

Abstract
The increasing risk of drug resistant bacterial infections indicates that there is a growing need for new and effective antimicrobial agents. One promising, but unexplored area in antimicrobial drug design is de novo purine biosynthesis. Recent research has shown that de novo purine biosynthesis is different in microbes than in humans. The differences in the pathways are centered around the synthesis of 4-carboxyaminoimidazole ribonucleotide (CAIR) which requires the enzyme N5-carboxyaminoimidazole ribonucleotide (N5CAIR) synthetase. Humans do not require and have no homologs of this enzyme. Unfortunately, no studies aimed at identifying small molecule inhibitors of N5CAIR synthetase have been published. To remedy this problem, we have conducted high-throughput screening (HTS) against Escherichia coli N5CAIR synthetase using a highly reproducible phosphate assay. HTS of 48,000 compounds identified 14 that inhibited the enzyme. The hits identified could be classified into three classes based upon chemical structure. Class I contains compounds with an indenedione core. Class II contains an indolinedione group, and Class III contains compounds that are structurally unrelated to other inhibitors in the group. We determined the Michaelis-Menten kinetics for five compounds representing each of the classes. Examination of compounds belonging to Class I indicate that these compounds do not follow normal Michaelis-Menten kinetics. Instead, these compounds inhibit N5CAIR synthetase by reacting with the substrate AIR. Kinetic analysis indicates that the Class II family of compounds are non-competitive with both AIR and ATP. One compound in Class III is competitive with AIR but uncompetitive with ATP, whereas the other is noncompetitive with both substrates. Finally, these compounds display no inhibition of the human AIR carboxylase:SAICAR synthetase indicating that these agents are selective inhibitors of N5CAIR synthetase
Introduction
Antibiotic resistant infections, once rare, are now becoming commonplace and pose a significant challenge for healthcare professionals1,2. A recent study of methicillin-resistant Staphylococcus aureus (MRSA) found that approximately 19,000 individuals die each year from this disease, a number greater than those that die from AIDS3. In addition to their lethality, resistance infections are responsible for longer hospital stays and higher healthcare costs4. Historically, MRSA infections have been associated with prolonged hospitalization; however, recent studies have shown an increase in community-acquired MRSA (CA-MRSA)5. CA-MRSA infections are especially troubling because they occur in otherwise healthy individuals, and some strains of CA-MRSA are extremely virulent5,6.
The standard for the treatment of antibiotic resistant infections is vancomycin. Vancoymcin is highly effective against most resistant bacteria; however, infections due to vancomycin-insensitive and vancomycin-resistant strains foreshadow the day in which this agent may no longer be useful7,8. Unfortunately, only two new classes of antibiotics have been brought to the clinic over the last 40 years. The first, linezolid is an oxazolidone which effects protein synthesis while the second, daptomycin, results in cell wall permeability9,10. While studies have shown that these agents are active against antibiotic-sensitive and resistant bacteria, resistance to linezolid has already been reported and daptomycin has potentially limiting side effects10,11. Clearly, neither agent represents a long term solution to the antibiotic crisis.
One unexploited target in antimicrobial drug design occurs in the de novo purine biosynthetic pathway where studies have revealed that purine biosynthesis is different in humans than microbes12–17. In humans, purines are synthesized from phosphoribosyl pyrophosphate (PRPP) by a 10-step metabolic pathway. However, in bacteria, yeast, and fungi, 11 steps are required, with the extra step being necessary for the synthesis of the intermediate, 4-carboxy-5-aminoimidazole ribonucleotide (CAIR, Figure 1)15,17.
Figure 1.

Differences between human and microbial de novo purine biosynthesis.
During purine synthesis in microbes, CAIR is synthesized from 5-aminoimidazole ribonucleotide (AIR) by the action of two enzymes. N5-carboxy-5-aminoimidazole ribonucleotide (N5CAIR) synthetase takes AIR, ATP and bicarbonate and produces the unstable intermediate N5CAIR17. N5CAIR mutase converts N5CAIR into CAIR by catalyzing the transfer of CO2 from the N5 position to C416. Humans do not produce N5CAIR and thus do not have a N5CAIR synthetase homolog. Instead, in humans, AIR and CO2 are directly converted to CAIR by the enzyme AIR carboxylase12,13. AIR carboxylase is evolutionarily related to N5CAIR mutase; however, AIR carboxylase cannot utilize N5CAIR as a substrate12.
