

Abstract
Enteric infections with attaching/effacing lesion-inducing bacterial pathogens are a worldwide health problem. A murine infection model with one such pathogen, Citrobacter rodentium, was used to elucidate the importance of the pleiotropic immune regulator, IL-6, in the pathogenesis of infection. IL-6 was strongly induced in colonic epithelial cells and macrophages upon C. rodentium infection and was required for effective host defense, because mice lacking IL-6 failed to control bacterial numbers 2–3 wk after infection and exhibited increased mortality. IL-6 was not needed for mounting effective T and B cell responses to the pathogens, nor was it important for induction of IFN-γ or TNF-α, cytokines involved in host defense against the bacteria, or the antibacterial effector, NO. Instead, IL-6 played a key role in mucosal protection, since its absence was associated with marked infection-induced apoptosis in the colonic epithelium and subsequent ulcerations. Cell culture studies confirmed that IL-6 protected colon epithelial cells directly against inducible apoptosis, which was accompanied by increased expression of an array of genes encoding anti-apoptotic proteins, including Bcl-xL, Mcl-1, cIAP-2, and Bcl-3. Ulcerations appeared to be pathogenetically important, because bacteria localized preferentially to those regions, and chemically induced colonic ulcerations promoted bacterial colonization. Furthermore, blood components likely present in ulcer exudates, particularly alanine, asparagine, and glycine, promoted bacterial growth. Thus, IL-6 is an important regulator of host defense against C. rodentium by protecting the mucosa against ulcerations which can act as a microbial niche for the bacteria.
Diarrheal disease caused by enteropathogenic Escherichia coli (EPEC)3 and enterohemorrhagic E. coli is a significant worldwide health problem (1). Insights into the pathogenesis of these infections are of great importance for improving prevention and treatment strategies, yet availability of reliable small animal models is limited. The murine bacterial pathogen, Citrobacter rodentium, shares functional and structural features with human EPEC strains, most important of which is the ability to attach intimately to the apical surface of the intestinal epithelium and induce localized microvillus destruction, a characteristic feature known as attaching/effacing lesion (2). The gene coding for the outer membrane protein responsible for intimate attachment, intimin, is functionally homologous in C. rodentium and clinical EPEC strains (3). C. rodentium, like EPEC, colonizes the colonic mucosa but does not invade deeper layers of the mucosa or spread systemically (4).
C. rodentium infection of mice causes epithelial hyperplasia in the colon and cecum, goblet cell loss, and mucosal infiltration with macrophages, lymphocytes, and neutrophils (5, 6). Immunocompetent mice eradicate infection after 3–4 weeks and develop immunity against secondary challenge (7, 8). Effective immune defense against C. rodentium requires CD4 T cells and B cells (8, 9). IgG Abs may play a role in controlling infection, whereas secretory IgA or IgM Abs have limited or no involvement in clearance (8). NO can kill the bacteria in vitro, and its production through inducible NO synthase (iNOS) has antimicrobial functions in vivo (10). Moreover, the cytokines IL-12, IFN-γ, TNF-α, and IL-17 are up-regulated in the colon of infected mice and are necessary for effective immune defense (11–13).
IL-6 was discovered some 20 years ago as a B cell activation and differentiation factor (14) but was subsequently shown to have other immune activities, as well as functions in hemopoietic, homeostatic, and neuroendocrine processes (15, 16). The cytokine enhances Ig secretion by activated B cells and causes their terminal differentiation into plasma cells (17). Mice deficient in IL-6 show reduced IgG2a production and may have attenuated mucosal IgA responses (18), although other studies have observed normal IgA responses in these mice (19). IL-6 promotes the acute phase response elicited by infection or tissue damage (18) and is required for the development of experimentally induced arthritis and colitis in mice (20, 21), indicating that it has proinflammatory functions. In contrast, IL-6 also inhibits the synthesis of the classical proinflammatory cytokines, TNF-α and IL-1, in monocytes/macrophages (22) and protects against tissue inflammation in some experimental models (23), which shows that it can be anti-inflammatory under certain conditions. IL-6 plays a role in systemic immune defense against the bacterial pathogens, Listeria monocytogenes and Mycobacterium tuberculosis, and the fungal pathogen, Candida albicans (24–26). Together, these observations indicate that the specific functions of IL-6 in regulating immune and inflammatory responses are variable and highly dependent on the pathophysiological context.
The goal of the present studies was to define the role of IL-6 in the pathogenesis of infection with C. rodentium as a prototypical attaching/effacing lesion-inducing bacterial pathogen. We show here that IL-6 expression was induced locally in epithelial cells and macrophages in the colon upon infection and that it was required for effective clearance of the bacteria. Moreover, IL-6 deficiency caused more severe infection-associated mucosal inflammation and ulceration, indicating that this cytokine was protective against colitis caused by C. rodentium infection.
Materials and Methods
Mice
All mice were obtained from The Jackson Laboratory and were maintained under specific pathogen-free conditions. IL-6-deficient mice were originally generated on a mixed C57BL/6 × 129 background (18), and subsequently back-crossed for >10 generations to a C57BL/6J background. B6129SF2/J and C57BL/6J mice, respectively, were used as controls. Initial studies showed that the genetic background had no significant impact on the differences in the inflammatory and immune responses to C. rodentium between wild-type and IL-6-deficient mice, allowing us to combine the data from these studies. Results from males and females are reported together, as no significant differences were detected between the genders in the course or severity of the infections. Mice with SCID were on a C57BL/6J background. Animal studies were approved by the University of California at San Diego Institutional Animal Care and Use Committee.
Bacterial infections
After overnight growth in Luria-Bertani broth at 37°C, bacteria were centrifuged and resuspended in fresh Luria-Bertani broth at a concentration of 2.5 × 109 CFU/ml. Adult (>10-wk-old) mice were infected with 200 μl of the bacterial suspension (5 × 108 bacteria) by oral gavage. Bacterial numbers in fecal and organ homogenates were determined as described before (8). Fecal pellets were collected over a 2- to 3-h period, weighed, and homogenized in 5 ml of saline. Spleens were homogenized in 2 ml of sterile saline. Serial dilutions of the homogenates were plated onto Mac-Conkey’s agar, and the number of CFU was determined after overnight incubation at 37°C. The detection limit of the assay was 103 CFU/g feces and 101 CFU per spleen. The identity of representative colonies was verified by PCR analysis (8).
Colitis induction with dextran sulfate sodium (DSS)
Mice were given 3% DSS (35–50 kDa; MP Biomedicals) in the drinking water for 5 days, with daily monitoring of body weight. For subsequent infections, mice were returned to normal drinking water for an additional 5 days. Mice not treated with DSS were used as controls. Mice were infected with 5 × 108 C. rodentium by oral gavage, and CFU numbers in the stool were determined at different times afterward. In parallel, representative mice were taken for histological analysis of the colon.
Histological analysis
Organs were removed and fixed overnight in Bouin’s solution at room temperature. Colons were opened longitudinally and processed as Swiss rolls before fixation. Fixed tissues were embedded in paraffin, and 5-μm sections were prepared and stained with H&E. Crypt depths were determined microscopically with a calibrated eyepiece reticle. Multiple sites were measured throughout each organ, and the 2 highest values obtained from different sites at least 10 crypts apart were used to calculate the maximal crypt depth for each sample. The extent of epithelial ulceration was determined by image analysis of digital photomicrographs of the entire colon. Colon infiltration with inflammatory cells was scored for mucosa (0, normal; 1, mild infiltration involving <10% of mucosal thickness; 2, moderate infiltration involving 10–50% of mucosal thickness; 3, severe infiltration involving >50% of mucosa), submucosa (0, normal; 1, mild to moderate infiltration; 2, severe infiltration), and muscularis and serosa (0, normal; 1, significant infiltration). Scores of the two most affected segments of the entire colon were averaged, resulting in a total range of 0–6 for in each animal. All histological scoring was performed in a blinded manner with regard to genotype and treatment.
For immunohistological studies, tissues were fixed in a zinc-formalin solution, and paraffin sections were prepared, deparaffinized, and incubated with 0.3% H2O2 for 20 min at room temperature to inactivate endogenous peroxidase. Sections were blocked with PBS containing 2% IgG-free BSA and 2% serum of the same species as the secondary Ab and incubated overnight at 4°C in the same buffer containing 1 μg/ml goat anti-mouse IL-6 Ab (R&D Systems), a 1/1,000 dilution of rabbit anti-C. rodentium (a gift from B. Vallance (British Columbia Children’s Hospital, Vancouver, Canada); Ref. 10), or 1 μg/ml monoclonal rat anti-mouse F4/80 Ag (BD Pharmingen) or, as respective controls, normal goat, rabbit, or rat IgG. Sections were washed twice for 10 min each with 0.3 M NaCl, 50 mM Tris (pH 7.6), 0.1% Tween 20 and further incubated for 1 h at room temperature with 3 μg/ml biotin-labeled donkey anti-goat IgG, 1 μg/ml HRP-labeled donkey anti-rabbit IgG, or HRP-labeled goat anti-rat IgG. If needed, sections were washed and incubated for 30 min at room temperature with HRP-labeled streptavidin. Sections were developed with 3,3′-diaminobenzidine and H2O2, and counterstained with Gill’s hematoxylin.
