratio index (p<0.001 for all parameters), whereas tyramine exerted only mild effects in the DEN group (INN vs. DEN p<0.05; Table 1, Figure 2). A significant negative correlation was found between the PEPI decrement and the septum-minus-chamber myocardial 6-[18F]fluorodopamine-derived radioactivity (r=−0.60, p<0.001; Figure 3). Similarly, a significant negative correlation was found between the decrement of systolic time ratio index and septum-minus-chamber myocardial 6-[18F] fluorodopamine-derived radioactivity (r=−0.54, p=0.002; Figure 3). Mean values for the velocity index and acceleration index increased significantly only in the INN group.
Figure 3.
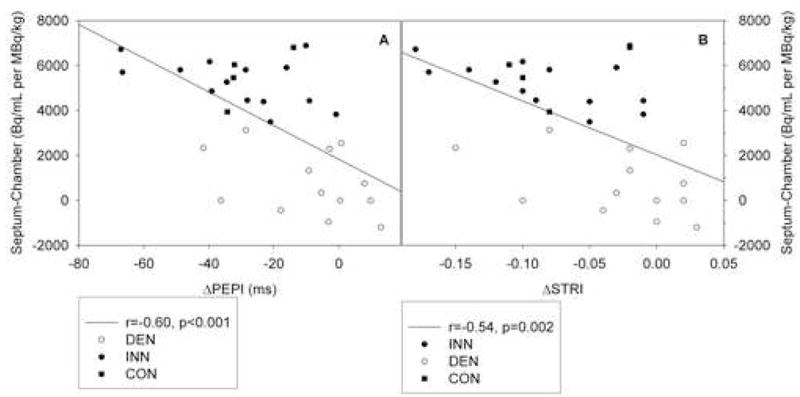
Correlations between myocardial 6-[18F] fluorodopamine-derived radioactivity in the interventricular septum subtracted by the radioactivity in the chamber and PEPI decrement (deltaPEPI; panel A) as well as between the former and STRI decrement (deltaSTRI; panel B) in response to TYR infusion in patients with cardiac sympathetic denervation (white circles), in patients with normal cardiac innervation (black circles), and in healthy controls (black squares).
Tyramine infusion increased plasma dihydroxyphenylglycol (p<0.001) and norepinephrine (p=0.003) levels in both the INN and DEN groups (Figure 4). These responses were smaller in the DEN than the INN group (p<0.001, Figure 4). The DEN group also tended to have a smaller plasma epinephrine response to tyramine than did the INN group (p=0.07; data not shown).
Isoproterenol Infusion
The mean isoproterenol infusion rate to attain the target heart rate increase in the DEN group was only about one-third that in the INN group (p=0.002; Figure 5). Isoproterenol plasma levels were assayed in 8 patients in the DEN group and 13 in the INN group. The mean isoproterenol concentration at the end of the infusion in the DEN group was also about one-third that in the INN group (Figure 4). The INN and DEN groups did not differ in systolic or diastolic blood pressure responses to isoproterenol infusion. In both groups, the acceleration index and velocity index increased during isoproterenol infusion.
Figure 5.
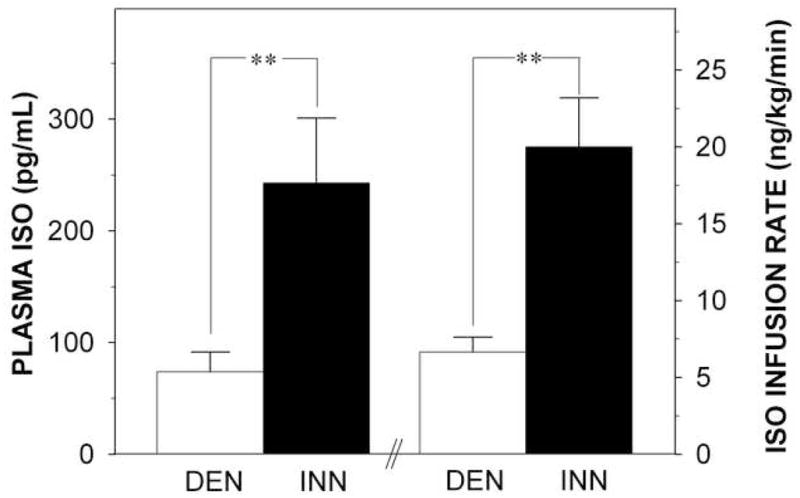
Concentration of isoproterenol (ISO) in plasma (left y-axis), ISO infusion rate (right y-axis), at ISO criterion dose i.e. approximately 25 beats per minute increase from baseline in patients with cardiac sympathetic denervation (DEN; white bars) and in patients with normal cardiac innervation (INN; black bars). **p<0.01.