Both N5CAIR synthetase and N5CAIR mutase are potential targets for the development of antimicrobial agents. However, N5CAIR synthetase is absent in humans indicating that this is an ideal target for the identification of selective inhibitors against microbial purine biosynthesis. Genetic studies on N5CAIR synthetase support the role of the enzyme in microbial growth and disease progression. Mutations of N5CAIR synthetase in E. coli, Salmonella typhimurium, Streptococcus pneumonia, Shigella, Pseudomonas aeruginosa, Bacillus anthracis, Cryptococcus neoformans and Candida albicans generate microorganisms that possess a significant reduction in growth in human serum and animals18–25.
The differences in de novo purine biosynthesis, coupled with existing genetic studies, suggest that de novo purine biosynthesis may be an ideal target for the development of antimicrobial drugs. Unfortunately, there are no known small molecule, drug-like inhibitors for this enzyme. In this report, we describe our efforts at identifying small molecule inhibitors of N5CAIR synthetase using high-throughput screening (HTS).
Results
High-Throughput Screening for N5CAIR Synthetase Inhibitors
In an effort to identify drug-like lead agents against N5CAIR synthetase, we initiated a high-throughput screen (HTS) at the Center for Chemical Genetics (CCG) at the University of Michigan. The published assay for N5CAIR synthetase relies upon the measurement of ADP using a system in which ADP production is coupled to the reduction of pyruvate14,17. While this assay is well known, it suffers from the problem that false hits could be generated by inhibiting the coupling enzymes rather than N5CAIR synthetase. Thus, additional screening would have to be done to identify the true target of any inhibitor identified in the assay.
To circumvent this problem, we utilized the well-known malachite green/phosphomolybdate assay to detect the AIR-dependent production of phosphate by the enzyme26. This assay has also been used in HTS of other enzyme systems27. In this assay, the phosphate produced complexes with molybdate to form a colorless complex which upon reaction with malachite green generates a blue color the can be measured at 600 nm26. We observed that this assay worked well for N5CAIR synthetase with the exception that the concentration of ATP had to be maintained at ≤100 μM (~Km) in order to avoid a significant AIR-independent ATPase activity. However, if the concentration of ATP is kept at 100 μM, we found that this assay could easily be run in 384-well plates and is highly reproducible (Z′ score of ~0.7).
Using this assay, 48,000 drug-like, commercially available compounds were screened at a single inhibitor concentration ranging from 5–27μM (depending upon supplier) and we found 212 potential inhibitors (hit rate: 0.44%). Dose response curves, using the phosphate assay, were generated for the most potent 107 compounds as determined from the primary screen. Of these, only 14 displayed a dose response relationship (hit rate: 0.03%). Importantly, none of these 14 were identified as hits in the PubChem database indicating that our compounds display some measure of selectivity.
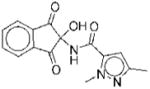
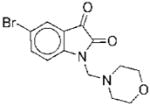
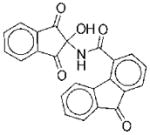
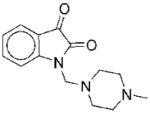
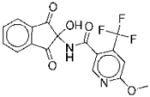
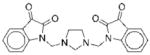
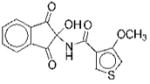
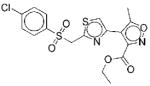
The structures of the 14 compounds are shown in Table I along with their IC50 values. The hits identified from HTS can be classified into three classes based upon chemical structure. Class I contains compounds with an indenedione core. Class II compounds contain an indolinedione group, and Class III are compounds which are structurally unrelated to any other. We determined the Michaelis-Menten kinetics for five compounds (1, 2, 7, 13, 14) using the standard, published N5CAIR synthetase assay based upon the PK/LDH coupled assay system. The results are given in Table I.
Table I.