Apoptotic cells in the colon were detected by the TUNEL technique. Briefly, paraffin sections of the colon were prepared and stained by reacting DNA ends with FITC-labeled dCTP using terminal transferase (In Situ Cell Death Detection Kit; Roche Applied Science) according to the manufacturer’s instructions. After TUNEL, sections were stained with rabbit anti-C. rodentium (10), washed, incubated with 3 μg/ml Cy3-labeled goat anti-rabbit IgG (Jackson ImmunoResearch) for 30 min at room temperature, and mounted with VECTASHIELD Mounting Media containing 4′,6′-diamidino-2-phenylindole (Vector Laboratories).
Myeloperoxidase analysis
Myeloperoxidase activity was determined by enzymatic assay. Colon segments were weighed and homogenized for 30 s on ice in an extraction buffer of 50 mM phosphate buffer (pH 6.0) and 0.5% hexadecyltrimethylammonium bromide. The resulting solution was centrifuged at 18,000 × g for 30 min at 4°C. Supernatants (10 μl) and 190 μl of a freshly prepared solution of 5 mM o-dianisidine dihydrochloride and 0.0005% H2O2 in extraction buffer were added to a microtiter plate, and color development was determined after 2–3 min at 450 nm in a microplate reader. Purified HRP of known enzymatic activity (Calbiochem) was used as a standard. Specific myeloperoxidase activity was normalized against tissue weight.
Cell culture
T84 human colon cancer cells were seeded into 12-well plates in 50% DMEM, 50% Ham’s F12 medium supplemented with 5% newborn calf serum. Upon reaching confluence, fresh medium was added, and cells were treated for 24 h with 100 ng/ml human IL-6 or were left untreated as controls. Apoptosis was induced by stimulation with 100 ng/ml anti-Fas Abs (clone CH-11; MBL International) for 18 h, after which apoptosis was assayed by ELISA for cytoplasmic nucleosome release (Cell Death Detection ELISA; Roche).
Real-time PCR analysis
Total RNA was extracted using Trizol reagent (Invitrogen). Reverse transcription and real-time PCR amplification, using the SYBR Green PCR Master Mix kit (Applied Biosystems). The primers, annealing temperatures, and expected PCR product sizes were as follows: mouse IL-6, 5′-ACAACCACGGCCTTCCCTAC-3′ (sense),5′-ACAATCAGAATTGCCA TTGCAC-3′ (antisense), 58°C, 168 bp; mouse iNOS, 5′-CCCAGAGTTC CAGCTTCTGG-3′ (sense), 5′-CCAAGCCCCTCACCATTATCT-3′ (antisense), 60°C, 51 bp; mouse IFN-γ, 5′-TGAACGCTACACACTGCA TCTTGG-3′ (sense), 5′-CGACTCCTTTTCCGCTTCCTGAG-3′ (anti-sense), 60°C, 460 bp; mouse TNF-α, 5′-ATGAGCACAGAAAGCATGA TC-3′ (sense), 5′-TACAGGCTTGTCACTCGAATT-3′ (antisense), 60°C, 276 bp; mouse GAPDH, 5′-ATCAACGACCCCTTCATTGACC-3′ (sense), 5′-CCAGTAGACTCCACGACATACTCAGC-3′ (antisense), 59°C, 209 bp; human Bcl-2, 5′-TGGTGGAGGAGCTCTTCAGGGA-3′ (sense), 5′-GGTTCAGGTACTCAGTCATCCACA-3′ (antisense), 60°C, 150 bp; human Bcl-xL, 5′AAGGGACTGAATCGGAGATGGAGA-3′ (sense), 5′-AGCTGGGATGTCAGGTCACTGAA-3′ (antisense), 60°C, 220 bp; human Mcl-1, 5′-CTG GTA ATA ACA CCA GTA CGG ACG-3′ (sense), 5′-TGCCCATTGGCTTTGTGTCCTT-3′ (antisense), 60°C, 152 bp; human cIAP-2, 5′-CATGGGTTCAACATGCCAAGTGGT-3′ (sense), 5′-TCCACGGCAGCATTAATCACAGGA-3′ (antisense), 60°C, 238 bp; human survivin, 5′-TTTCTCAAGGACCACCGCATCTCT-3′ (sense), 5′-CTTGAAGCAGAAGAAACACTGGGC-3′ (antisense), 60°C, 150 bp; human Bcl-3, 5′-CAGTGAACACCGAGTGCCAAGAAA-3′ (sense), 5′-GG AGTACATTTGCGCGTTCACGTT-3′ (antisense), 60°C, 179 bp. Amplification of the expected single products was confirmed on 1% agarose gels stained with ethidium bromide. Relative changes in target mRNA levels were calculated as 2ΔCt, where ΔCt = (Ct target control − Ct GAPDH control) − (Ct target stimulated − Ct GAPDH stimulated) and Ct designates the cycle number at which specific fluorescence crossed the detection threshold.
Epithelial cell isolation
Colons were removed, cut open longitudinally, and washed with cold PBS. For mucus removal, the tissue was incubated for 10 min at room temperature in 1 mM DTT in PBS. The tissue was rinsed, cut into 0.5-cm pieces, and incubated for 20 min at 37°C with gentle shaking in prewarmed HBSS (Ca+ and Mg+ free) containing 5 mM EDTA, 5% FCS, 15 mM HEPES (pH 7.3), and 0.5 mM DTT. The supernatant was collected and the remaining tissue was washed three times with PBS. The supernatant and washes were pooled together and passaged through a 100-μm pore size nylon mesh strainer. The collected epithelial cells were centrifuged at 300 × g for 10 min at 4°C, and the pellet was resuspended in Trizol reagent.
Analysis of colonic cytokine and NO production
For the analysis of cytokine and NO production in explant cultures, the colon was cut into 2- × 2-mm2 pieces, which were placed into a 12-well plate in 500 μl of RPMI 1640 with 100 μg/ml gentamicin and 50 μg/ml each streptomycin and penicillin and incubated for 6 h at 37°C. Supernatants were centrifuged to remove debris and were stored at − 80°C. The remaining tissue pieces were subsequently dried and weighed to allow normalization of cytokine secretion. Cytokine levels in the supernatants were assayed by ELISA (R&D Systems). Levels of the stable NO breakdown product, nitrite, were determined with the Griess reaction.
Determination of Ab titers
Ab titers against C. rodentium were determined by ELISA (8). Briefly, 50 μl/well of a C. rodentium suspension (2 × 109 cells/ml in water) were added to 96-well polystyrene plates, air-dried overnight at room temperature, and fixed for 5 min at room temperature with 0.15% glutaraldehyde in 0.15 M phosphate buffer (pH 7.0). Unreacted aldehyde groups were blocked with 0.15 M glycine in 15 mM phosphate buffer (pH 7.0), and plates were incubated overnight at 4°C with PBS containing 5% nonfat dry milk and 0.5% Tween 20. Serum samples were added, and plates were incubated for 2 h at room temperature, followed by 1 h of incubation at room temperature with peroxidase-conjugated goat Abs against mouse IgG, IgG1, IgG2a, or IgM. Bound peroxidase was visualized with tetramethylbenzidine-H2O2 in acetate buffer, and reactions were stopped with sulfuric acid and read at 450 nm.
Bacterial growth studies
Blood from normal mice was obtained by cardiac puncture. Serum and blood cells were separated by centrifugation at 1000 × g for 10 min at room temperature. C. rodentium was grown overnight in Luria-Bertani broth, washed in PBS, and suspended 105/ml in 1% DMEM in PBS in the absence or presence of whole blood, blood cell lysate, or serum. Final concentrations were adjusted to 25 mg/ml total protein, as determined by Bradford assay with BSA as a standard. To define serum factors important in promoting bacterial growth, fresh serum was first depleted of critical nutrients by growing C. rodentium in 100% serum overnight. Depleted and fresh sera were diluted to 30% in PBS and supplemented with specific amino acids at different concentrations, as expressed by a percentage (1–100%) of their physiological levels in normal serum. Bacteria were then added at 104–105/ml, cultures were grown at 37°C for different times, and CFUs were determined as described above.
Data analysis
Colony counts were log10 transformed, and means and SEs of the mean were calculated from the log values. Samples without detectable C. rodentium colonies were assigned a log10 value equivalent to one-half of the detection limit of the CFU assay. Differences between groups of mice were evaluated by Mann-Whitney rank sum test or Student’s t test, as appropriate. Survival data were analyzed by Kaplan-Meier survival statistics. Differences with a p value of <0.05 were considered significant.