Isoproterenol increased plasma norepinephrine levels in the INN group only (406±75 to 572±106 pg/ml, p<0.05; 175±37 to 191±48 pg/ml, p=ns in the DEN group). Plasma dihydroxyphenylglycol and epiephrine levels did not change during ISO infusion in either group.
DISCUSSION
In this study, patients with neurogenic orthostatic hypotension and neuroimaging evidence of cardiac sympathetic denervation failed to increase values for indices of cardiac contractility during i.v. tyramine infusion, whereas patients with neurogenic orthostatic hypotension and intact cardiac innervation had normal inotropic responses.
Systemic administration of the indirectly acting sympathomimetic amine, tyramine, has been used for many years as a pharmacological tool to assess sympathetic nervous system function.[16–20] Sympathetic nerves take up tyramine via the cell membrane norepinephrine transporter. Tyramine in the axoplasm is then taken up into vesicles via the vesicular monoamine transporter, and in the vesicles tyramine displaces norepinephrine. Much of the norepinephrine is displaced into the axoplasm, where it is deaminated to form dihydroxyphenylglycol, the main neuronal metabolite of norepinephrine; a proportion is released into the extracellular fluid by a calcium-independent mechanism, where the norepinephrine can bind to adrenoceptors on cardiovascular smooth muscle cells. Because inotropic effects of tyramine depend on this displacement of norepinephrine from cardiac sympathetic nerves, attenuation of cardiac inotropic responses to tyramine probably reflects decreased vesicular norepinephrine stores. Our findings therefore suggest that cardiac sympathetic denervation results in a failure to increase contractility in response to a stimulus that normally increases norepinephrine release from sympathetic nerves.
Pressor responses to tyramine depend on the release of endogenous norepinephrine from sympathetic nerves both in the heart and vasculature. Although one might presume that tyramine increases blood pressure via generalized vasoconstriction and increased total peripheral resistance, it appears that most of the pressor response to tyramine depends on increased cardiac output, from increased stroke volume, in turn from increased myocardial contractility or increased venous return to the heart.[18, 20] Others have reported forearm vasodilation in response to systemic tyramine administration, which would tend to decrease total peripheral resistance. Dopamine contamination of infused tyramine probably confounded the previously published results[19, 21]; however, even infusion of uncontaminated tyramine seems to increase blood pressure via an increase in cardiac output.[20] The findings of the present study lead us to propose that failure to augment delivery of norepinephrine to its receptors may explain complaints such as fatigue and exercise intolerance in patients with cardiac sympathetic denervation in the setting of neurogenic orthostatic hypotension.[22, 23]
The second finding of the study was that to attain the target increment in heart rate (about 25 bpm), patients with cardiac sympathetic denervation required lower doses of infused isoproterenol and had lower plasma isoproterenol concentrations than did patients with intact cardiac innervation. These findings suggest supersensitivity of cardiac adrenoceptor-mediated processes in denervated hearts, consistent with previous findings.[24]
Although the INN and DEN groups differed in contractility responses to tyramine, the differences were not as dramatic as one would have expected from the marked differences in cardiac innervation and in norepinephrine release. One explanation for this finding is that beta-adrenoceptor supersensitivity may offset effects of decreased ability to release norepinephrine. Consistent with this concept, the DEN and INN groups had similar pressor responses to tyramine, despite smaller plasma norepinephrine responses than in the INN group. Previous reports have also described evidence of denervation supersensitivity of vascular alpha-adrenoceptors in multiple system atrophy patients with sympathetic degeneration.[25]
Sympathetic nerves possess beta-2 adrenoceptors, which when stimulated augments norepinephrine release for a given amount of nerve traffic.[26] In the present study isoproterenol infusion increased plasma norepinephrine levels much more in the INN than the DEN groups, consistent with fewer extra-cardiac sympathetic nerves in the DEN group. Isoproterenol may cause vasodilation via stimulation of peripheral beta-adrenoceptors, thereby stimulating sympathetic outflows indirectly through baroreflex inhibition. The latter process cannot explain the differences in the norepinephrine response to isoproterenol in the present study, since the DEN and INN groups had similarly severe baroreflex failure.