Kinetic information for inhibitors identified by high-throughput screening.
| No. | Structure | Classa | IC50b | Kic | Typed | No. | Structure | Classa | IC50b | Kic | Typed |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
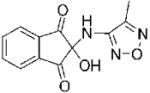
|
I | 2.3 | NAe | R | 8 |
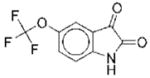
|
II | 19 | ND | NA |
| 2 |
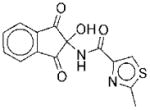
|
I | 5.6 | NAe | R | 9 |
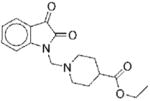
|
II | 22 | ND | NA |
| 3 |
|
I | 7.8 | ND | NA | 10 |
|
II | 23 | ND | NA |
| 4 |
|
I | 8.7 | ND | NA | 11 |
|
II | 37 | ND | NA |
| 5 |
|
I | 17 | ND | NA | 12 |
|
II | 69 | ND | NA |
| 6 |
|
I | 17 | ND | NA | 13 |
|
III | 10 | 62f 43g |
Cf Ug |
| 7 |
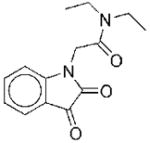
|
II | 10.5 | 29f 40g |
NCf NCg |
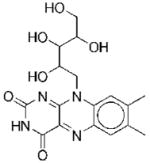
14 |
|
III | 20 | 81f 122g |
NCf NCg |
compound class was determined based upon structural similarity to other hits identified in the screen.
IC50 values determined from dose response curves using the phosphate assay.
Ki values determined using the ATPase assay with variable amounts of either AIR or ATP at a fixed concentration of bicarbonate. Values determined by curve fitting data using various models of inhibition.
type of inhibition determined by fitting data to the modified Michaelis-Menten equations for the presence of different types of inhibitors. Equations which produced the best fit to the data were used to determine the type of inhibition. Results of these curve fits were validated by Lineweaver-Burke plots. R: reacts with substrate; NC: non-competitive; C: competitive; U:uncompetitive.
the compounds react with substrate and thus do not follow normal Michaelis-Menten kinetics.
obtained using variable concentrations of AIR and fixed ATP and bicarbonate concentrations.
obtained using variable concentrations of ATP and fixed AIR and bicarbonate concentrations.
Class I compounds inhibit N5CAIR synthetase by reacting with the substrate
The Michaelis-Menten plot of 1 and 2 versus AIR concentration revealed a biphasic relationship in which there was a lag followed by an increase in velocity as the concentration of AIR increased (Figure 2, compound 2 not shown). Examination of the data revealed that the breakpoint between the lag versus turnover always occurred when the concentration of AIR was twice that of the inhibitor. This suggested that the inhibitor reacts with AIR. Once the inhibitor is consumed by AIR, additional free AIR is then available for turnover by the enzyme.
Figure 2.

Michaelis-Menten plot for N5CAIR synthetase in the presence (■) and absence (●) of 1.
Such a result is odd for two reasons. First, in these reactions, the substrate and the inhibitor are added at the same time to the enzyme. Thus, the reaction between the inhibitor and AIR had to occur faster than the conversion of AIR to N5CAIR. Second, the structures of the Class I inhibitors do not display any obvious reactive groups.
A clue as to what could be occurring came with an examination of the synthesis of compounds related to Class I. Synthesis of these agents is accomplished by reacting amines and amides with ninhydrin to form the hemiaminal or the 1-hydroxy acetamide. We hypothesized that such molecules may be unstable in an aqueous solution where they could decompose to the 1,2,3-triketoindane (the dehydrated form of ninhydrin) and the amine or amide. If such a reaction occurred, the 1,2,3-triketoindane present in the reaction mixture could react with the amine of AIR thereby preventing AIR from interacting with the enzyme (Figure 3).
Figure 3.
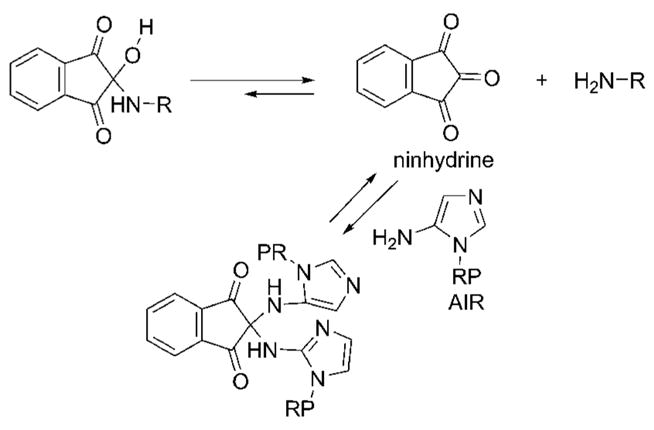
Proposed mechanism for the reaction of Class I inhibitors with the substrate AIR. RP is ribose phosphate.