Results
Increased colonic IL-6 expression after C. rodentium infection
To investigate the role of IL-6 in mucosal host defense against C. rodentium, we first examined the expression of the cytokine during the course of infection. Oral inoculation of normal mice with C. rodentium leads to maximal colonic and cecal colonization within 1 wk, followed by bacterial clearance within 3–4 wk (Ref. 8; see Fig. 3). Analysis of colonic IL-6 mRNA levels by real-time PCR revealed increased expression at 1 wk, peak levels by 2 wk with a ~100-fold increase, and a decrease to baseline by 3–4 wk (Fig. 1A). Enhanced IL-6 mRNA expression was not observed in the small intestine, mesenteric lymph nodes, liver, or spleen of infected mice (Fig. 1B), indicating that the IL-6 response was localized to the site of infection in the colon. Increased mRNA levels were paralleled by greater IL-6 secretion in colonic explant cultures 2 wk after infection (Fig. 1C). Addition of the protein synthesis inhibitor, cycloheximide, abolished IL-6 production in the cultures (Fig. 1C), indicating that the cytokine was actively produced and secreted under these conditions rather than merely present in the tissue at elevated levels upon explantation.
FIGURE 3.
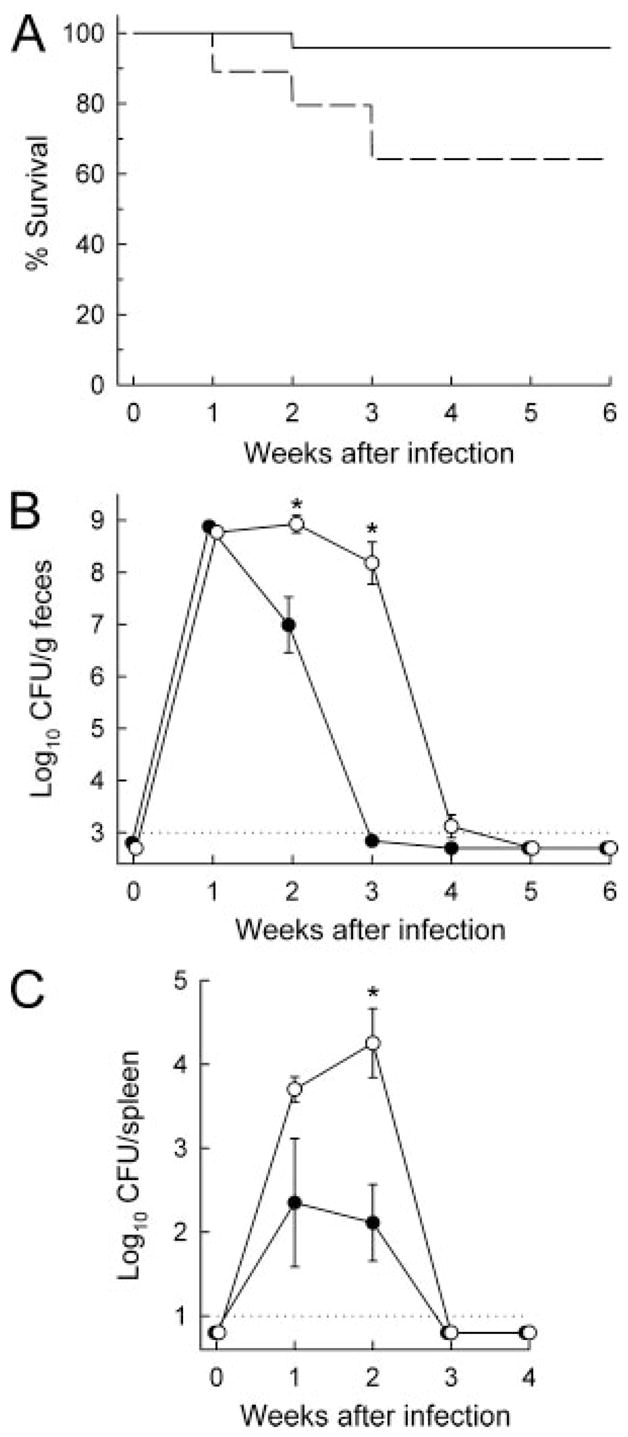
C. rodentium infection of IL-6-deficient mice. IL-6-deficient (○, – – – –; n = 21) and wild-type mice (●, —; n = 14) were infected orally with C. rodentium and observed for survival for up to 6 wk (A). IL-6-deficient mice showed significantly lower survival than controls (p < 0.01 by Kaplan-Meier survival statistics). Bacterial numbers in fecal and spleen homogenates were determined weekly (B and C). Data are mean ± SEM of at least eight mice for each data point (B and C). *, p < 0.05.
FIGURE 1.
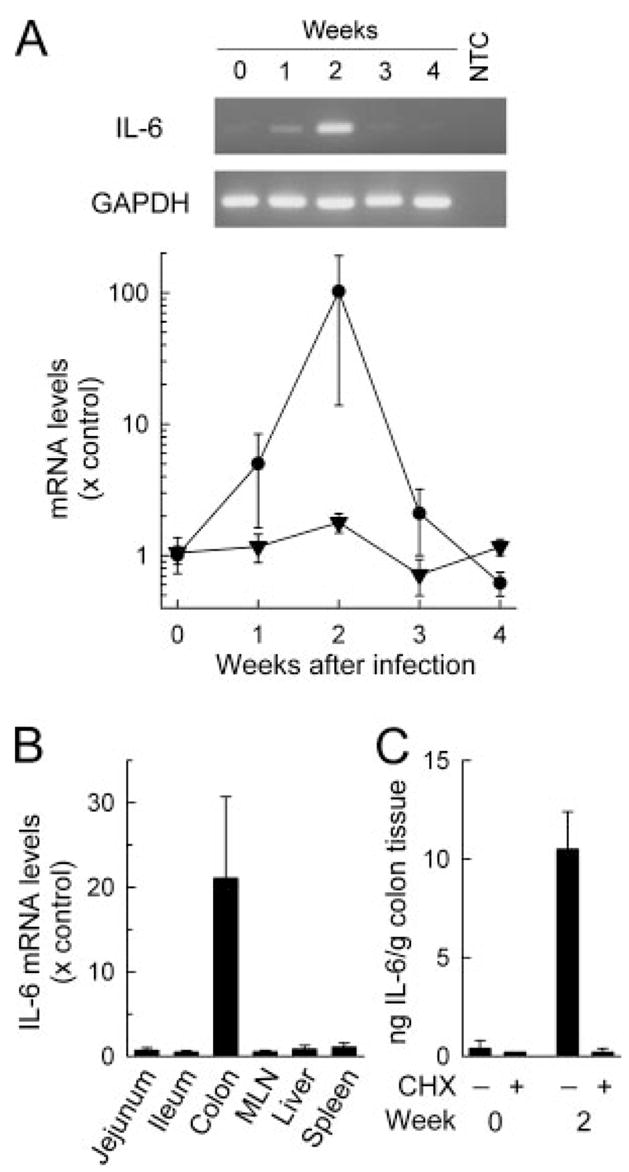
Increased colonic IL-6 expression after C. rodentium infection. Wild-type (C57BL/6J) mice were infected orally with 5 × 108 C. rodentium, and organs were removed at the indicated times (A) or after 2 wk (B and C). Uninfected mice were used as controls (wk 0). Total RNA was extracted and analyzed for mRNA expression of IL-6 (●) or GAPDH (▼) by qualitative (A, top) or real-time PCR (A, bottom; and B). As a PCR negative control, RNA was omitted from the reactions (no template control; NTC). Levels of mRNA are expressed relative to those of uninfected controls. Colon fragments were cultured for 6 h with or without cycloheximide (CHX), and IL-6 levels in the supernatants were determined by ELISA (C). Data represent the mean ± SEM of three or more mice.
We next used an immunohistological approach to determine the cellular source of IL-6 after infection. Colon sections stained with an Ab against IL-6 showed increased levels of the cytokine in the epithelium and in mononuclear cells in the lamina propria at 2 wk, whereas noninfected controls exhibited only low levels of expression (Fig. 2A). Staining of adjacent sections with an Ab directed against the macrophage marker F4/80 indicated that many of the IL-6-positive cells in the lamina propria were macrophages (Fig. 2B). To confirm the staining results with an independent method, real-time PCR analysis was performed on isolated epithelial cells. Infection induced a marked (9-fold) increase in epithelial IL-6 mRNA expression when compared with uninfected controls. Thus, IL-6 is produced at increased levels after C. rodentium infection by at least two different cell types in the colon, epithelial cells, and macrophages.
FIGURE 2.