In our study we included a small control group of healthy subjects, to demonstrate normal responses to tyramine. Since denervated patients had neurogenic orthostatic hypotension and blunted baroreflex function, comparison with a group who had neurogenic orthostatic hypotension and decreased baroreflex function but normal innervation (i.e., multiple system atrophy) would be more appropriate.
Overall the results illustrate both direct and indirect effects and complex adaptive capacities of neurocirculatory regulation in response to degenerative loss of sympathetic nerves. Despite substantial alterations in neurochemical profiles reflecting generalized sympathetic denervation in Parkinson disease with neurogenic orthostatic hypotension and pure autonomic failure, tyramine infusion reveals smaller differences in cardiac contractility between DEN and INN patients. The compensatory adjustments seem to include augmented responses to occupation of cardiovascular adrenoceptors.
Acknowledgments
The research reported here was supported by the intramural research program of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marti MJ, Tolosa E, Campdelacreu J. Clinical overview of the synucleinopathies. Mov Disord. 2003;18 (Suppl 6):S21–7. doi: 10.1002/mds.10559. [DOI] [PubMed] [Google Scholar]
- 2.Braune S, Reinhardt M, Schnitzer R, Riedel A, Lucking CH. Cardiac uptake of [123I]MIBG separates Parkinson’s disease from multiple system atrophy. Neurology. 1999;53:1020–5. doi: 10.1212/wnl.53.5.1020. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO., 3rd Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med. 2000;133:338–47. doi: 10.7326/0003-4819-133-5-200009050-00009. [DOI] [PubMed] [Google Scholar]
- 4.Takatsu H, Nishida H, Matsuo H, Watanabe S, Nagashima K, Wada H, et al. Cardiac sympathetic denervation from the early stage of Parkinson’s disease: clinical and experimental studies with radiolabeled MIBG. J Nucl Med. 2000;41:71–7. [PubMed] [Google Scholar]
- 5.Holmes C, Eisenhofer G, Goldstein DS. Improved assay for plasma dihydroxyphenylacetic acid and other catechols using high-performance liquid chromatography with electrochemical detection. J Chromatogr B Biomed Appl. 1994;653:131–8. doi: 10.1016/0378-4347(93)e0430-x. [DOI] [PubMed] [Google Scholar]
- 6.Weissler AM, Harris WS, Schoenfeld CD. Systolic time intervals in heart failure in man. Circulation. 1968;37:149–59. doi: 10.1161/01.cir.37.2.149. [DOI] [PubMed] [Google Scholar]
- 7.Atkins CE, Snyder PS. Systolic time intervals and their derivatives for evaluation of cardiac function. J Vet Intern Med. 1992;6:55–63. doi: 10.1111/j.1939-1676.1992.tb03152.x. [DOI] [PubMed] [Google Scholar]
- 8.Berntson GG, Lozano DL, Chen YJ, Cacioppo JT. Where to Q in PEP. Psychophysiology. 2004;41:333–7. doi: 10.1111/j.1469-8986.2004.00156.x. [DOI] [PubMed] [Google Scholar]
- 9.Cacioppo JT, Berntson GG, Binkley PF, Quigley KS, Uchino BN, Fieldstone A. Autonomic cardiac control. II. Noninvasive indices and basal response as revealed by autonomic blockades. Psychophysiology. 1994;31:586–98. doi: 10.1111/j.1469-8986.1994.tb02351.x. [DOI] [PubMed] [Google Scholar]
- 10.Graboys TB, Forlini FJ, Jr, Michaelson ED. Systolic time intervals during lower body negative pressure. J Appl Physiol. 1974;37:329–32. doi: 10.1152/jappl.1974.37.3.329. [DOI] [PubMed] [Google Scholar]
- 11.Lewis RP, Boudoulas H, Welch TG, Forester WF. Usefulness of systolic time intervals in coronary artery disease. Am J Cardiol. 1976;37:787–96. doi: 10.1016/0002-9149(76)90376-3. [DOI] [PubMed] [Google Scholar]