We examined aqueous solutions of 1 and 2 by TLC and found that the compounds decomposed to generate ninhydrin. The rate of decomposition was dependent upon the chemical nature of the compound with compound 1 completely decomposing to ninhydrin after approximately 30 minutes while decomposition of 2 took around 45 minutes. This decomposition was not observed in DMSO solutions of the compounds indicating that a proton (presumably from water) was necessary for the reverse reaction.
Incubation of ninhydrin with AIR resulted in a very rapid reaction (≪ 20 s) as determined by TLC analysis. Experiments utilizing TLC and UV spectroscopy indicated that the enzyme did not appear to catalyze a reaction between AIR and ninhydrin. To determine if the reaction between AIR and ninhydrin was responsible for the kinetics observed for 1 and 2, we determined the Michaleis-Menten plot with variable AIR and ninhydrin. Once again, we observed the same kinetic results as for 1 and 2; namely, no reaction until the concentration of AIR exceeded that of ninhydrin. This suggested that ninhydrin was responsible for the inhibition of the reaction. To confirm this, we examined whether DMSO solutions of 1 or 2 were capable of inhibiting the N5CAIR synthetase. We found that upon addition of these non-aqueous solutions, neither compound resulted in inhibition of the enzyme. Thus, our data suggest that compounds belonging to Class I are not true inhibitors of N5CAIR synthetase, but rather inhibit the reaction by reacting with the substrate and preventing it from being converted by the enzyme. Given this mechanism of action, Class I compounds do not represent viable lead agents for the development of antimicrobial agents.
Class II compounds are non-competitive inhibitors of N5CAIR synthetase
We determined the Michaelis-Menten kinetics of compound 7, which is the most potent member of the Class II family of inhibitors. Kinetic analysis reveals that 7 is non-competitive with both AIR and ATP (Figure 4). N5CAIR synthetase has no known allosteric regulators, however, HTS of other enzymes related to N5CAIR synthetase have identified non-competitive inhibitors as well 28,29. Given the structural similarity between 7 and the other Class II inhibitors, we anticipate that all compounds belonging to this class will be non-competitive inhibitors of the enzyme. We are currently working to identify the binding site for the Class II inhibitors.
Figure 4.

Lineweaver-Burke plots for the inhibition of N5CAIR synthetase by 7. A. Lineweaver-Burke plot with varied concentration of ATP, fixed concentration of AIR and various concentrations of 7 ((●) 0 μM, (■) 5μM, (▲) 10μM, (◆) 25μM). B. Lineweaver-Burke plot with varied concentration of AIR, fixed concentration of ATP and various concentrations of 7 ((●) 0μM, (■) 5μM, (▲) 10μM, (◆) 25μM).
Investigation of Class III compounds
We have identified two compounds that are chemically unrelated to any other hit identified in the screen. Compound 14 is riboflavin. Although this compound is unlikely to be a lead agent, we conducted kinetic analysis to determine the mechanism of inhibition. Steady-state kinetic analysis indicates that riboflavin is noncompetitive with both AIR and ATP (Figure 5).
Figure 5.

Lineweaver-Burke plots for the inhibition of N5CAIR synthetase by 14. A. Lineweaver-Burke plot with varied concentration of AIR, fixed concentration of ATP and various concentrations of 14 ((●) 0 μM, (■) 5 μM, (▲) 10 μM, (◆) 25 μM, (▼) 50 μM). B. Lineweaver-Burke plot with varied concentration of ATP, fixed concentration of AIR and various concentrations of 14 ((●) 0 μM, (■) 5 μM, (▲) 10 μM, (◆) 25 μM, (▼) 50 μM).
Steady-state kinetics on 13 indicate that the compound is competitive with AIR but uncompetitive with ATP (Figure 6). This suggests that the compound binds to the AIR binding site of the enzyme only after ATP binds to the enzyme. Since there are no crystal structures of N5CAIR synthetase with bound AIR, information gleaned from binding studies of 13 with the enzyme could be helpful in identifying important substrate binding features on the enzyme.
Figure 6.