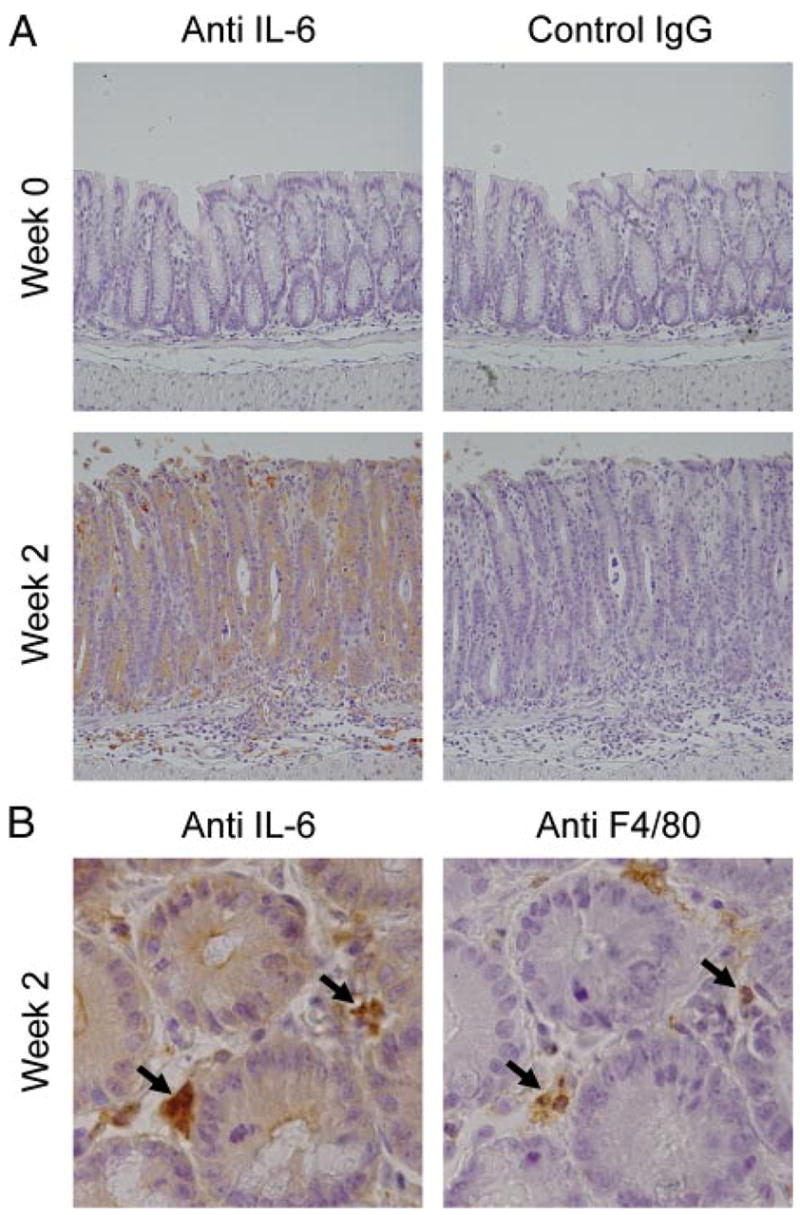
Immunohistological analysis of colonic IL-6 expression. Wild-type mice 2 wk after oral C. rodentium infection and uninfected control mice were analyzed by indirect immunoperoxidase staining for expression of IL-6 (A and B) and the macrophage marker, F4/80 (B). As staining controls, sections were incubated with an isotype-matched control Ab (control IgG). Arrows in B depict cells that costain for IL-6 and F4/80.
IL-6 is required for effective host defense against C. rodentium
The strong colonic IL-6 response to C. rodentium infection suggested a potential role of this cytokine in host defense against the bacteria. To define the physiological importance of IL-6 in this process, we conducted oral challenge studies in mice with a targeted mutation in the IL-6 gene (18) and their wild-type controls. Very few (<5%) control mice died during the 6-wk course of the infection, while infection-related mortality reached 35% in IL-6-deficient mice by 3 wk (Fig. 3A). Analysis of the bacterial burden revealed that both groups of mice had similar infectious loads after 1 wk, as indicated by comparable bacterial numbers in fecal homogenates (Fig. 3B), a measure reflective of colonic and cecal bacterial colonization (7, 8). Control mice cleared the infection rapidly thereafter, with few or no detectable fecal bacteria after 3–4 wk (Fig. 3B). In contrast, IL-6-deficient mice showed a marked and highly reproducible defect in bacterial clearance, with 100- to >1,000-fold increased fecal bacterial numbers compared with wild-type mice after 2–3 wk (Fig. 3B). In parallel, bacterial colonization of the spleen was 50- to 100-fold higher in IL-6-deficient relative to wild-type mice at 1–2 wk (Fig. 3C). Despite the important function of IL-6 in host defense, the cytokine was ultimately not necessary for eradication, because bacterial numbers were below detectable levels after 6 wk in IL-6-deficient mice (Fig. 3B). These results show that IL-6 plays an essential role in the early phase of developing an effective host defense against C. rodentium.
IL-6 deficiency exacerbates mucosal inflammation and damage caused by C. rodentium infection and chemical irritants
Infection with C. rodentium causes mucosal inflammation in the colon (8). Because IL-6 can promote intestinal inflammation (21), we asked whether IL-6 deficiency had any impact on the severity of the infection-associated colitis. Wild-type mice exhibited marked epithelial hyperplasia and modest infiltration with mononuclear and neutrophils into mucosa and submucosa at 2–3 wk (Fig. 4). Epithelial erosions and ulcerations were only rarely observed in these mice (Fig. 4C). In comparison, IL-6-deficient mice exhibited a similar degree of epithelial hyperplasia (Fig. 4, A and B) but had extensive epithelial ulcerations and marked mucosal infiltration with inflammatory cells at 2–3 wk (Fig. 4, A, C, and D), which was accompanied by increased levels of the neutrophil marker myeloperoxidase in colon homogenates at 2 wk (Fig. 4E). Hyperplasia and inflammatory infiltration were transient in both strains of mice, with resolution coinciding with bacterial clearance (Figs. 3B and 4, B and D). Thus, the absence of IL-6 enhanced, rather than abrogated, the mucosal inflammatory response to C. rodentium.
FIGURE 4.
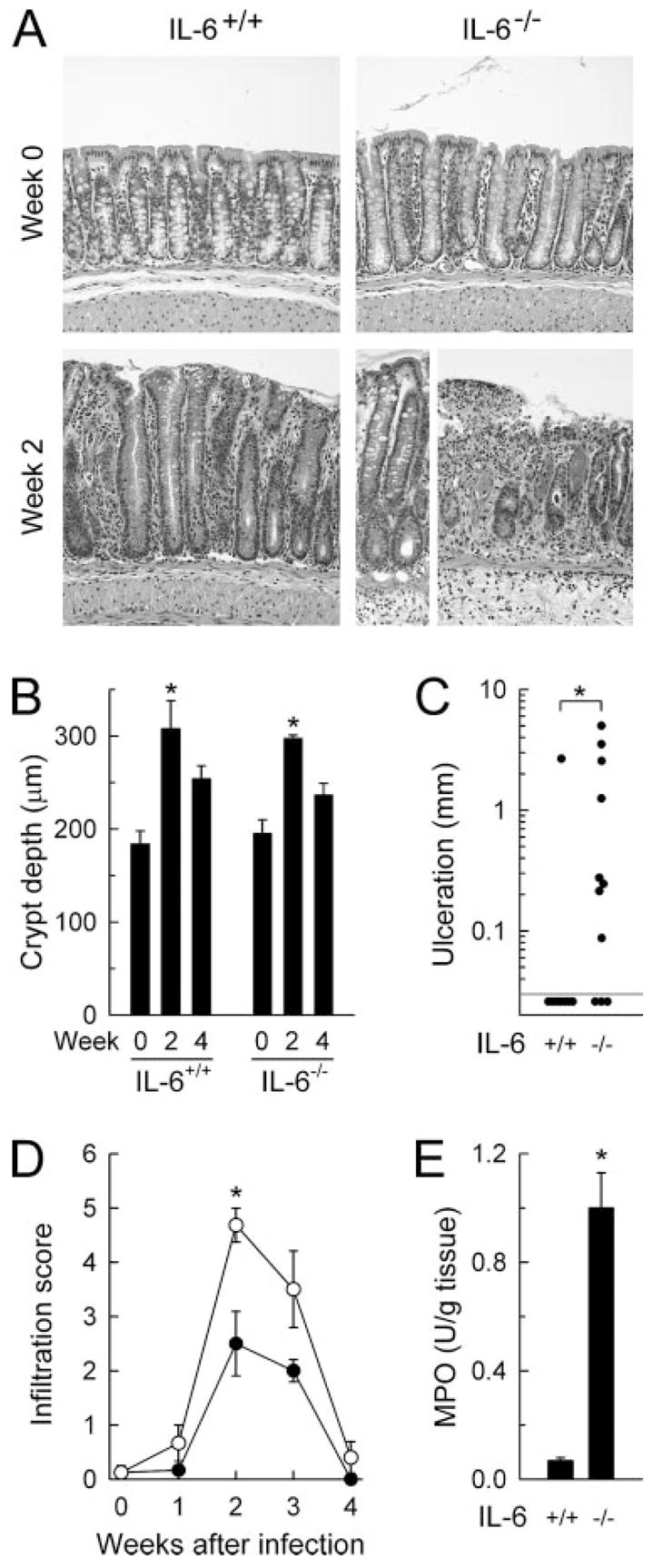
Histological analysis of infection-associated colitis in IL-6-deficient mice. IL-6-deficient mice (IL-6−/−, ○) and wild-type controls (IL-6+/+, ●) were infected orally with C. rodentium or left uninfected as controls (wk 0). The colon was removed at the indicated times, paraffin sections were prepared, stained with H&E, and assessed for damage and inflammatory cell infiltration. A, Representative colon areas, showing similar hyperplasia but greater epithelial ulceration and mucosal infiltration with inflammatory cells after infection in IL-6-deficient mice compared with wild-type controls. Crypt depths (B) and epithelial ulceration (C) were determined morphometrically, and infiltration of mucosa and submucosa with inflammatory cells was scored semiquantitatively (D). Colon homogenates were analyzed for neutrophil infiltration by enzymatic assay for myeloperoxidase (MPO) activity (E). Uninfected mice had no detectable myeloperoxidase activity in the colon (<0.01 U/g tissue). Data are mean ± SEM of three or more mice (B, D, and E) or are results from individual mice (C). *, p < 0.05.