- 12.Randazzo A, Sardella F, Martinotti R, Daffara G, Parziale M, Cremonesi G. Evaluation of systolic time intervals in patients with angina pectoris. Am Heart J. 1983;105:756–62. doi: 10.1016/0002-8703(83)90237-5. [DOI] [PubMed] [Google Scholar]
- 13.Sutton R, Hodd WP, Jr, Koch GG. Noninvasive assessment of left ventricular function in chronic heart disease. Am Heart J. 1977;93:289–97. doi: 10.1016/s0002-8703(77)80246-9. [DOI] [PubMed] [Google Scholar]
- 14.Schachinger H, Weinbacher M, Kiss A, Ritz R, Langewitz W. Cardiovascular indices of peripheral and central sympathetic activation. Psychosom Med. 2001;63:788–96. doi: 10.1097/00006842-200109000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein DS, Horwitz D, Keiser HR. Comparison of techniques for measuring baroreflex sensitivity in man. Circulation. 1982;66:432–9. doi: 10.1161/01.cir.66.2.432. [DOI] [PubMed] [Google Scholar]
- 16.Bianchetti MG, Minder I, Beretta-Piccoli C, Meier A, Weidmann P. Effects of tyramine on blood pressure and plasma catecholamines in normal and hypertensive subjects. Klin Wochenschr. 1982;60:465–70. doi: 10.1007/BF01720361. [DOI] [PubMed] [Google Scholar]
- 17.Charkoudian N, Joyner MJ, Sokolnicki LA, Johnson CP, Eisenach JH, Dietz NM, et al. Vascular adrenergic responsiveness is inversely related to tonic activity of sympathetic vasoconstrictor nerves in humans. J Physiol. 2006;572:821–7. doi: 10.1113/jphysiol.2005.104075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacob G, Costa F, Vincent S, Robertson D, Biaggioni I. Neurovascular dissociation with paradoxical forearm vasodilation during systemic tyramine administration. Circulation. 2003;107:2475–9. doi: 10.1161/01.CIR.0000065605.37863.C0. [DOI] [PubMed] [Google Scholar]
- 19.Jacob G, Gamboa A, Diedrich A, Shibao C, Robertson D, Biaggioni I. Tyramine-induced vasodilation mediated by dopamine contamination: a paradox resolved. Hypertension. 2005;46:355–9. doi: 10.1161/01.HYP.0000172353.62657.8b. [DOI] [PubMed] [Google Scholar]
- 20.Meck JV, Martin DS, D’Aunno DS, Waters WW. Pressor response to intravenous tyramine is a marker of cardiac, but not vascular, adrenergic function. J Cardiovasc Pharmacol. 2003;41:126–31. doi: 10.1097/00005344-200301000-00016. [DOI] [PubMed] [Google Scholar]
- 21.Holmes C, Moak J, Eldadah B, Zimmerly E, Sharabi Y, Goldstein DS. Dopamine contamination of infused tyramine. Clin Chem. 2005;51:1733–5. doi: 10.1373/clinchem.2005.054361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman JH, Brown RG, Comella C, Garber CE, Krupp LB, Lou JS, et al. Fatigue in Parkinson’s disease: a review. Mov Disord. 2007;22:297–308. doi: 10.1002/mds.21240. [DOI] [PubMed] [Google Scholar]
- 23.Werner WG, DiFrancisco-Donoghue J, Lamberg EM. Cardiovascular response to treadmill testing in Parkinson disease. J Neurol Phys Ther. 2006;30:68–73. doi: 10.1097/01.npt.0000282570.78544.00. [DOI] [PubMed] [Google Scholar]
- 24.Polinsky RJ, Kopin IJ, Ebert MH, Weise V. Pharmacologic distinction of different orthostatic hypotension syndromes. Neurology. 1981;31:1–7. doi: 10.1212/wnl.31.1.1. [DOI] [PubMed] [Google Scholar]
- 25.Davies B, Sudera D, Sagnella G, Marchesi-Saviotti E, Mathias C, Bannister R, et al. Increased numbers of alpha receptors in sympathetic denervation supersensitivity in man. J Clin Invest. 1982;69:779–84. doi: 10.1172/JCI110516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Majewski H. Modulation of noradrenaline release through activation of presynaptic beta-adrenoreceptors. J Auton Pharmacol. 1983;3:47–60. doi: 10.1111/j.1474-8673.1983.tb00496.x. [DOI] [PubMed] [Google Scholar]