Lineweaver-Burke plots for the inhibition of N5CAIR synthetase by 13. A. Lineweaver-Burke plot with varied concentration of AIR, fixed concentration of ATP and various concentrations of 13 ((●) 0 μM, (■) 5 μM, (▲) 10 μM, (◆) 25 μM, (▼) 50 μM). B. Lineweaver-Burke plot with varied concentration of ATP, fixed concentration of AIR and various concentrations of 13 ((●) 0 μM, (■) 5 μM, (▲) 10 μM, (◆) 25 μM).
Since our screen was unable to identify other compounds related to 13, we conducted a structure-based search of all compounds available at the CCG and assayed eight compounds, which had structural similarity to 13. All eight failed to inhibit N5CAIR up to a concentration of 100 μM. Examination of a subset of these compounds demonstrates the importance of substituents in the molecule (Figure 7). The isoxazole-thiazole rings are necessary but not sufficient for activity as demonstrated by the fact that 15 does not inhibit the enzyme. The relative orientation of the substituents on the isoxazole and thiazole are also important for activity. Compound 16 is similar to 13 except that the thiazole is moved from the C4 to C5 position of the isoxazole. The presence of a sulfonyl group beta to the thiazole is not required for inhibition since 15, 17, and 18 all have this group but are inactive. The presence of an ester at C3 of isoxazole appears to be necessary since 18 is inactive. Finally, there appears to be a requirement for a particular distance between the thiazole and the phenyl ring on the sulfonyl group. Compound 17, which has an added carbon, is inactive.
Figure 7.

Structurally related analogs of 13 that are not inhibitors of N5CAIR synthetase.
The compounds do not inhibit human AIR carboxylase or SAICAR synthetase
We were interested to determine if these agents had any inhibitory activity against the human bifunctional enzyme AIR carboxylase:SAICAR synthetase. We assayed both activities simultaneously using the coupled assay system as previously described for the chicken AIR carboxylase:SAICAR synthetase13. We found that inclusion 200 μM of 7, 13, or 14 resulted in no inhibition of either enzymatic activity. Incubation with higher concentrations of the compounds resulted in precipitation. These result indicates that these agents are selective for the microbial enzyme.
Discussion
In the last 40 years, only two new antibacterial agents have been brought to market, and most major pharmaceutical companies have drastically reduced or eliminated their programs on antimicrobials2,30. De novo purine biosynthesis represents an undervalued pathway for antimicrobial drug design, although a recent study has identified an agent selective for microbial guanosine monophosphate synthase (GMPS)31. In this paper, we have described our efforts at identifying inhibitors of the enzyme N5CAIR synthetase. We have identified, for the first time, selective, non-nucleotide inhibitors of this enzyme. These compounds inhibit the enzyme in the range of 30–120 μM and follow Lipinski’s rules. The compounds were classified into three distinct classes based upon structural similarity. Members of Class I contained an indenedione group, which upon incubation in water resulted in the production of ninhydrin that reacted with the substrate AIR. Since these compounds are present in commercial chemical libraries, researchers conducting HTS should be aware of the potential of these compounds to decompose and react with amines present in the substrate.
Compounds belonging to Class II are ideal lead agents in that they display non-competitive kinetics with respect to both AIR and ATP. Such a feature is necessary for agents targeting an enzyme in the middle of a biosynthetic pathway since increases in the concentration of the substrate could not overcome inhibition by the drug. Class II compounds are small (~250 MW) and thus provide flexibility to build additional binding features into the molecule to enhance potency.
Although the kinetic mechanism of the enzyme remains to be determined, the non-competitive kinetics of Class II molecules could indicate that these agents recognize a unique binding pocket on the enzyme. Interestingly, HTS of other members of the N5CAIR synthetase superfamily have identified compounds which regulate activity and bind to sites distinct from the active site. For example, the natural product soraphen A inhibits acetyl-CoA carboxylase by binding to a pocket some 25Ǻ away from the active site28. Binding to this location is thought to disrupt protein dimer formation resulting in inactivation of the enzyme. HTS against Staphylococcus aureus D-ala:D-ala ligase also identified a non-competitive inhibitor which bound very near the active site and precluded binding of both ATP and D-alanine29. Crystallographic studies are currently underway to determine the structure of the 7:synthetase complex. Such information will be invaluable for identifying the binding site for this compound, and these investigations will have important ramifications for future structure guided drug design efforts aimed at improving the potency of these agents.