To examine the mechanisms responsible for increased mucosal damage in infected IL-6-deficient mice, we tested the possibilities that either increased bacterial load or greater general host susceptibility to inflammatory challenges were important. C. rodentium infection of SCID mice, which are devoid of T and B cells and have very high bacterial numbers upon C. rodentium infection (log10 CFU/g feces at 2 wk: 9.8 ± 0.2 in SCID mice, n = 7, compared with 8.9 ± 0.1 in IL-6-deficient mice, n = 18), did not experience mucosal ulceration after 2 wk (Fig. 5A), suggesting that a greater bacterial load alone was not responsible for increased mucosal damage in IL-6-deficient mice. In contrast, challenge of IL-6-deficient mice with a chemical irritant, the colitis-inducing agent DSS (27), caused greater mucosal inflammation and ulceration compared with controls, which was paralleled by enhanced body weight loss as a clinical marker of overall disease severity (Fig. 5, B–D). Taken together, these results indicate that IL-6 deficiency renders the host more susceptible to mucosal damage caused by different agents, both infectious and noninfectious ones.
FIGURE 5.
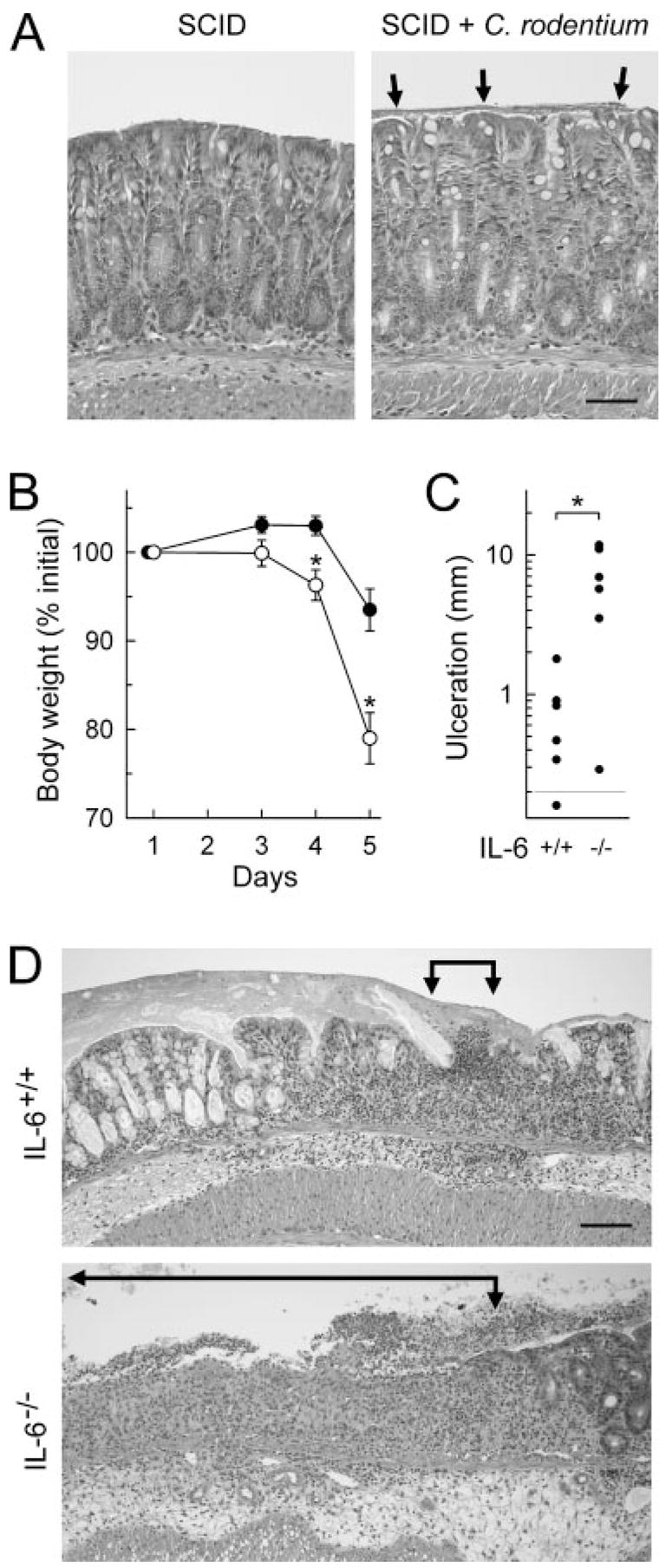
Increased susceptibility of IL-6-deficient mice to chemically induced colitis. A, Adult SCID mice were infected orally with C. rodentium or left uninfected as a control. After 2 wk, paraffin sections of the colon were prepared and stained with H&E. Arrows indicate a bacterial layer on the mucosal surface in infected mice, most likely consisting of C. rodentium (Fig. 8). Mucosal ulcerations were not observed after infection. B–D, IL-6-deficient mice (IL-6−/−, ○) and wild-type controls (IL-6+/+, ●) were given 3% DSS in the drinking water for 5 days. Body weights were determined at the indicated times (B), and colon histology was evaluated on H&E-stained paraffin sections on day 5. Total colonic ulceration was determined morphometrically (C). Representative sections are shown in D, with ulcerated areas indicated by arrows. Results are mean ± SEM (B) or are values from individual mice (C). Bars, 50 and 100 μm, respectively, in A and D. *, p < 0.05 compared with wild-type mice.
IL-6 protects colon epithelial cells from apoptosis
Because IL-6-deficient mice exhibited greater infection-induced mucosal ulceration, we explored potential mechanisms that might account for IL-6-dependent mucosal protection. Formation of frank epithelial ulcerations can be preceded by focal apoptosis of epithelial cells in the colon (28). We therefore evaluated the occurrence of apoptosis after C. rodentium infection in IL-6-deficient and wild-type mice by the TUNEL technique. Uninfected mice showed little apoptosis and no difference between the groups (Fig. 6A). In contrast, 7–10 days after infection, a time at which bacterial loads are comparable between the groups (Fig. 3B), IL-6-deficient mice displayed markedly more apoptosis in localized regions of colonic surface epithelium, as well as scattered throughout the crypt regions (Fig. 6A). Furthermore, apoptosis of surface epithelial cells was readily apparent by H&E staining in IL-6-deficient mice by 10 days after infection, with detachment and nuclear condensation of numerous epithelial cells (Fig. 6A).
FIGURE 6.
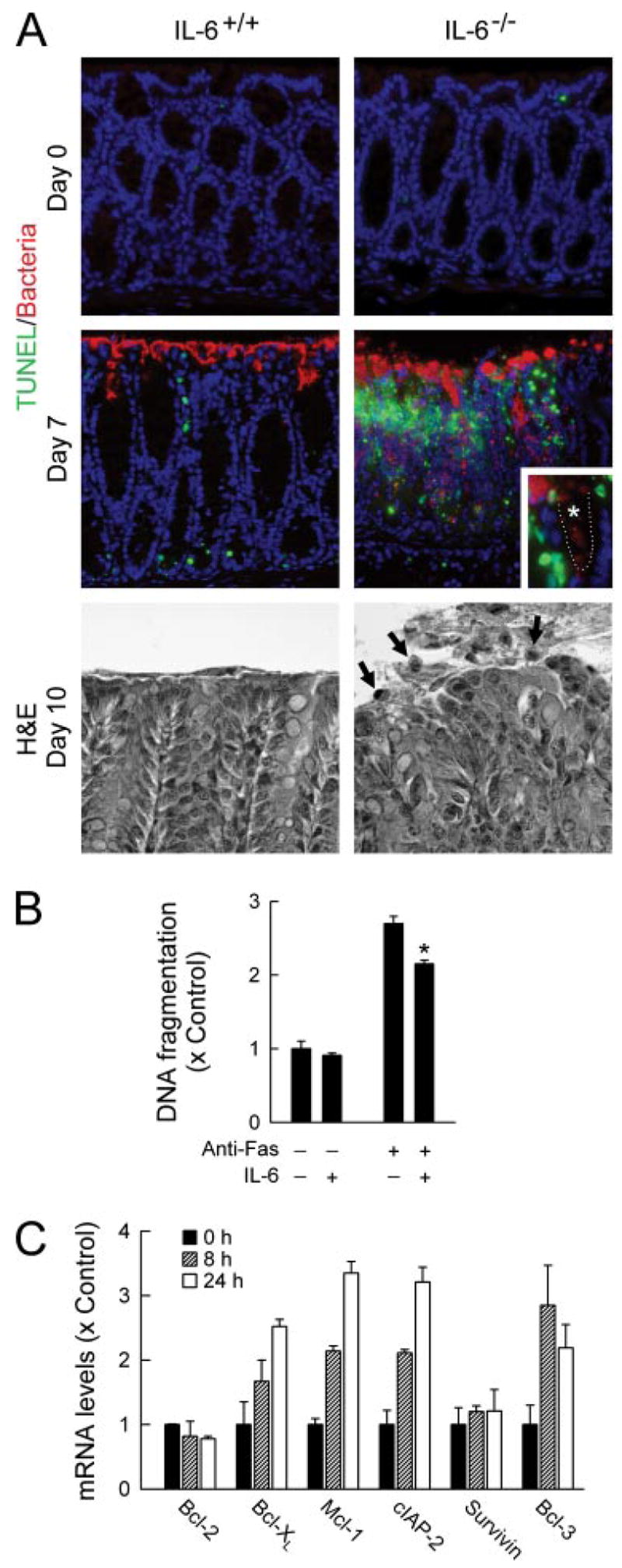
IL-6-dependent protection of colon epithelial cells against apoptosis. A, IL-6-deficient mice (IL-6−/−) and wild-type controls (IL-6+/+) were infected orally with C. rodentium or left uninfected as controls (day 0). The colon was removed at the indicated times, paraffin sections were prepared, double-stained for apoptotic cells by the TUNEL technique (green) and for C. rodentium by specific Abs (red), and counterstained with 4′,6′-diamidino-2-phenylindole (A, top). Inset, crypt region of an IL-6-deficient mouse at higher magnification. ....., Epithelial surface; *, crypt lumen. Parallel sections were stained with H&E (A, bottom). Arrows, apoptotic cells. B, T84 colon epithelial cells were incubated with or without IL-6 for 24 h and treated with anti-Fas Abs (clone CH-11) for an additional 18 h. Apoptosis was assayed with a nucleosome release ELISA. Values are mean ± SD (n = 3) and are expressed relative to unstimulated controls. *, p < 0.05 compared with cells not treated with IL-6. C, T84 cells were stimulated with 100 ng/ml IL-6 for 8 or 24 h or were left unstimulated (0 h), and mRNA levels for the indicated genes were determined by real-time RT-PCR. Values are mean ± SD (n = 3) and are expressed relative to unstimulated controls.
To determine whether IL-6 can directly protect intestinal epithelial cells against apoptosis, we used the T84 human colon epithelial cell culture model (29). Ab-mediated cross-linking of the Fas death receptor increased apoptosis of the cells, which was significantly reversed by IL-6 stimulation (Fig. 6B). Protection against apoptosis was accompanied by IL-6-induced up-regulation of several genes encoding proteins with antiapoptotic functions, including the Bcl family members Bcl-xL and Mcl-1 (but not Bcl-2), the IAP family member cIAP-2 (but not survivin), and the NF-κB family member, Bcl-3 (Fig. 6C). These data suggest that IL-6 can directly protect the colonic epithelium against microbe-induced apoptosis through the induction of antiapoptotic proteins.
IL-6 is dispensable for the induction of a specific Ab response to C. rodentium
We next turned our attention to the mechanisms that govern IL-6-dependent bacterial clearance. B cells play a central role in clearance of C. rodentium, a function that may be mediated, at least in part, by antibacterial IgG or IgM Abs (8). Furthermore, IL-6 is a growth and differentiation factor for B cells (17), so its deficiency could affect the development of an effective Ab response to C. rodentium. To evaluate this possibility, we determined antibacterial Ab titers in the serum of normal and IL-6-deficient mice during the course of infection. In both groups of mice, antibacterial IgM and IgG Abs were first observed at 1 wk and reached maximal levels 2–3 wk after infection (Fig. 7A). IL-6-deficient mice had comparable IgM titers throughout the infection, but moderately higher IgG titers. The latter was also reflected in higher titers of antibacterial IgG1 and IgG2a in IL-6-deficient mice, as shown by further IgG subclass analysis (data not shown). IgG3 and IgA titers were not determined because our prior studies had shown that these isotypes play no role in bacterial clearance (8). Thus, IL-6 was not required for mounting a specific Ab response to C. rodentium, indicating that specific Abs do not mediate the protective effects of IL-6 in antibacterial host defense. These data also suggest that the interactions among T cells, B cells, and dendritic cells needed for the development of a specific Ab response were not impaired in the absence of IL-6 (30, 31).
FIGURE 7.
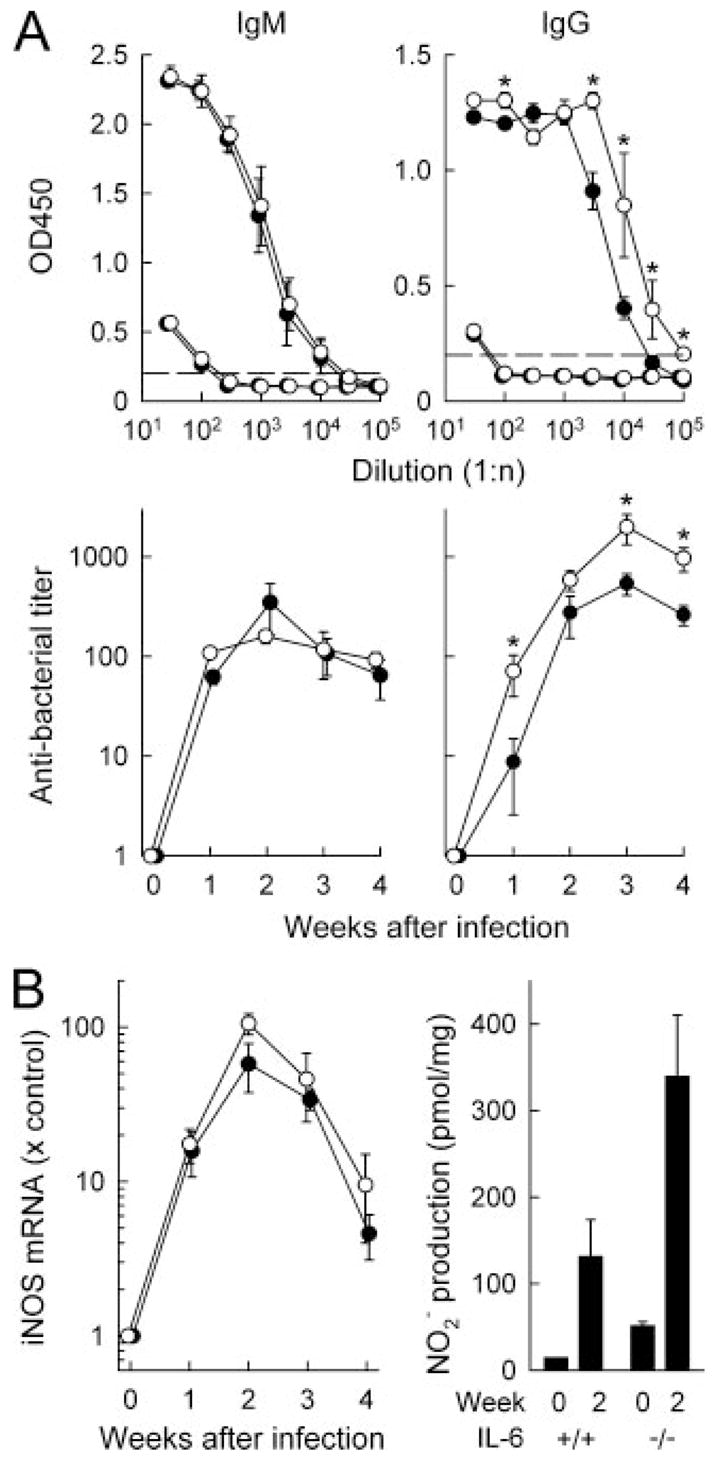
IL-6 deficiency does not compromise development of antibacterial Abs or NO production. A, Serum samples were collected from IL-6-deficient (○) and wild-type mice (●) 4 wk (top) or 1–4 wk (bottom) after C. rodentium infection. Uninfected mice were used as controls. Anti-C. rodentium titers of IgM (left) and IgG (right) were determined by ELISA. Results are shown as mean ± SEM of the data from six or more mice per group. *, p < 0.05 compared with wild-type mice. B, The colon of IL-6-deficient (○, IL-6−/−) and wild-type mice (●, IL-6+/+) were collected at the indicated times after C. rodentium infection. Total RNA was extracted and analyzed by real-time PCR for iNOS mRNA expression. Data were normalized against GAPDH mRNA expression and are shown as fold change relative to the mRNA levels in uninfected mice. In parallel, colon fragments were cultured for 6 h, and levels of the stable NO breakdown product, nitrite (NO2−), were assayed in the supernatants by the Griess reaction and normalized against dry weight of the tissue fragments. All data are mean ± SEM of four or more mice per group.