Compound 13, which belongs to Class III inhibitors, displays competitive kinetics with respect to AIR. This suggests that 13 binds at or near the AIR binding site. To date, no structure of N5CAIR synthetase with AIR has been solved, however, the binding site for AIR can be postulated based upon a homology model between N5CAIR synthetase and a related enzyme PurT32. PurT catalyzes an alternative formylation of the purine intermediate glycinamide ribonucleotide (GAR) and the structure of this enzyme complexed with GAR has been solved33. The location of GAR binding in PurT corresponds to a region in N5CAIR synthetase with conserved amino acids suggesting the location of the AIR binding site. Using this information, preliminary molecular modeling of 13 into the presumed AIR binding site is being conducted and structural studies are under way to determine the structure of the protein:13 complex.
The uncompetitive nature of 13 with respect to ATP is interesting because it indicates that the inhibitor binds only to the enzyme:ATP complex. Given that 13 is competitive with AIR and thus likely binds to the AIR binding site, this suggests that N5CAIR synthetase may function via an ordered kinetic mechanism with ATP binding first followed by AIR. Once both substrates are bound, catalysis occurs. Additional kinetic studies are necessary to validate or refute this conclusion.
Conclusion
There is little doubt that the increases in antibiotic resistant infections pose a significant public health threat. Unfortunately, there are a limited number of antibiotics that are available for treating resistant infections. In this paper, we have described the identification of compounds which inhibit N5CAIR synthetase. This enzyme is involved in de novo purine biosynthesis and is unique to microbes. These compounds follow Lipinski’s rules and although they possess modest potencies, they represent ideal lead agents for the development of novel antimicrobial agents. Additional studies aimed at determining the binding sites of these molecules and increasing the potency of these compounds is underway. The results of these studies will be reported in due course.
Materials and Methods
Compound Library
The 48,000-member library was collected from the following commercial sources: 16,000 compounds from the Maybridge HitFinder library (Maybridge), 20,000 compounds from ChemDiv, 10,000 compounds from Chembridge and 2,000 compounds from the MS Spectrum library. Each library member was dissolved in DMSO and stored at an initial concentration of 0.73–4 mM at −20 °C before use. Individual compounds were purchased from suppliers for retesting.
High-throughput screening
E. coli N5CAIR synthetase was prepared as previously stated15. Reactions were conducted in a 384-well plate (Corning 3701). Buffer was added to each well of the plate (50 mM HEPES, pH 7.5, 20 mM KCl, 6.0 mM MgCl2, 100 μM ATP, supplemented with a 1/100 dilution of saturated NaHCO3) and 0.2 μL of the DMSO stock of each compound was added to the plate using a Biomek FX pin tool such that the final concentration of the compound in each well was between 5–27 μM. Each plate contained a column of positive controls (enzyme + AIR + 0.2 μL of DMSO) and negative controls (enzyme − AIR + 0.2 μL of DMSO). AIR (50 μM) was added to the appropriate wells using a Thermo Labsystems Multidrop Micro plate dispenser followed by 168 ng of N5CAIR synthetase. The reaction was incubated at room temperature for 5 minutes and was quenched by the addition of 6 μL the malachite green reagent (Bioassay Systems) to each well. The plates were incubated at room temperature for at least 15 minutes and then the absorbance (600 nm) of each at was determined using a PHERAstar (BMG Labs) plate reader.
Dose-response assays
The eight-point dose response assay was using the same protocol as the high-throughput assay except that the concentration of each compound assayed was varied from 0.78–100 μM. Each assay contained the same positive and negative controls used for the high-throughput screen. Data for compounds displaying a dose-response relationship were fitted to equation 1 where R is the response, N is the value of the negative control, P is the value of the positive control, H is the Hill slope and pIC50 is the negative logarithm of the IC50.