Infection-associated production of NO and critical immune regulators is not IL-6 dependent
In addition to Abs, other effector molecules have been shown or proposed to play a role in controlling C. rodentium infection. In particular, NO kills C. rodentium in vitro, and its production through inducible NO synthase (iNOS) contributes to antibacterial host defense (10). We therefore assayed iNOS expression and NO production after infection of normal and IL-6-deficient mice. Levels of iNOS mRNA were increased in the colon within 1 wk after C. rodentium infection and reached maximal levels by 2 wk in both groups, with slightly, but not significantly, higher expression in IL-6-deficient mice throughout the infection (Fig. 7B). In parallel, NO production in colonic explant cultures was modestly higher in C. rodentium-infected IL-6-deficient mice than in infected wild-type mice (Fig. 7B). These data demonstrate that colonic NO production is not compromised in IL-6-deficient mice after C. rodentium infection and therefore is not likely to account for the actions of IL-6 in host defense against the bacteria.
Several immunoregulatory cytokines, including IFN-γ and TNF-α, are required for effective immune defense against C. rodentium (11, 12). To evaluate whether the function of IL-6 might be mediated by these cytokines, we determined their expression levels during the course of infection. Real-time PCR analysis of mRNA levels in total colon RNA revealed increased expression of IFN-γ and TNF-α in IL-6-deficient and wild-type mice within 1 week after infection and maximal levels by 2 wk, but no evidence for attenuated expression in the absence of IL-6 (data not shown). Consistent with this, IL-6-deficient mice showed no defect in TNF-α secretion in colonic explant cultures 2 wk after infection (4.3 ± 1.9 pg/mg tissue in IL-6-deficient mice vs 3.8 ± 0.9 pg/mg in controls, whereas uninfected mice had <1 pg/mg in both groups). We could not detect IFN-γ production in any of the groups under these conditions. These data demonstrate that IL-6 is not required for increased colonic IFN-γ or TNF-α expression after infection and suggest that these cytokines are not likely to mediate the function of IL-6 in mucosal defense against C. rodentium.
Colonic ulcerations form a microbial niche for C. rodentium
Because the targeted analysis of immune mediators did not reveal any apparent mechanisms of IL-6-dependent immune defense against C. rodentium, we examined the interaction of the bacteria with the host morphologically to gain potential clues as to the mucosal defense defects caused by IL-6 deficiency. Immunohistological staining revealed greater C. rodentium colonization in the colon of IL-6-deficient than in wild-type mice at 2 wk (Fig. 8A), which was consistent with the fecal bacterial counts (Fig. 3B). Importantly, whereas control mice exhibited only surface colonization, bacteria were found deep in the colonic mucosa of IL-6-deficient mice, particularly at the edges of and within ulcerated regions (Fig. 8A). By comparison, SCID mice showed heavy surface colonization but little bacterial penetration into the mucosa (Fig. 8B). These findings suggested that colonic ulcers may provide a microbial niche for bacterial colonization.
FIGURE 8.
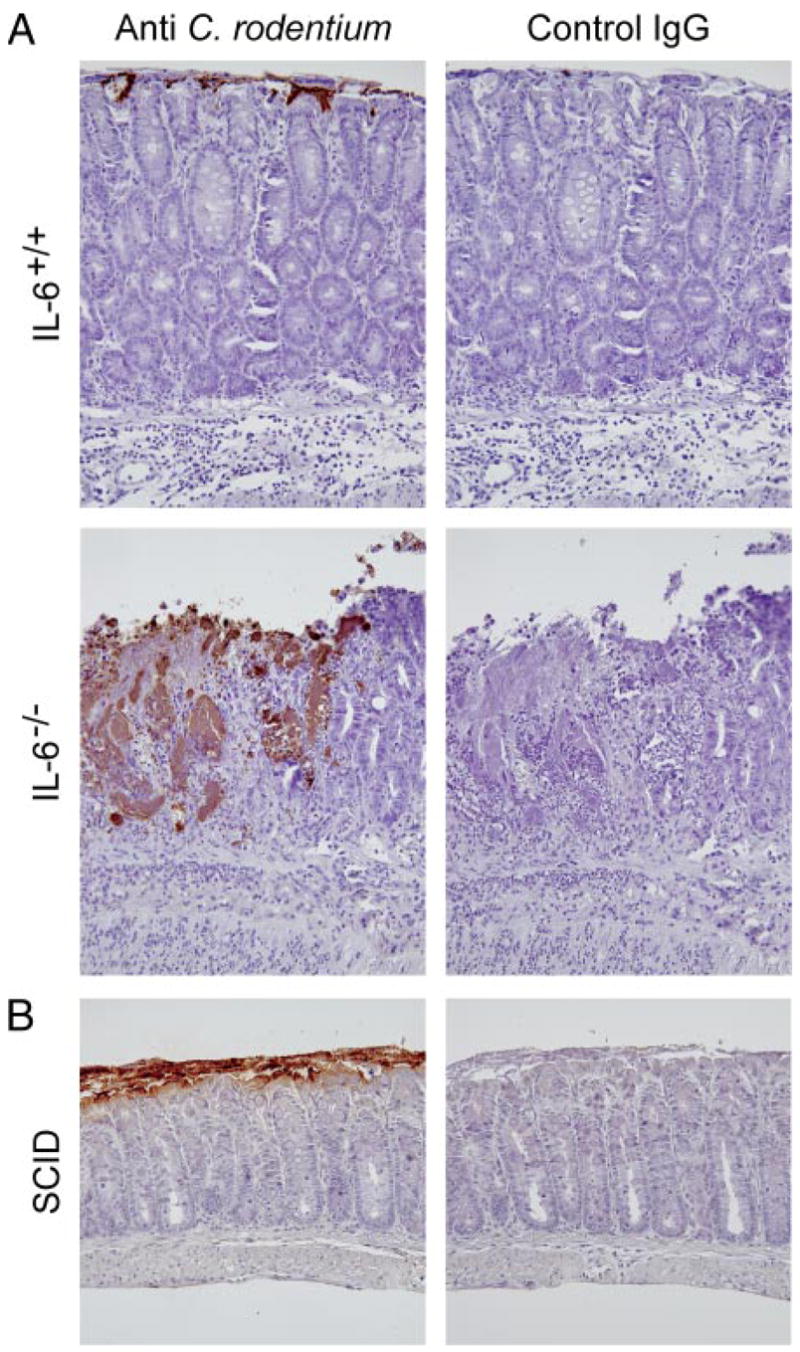
Bacterial localization in the colon of IL-6-deficient mice. Colon segments from IL-6-deficient (IL-6−/−, A) and wild-type mice (IL-6+/+, A) or SCID mice (B) were collected 2 wk after C. rodentium infection, and paraffin sections were stained by an indirect immunoperoxidase method with Abs against C. rodentium or with control IgG Abs. No specific staining was observed in uninfected mice in any of the groups.
To test this possibility, we adopted a two-step protocol in which we first induced colonic ulceration in normal mice by DSS administration for 5 days in the drinking water followed by 5 days of regular drinking water, and we subsequently challenged the mice with C. rodentium and examined the time course of bacterial colonization. Ulcerated mice were colonized more effectively with the bacteria than nonulcerated controls and had significantly higher bacterial numbers for the first week after infection (Fig. 9A). Furthermore, bacteria were found to localize preferentially to ulcerated regions in DSS-treated mice, whereas colonization was limited to the colon surface in nonulcerated controls (Fig. 9B). Once ulcers had healed by 2–3 wk after infection, bacterial numbers were not significantly different between DSS-treated and control mice (data not shown). Thus, mucosal ulcerations can provide a colonization niche for C. rodentium in the colon.
FIGURE 9.