| (1) |
Kinetic analysis of inhibitors against N5CAIR synthetase
In a 1 mL cuvette, 50 mM HEPES, pH 7.5 buffer, containing 20 mM KCl and 6.0 mM MgCl2, was added followed by 0.2 mM NADH, 2.0 mM PEP, 1.0 mM NaHCO3, 4 units of pyruvate kinase and 17 units of lactate dehydrogenase. To the cuvette, 168 ng of N5CAIR synthetase was added, followed by ATP and the cuvette was incubated at 37 °C for 3 minutes. For experiments in which ATP was held constant, the ATP concentration was 1.1 mM while the concentration of AIR was varied from 5 to 250 μM. For experiments in which AIR was held constant, the AIR concentration was held at 100 μM while ATP was varied from 5 to 250 μM. Background levels of ATP hydrolysis were measured, and the reaction was initiated by the addition of AIR and the inhibitor to be tested (0–50 μM). The reaction was incubated at 37 °C and NADH oxidation was monitored at 340 nm. The background ATPase activity was very low (<0.5%) and thus was not subtracted from the initial velocity. The initial velocity of each reaction was determined during the first two minutes, and a plot of initial velocity versus varied substrate was generated. Lineweaver-Burke plots of each experiment were conducted to determine the likely mode of inhibition. Secondary plots of the slope versus inhibitor concentration were used to determine the Ki value for each inhibitor. All graphing and curve fitting was done using KaleidaGraph.
Kinetic analysis of inhibitors against Human AIR Carboxylase:SAICAR Synthetase
His-tagged human AIR carboxylase:SAICAR synthetase was produced from a pET22b expression vector and purified using a cobalt-column (Pierce) according to the manufactures protocol. The protein was judged to be greater than 95% pure based upon SDS-PAGE analysis. Compounds 7, 13 and 14 were analyzed as inhibitors against the human bifunctional enzyme using the coupled assay as previously described for the G. gallus bifunctional AIR carboxylase:SAICAR synthetase13.
Acknowledgments
We thank Wayne State University, the American Heart Association (AHA 0855712G to SMF) and NIH (DK47814 to HMH) for funding. We also thank Dr. W. W. Cleland for his help with the interpretation of the kinetic data for compounds 1 and 2. We thank Martha Larsen and Dr. Robert Neubig of the CCG at the University of Michigan for their help with this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Appelbaum PC. Clin Microbiol Infect. 2006;12(Suppl 2):3–10. doi: 10.1111/j.1469-0691.2006.01402.x. [DOI] [PubMed] [Google Scholar]
- 2.Alekshun MN. Expert Opin Investig Drugs. 2005;14:117–34. doi: 10.1517/13543784.14.2.117. [DOI] [PubMed] [Google Scholar]
- 3.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. JAMA. 2007;298:1763–71. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 4.Lode H. Clin Microbiol Infect. 2005;11:778–87. doi: 10.1111/j.1469-0691.2005.01220.x. [DOI] [PubMed] [Google Scholar]
- 5.Carleton HA, Diep BA, Charlebois ED, Sensabaugh GF, Perdreau-Remington F. J Infect Dis. 2004;190:1730–8. doi: 10.1086/425019. [DOI] [PubMed] [Google Scholar]
- 6.Naimi TS, LeDell KH, Como-Sabetti K, Borchardt SM, Boxrud DJ, Etienne J, Johnson SK, Vandenesch F, Fridkin S, O’Boyle C, Danila RN, Lynfield R. JAMA. 2003;290:2976–84. doi: 10.1001/jama.290.22.2976. [DOI] [PubMed] [Google Scholar]
- 7.Hiramatsu K. Lancet Infect Dis. 2001;1:147–55. doi: 10.1016/S1473-3099(01)00091-3. [DOI] [PubMed] [Google Scholar]
- 8.Eliopoulos GM. Eur J Clin Microbiol Infect Dis. 2005;24:826–31. doi: 10.1007/s10096-005-0055-1. [DOI] [PubMed] [Google Scholar]
- 9.Stevens DL, Herr D, Lampiris H, Hunt JL, Batts DH, Hafkin B. Clin Infect Dis. 2002;34:1481–90. doi: 10.1086/340353. [DOI] [PubMed] [Google Scholar]
- 10.Arbeit RD, Maki D, Tally FP, Campanaro E, Eisenstein BI. Clin Infect Dis. 2004;38:1673–81. doi: 10.1086/420818. [DOI] [PubMed] [Google Scholar]
- 11.Herrero IA, Issa NC, Patel R. N Engl J Med. 2002;346:867–9. doi: 10.1056/NEJM200203143461121. [DOI] [PubMed] [Google Scholar]
- 12.Firestine SM, Poon SW, Mueller EJ, Stubbe J, Davisson VJ. Biochemistry. 1994;33:11927–34. doi: 10.1021/bi00205a031. [DOI] [PubMed] [Google Scholar]
- 13.Firestine SM, Davisson VJ. Biochemistry. 1994;33:11917–26. doi: 10.1021/bi00205a030. [DOI] [PubMed] [Google Scholar]
- 14.Firestine SM, Misialek S, Toffaletti DL, Klem TJ, Perfect JR, Davisson VJ. Arch Biochem Biophys. 1998;351:123–34. doi: 10.1006/abbi.1997.0512. [DOI] [PubMed] [Google Scholar]
- 15.Meyer E, Leonard NJ, Bhat B, Stubbe J, Smith JM. Biochemistry. 1992;31:5022–32. doi: 10.1021/bi00136a016. [DOI] [PubMed] [Google Scholar]
- 16.Meyer E, Kappock TJ, Osuji C, Stubbe J. Biochemistry. 1999;38:3012–8. doi: 10.1021/bi9827159. [DOI] [PubMed] [Google Scholar]
- 17.Mueller EJ, Meyer E, Rudolph J, Davisson VJ, Stubbe J. Biochemistry. 1994;33:2269–78. doi: 10.1021/bi00174a038. [DOI] [PubMed] [Google Scholar]
- 18.McFarland WC, Stocker BA. Microb Pathog. 1987;3:129–41. doi: 10.1016/0882-4010(87)90071-4. [DOI] [PubMed] [Google Scholar]
- 19.Polissi A, Pontiggia A, Feger G, Altieri M, Mottl H, Ferrari L, Simon D. Infect Immun. 1998;66:5620–9. doi: 10.1128/iai.66.12.5620-5629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samant S, Lee H, Ghassemi M, Chen J, Cook JL, Mankin AS, Neyfakh AA. PLoS Pathog. 2008;4:e37. doi: 10.1371/journal.ppat.0040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Mushegian A, Lory S, Jin S. Proc Natl Acad Sci U S A. 1996;93:10434–9. doi: 10.1073/pnas.93.19.10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cersini A, Martino MC, Martini I, Rossi G, Bernardini ML. Infect Immun. 2003;71:7002–13. doi: 10.1128/IAI.71.12.7002-7013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirsch DR, Whitney RR. Infect Immun. 1991;59:3297–300. doi: 10.1128/iai.59.9.3297-3300.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donovan M, Schumuke JJ, Fonzi WA, Bonar SL, Gheesling-Mullis K, Jacob GS, Davisson VJ, Dotson SB. Infect Immun. 2001;69:2542–8. doi: 10.1128/IAI.69.4.2542-2548.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perfect JR, Toffaletti DL, Rude TH. Infect Immun. 1993;61:4446–51. doi: 10.1128/iai.61.10.4446-4451.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baykov AA, Evtushenko OA, Avaeva SM. Anal Biochem. 1988;171:266–70. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- 27.Rowlands MG, Newbatt YM, Prodromou C, Pearl LH, Workman P, Aherne W. Anal Biochem. 2004;327:176–83. doi: 10.1016/j.ab.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 28.Shen Y, Volrath SL, Weatherly SC, Elich TD, Tong L. Mol Cell. 2004;16:881–91. doi: 10.1016/j.molcel.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 29.Liu S, Chang JS, Herberg JT, Horng MM, Tomich PK, Lin AH, Marotti KR. Proc Natl Acad Sci U S A. 2006;103:15178–83. doi: 10.1073/pnas.0604905103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spellberg B, Powers JH, Brass EP, Miller LG, Edwards JE., Jr Clin Infect Dis. 2004;38:1279–86. doi: 10.1086/420937. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Suarez R, Xu D, Veillette K, Davison J, Sillaots S, Kauffman S, Hu W, Bowman J, Martel N, Trosok S, Wang H, Zhang L, Huang LY, Li Y, Rahkhoodaee F, Ransom T, Gauvin D, Douglas C, Youngman P, Becker J, Jiang B, Roemer T. Chem Biol. 2007;14:1163–75. doi: 10.1016/j.chembiol.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Thoden JB, Firestine S, Nixon A, Benkovic SJ, Holden HM. Biochemistry. 2000;39:8791–802. doi: 10.1021/bi000926j. [DOI] [PubMed] [Google Scholar]
- 33.Marolewski A, Smith JM, Benkovic SJ. Biochemistry. 1994;33:2531–7. doi: 10.1021/bi00175a023. [DOI] [PubMed] [Google Scholar]