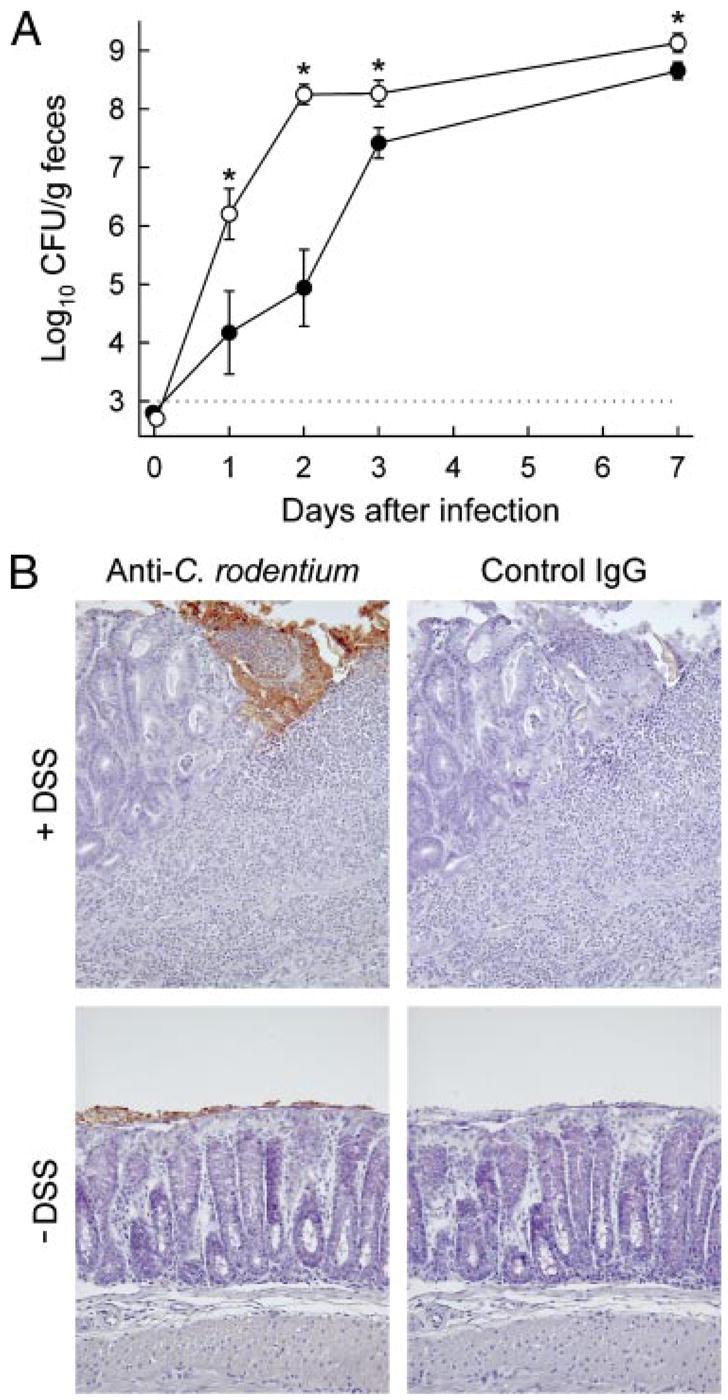
Colonic ulceration promotes C. rodentium colonization. Adult C57BL/6J mice were given 3% DSS in the drinking water for 5 days, followed by 5 days of regular drinking water (○, +DSS) or were left untreated as a control (●, − DSS). Body weights showed maximal loss 7 days after the beginning of DSS administration with 20 ± 3% (mean ± SEM) of the initial weight and recovered thereafter. Mice were subsequently infected with C. rodentium. A, Bacterial numbers in the feces were determined by CFU assay at the indicated times. Results are mean ± SEM of 10 or more mice per group. *, p < 0.05 compared with non-DSS treated mice at the same time point. (B) The colon was collected 3 days after infection, and paraffin sections were prepared and stained by an indirect immunoperoxidase method with Abs against C. rodentium or with control IgG Abs.
Serum factors promote C. rodentium growth
To determine potential mechanisms by which mucosal ulceration promotes bacterial colonization, we reasoned that factors present in the blood or extracellular mucosal space, which probably exude into the lumen in the absence of an effective epithelial barrier, might affect bacterial growth. For these studies, bacteria were grown under limiting nutrient conditions, a situation that might be expected to prevail in the colonic lumen due to intense bacterial competition (32), in the absence or presence of blood or its components. Addition of whole mouse blood significantly enhanced the growth of C. rodentium in log phase, and increased bacterial density in stationary phase (data not shown). Crude blood fractionation revealed that the growth-promoting effect was mainly mediated by serum components, whereas cellular elements (i.e., RBC) had a more modest impact on bacterial growth (Fig. 10A).
FIGURE 10.
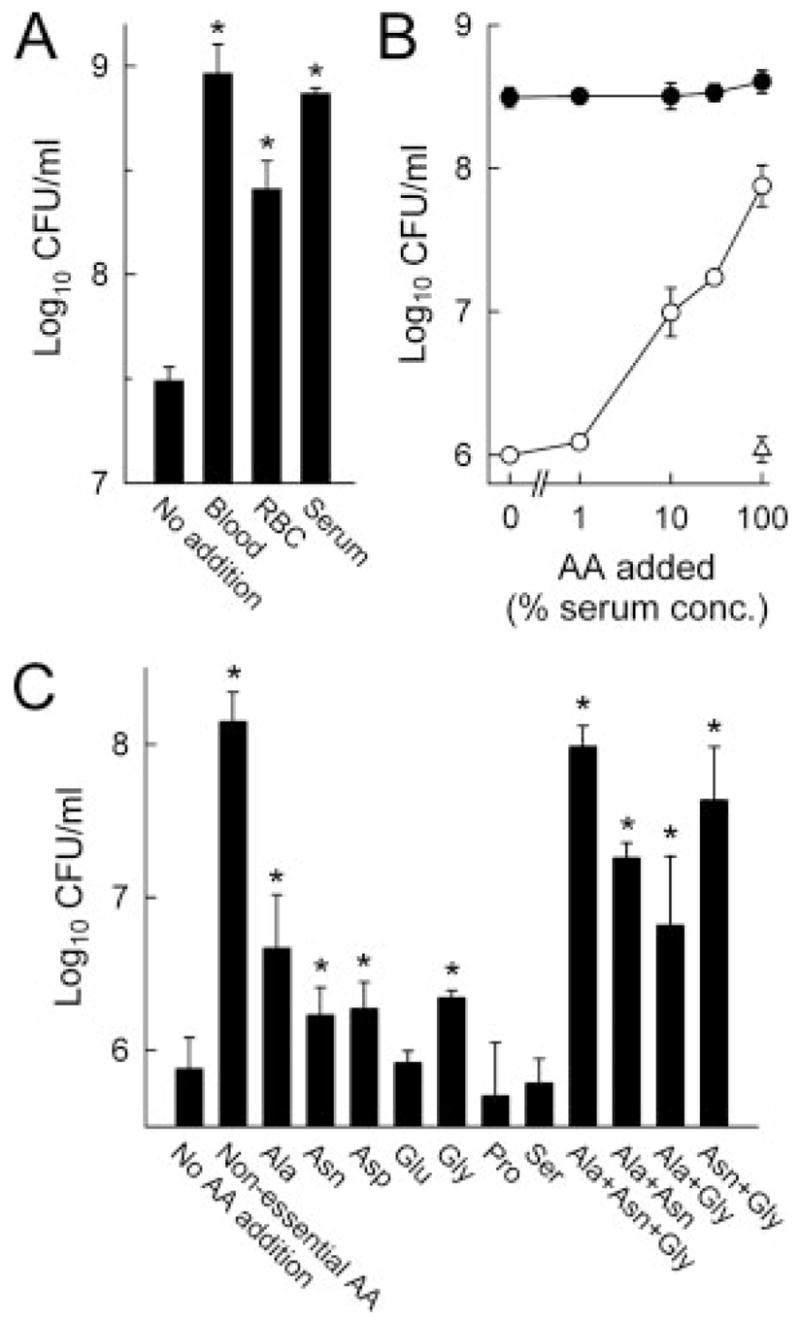
Serum components promote C. rodentium growth. A, C. rodentium (105/ml) were cultured in 1% DMEM in PBS in the absence or presence of whole blood or the indicated blood components at 37°C for 24 h, after which bacterial numbers were determined by CFU assay. Results are mean ± SEM (n = 4); *, p < 0.05 compared with cultures without additions. B, C. rodentium (104/ml) was cultured with different amounts of a mixture of nonessential amino acids in PBS containing 30% depleted (○) or nondepleted (●) mouse serum, or with essential amino acids (AA) in depleted serum (△). C, C. rodentium (104/ml) was cultured in the presence of the indicated individual or combinations of nonessential amino acids at physiological serum levels.
Further characterization of the growth-promoting serum factors revealed that they were heat stable, because heat inactivation did not diminish bacterial growth and had a low molecular mass, because substantial activity was retained after passage through a filter with a 3-kDa molecular mass cut-off. Because amino acids are likely present in ulcer exudates, have a low molecular mass, are heat stable, and have been shown to enhance bacterial growth in other systems, experiments were undertaken to determine whether they might account for the ability of serum to promote C. rodentium growth. Supplementation of growth medium containing depleted mouse serum (generated by prior overnight growth of the bacteria in the serum) with a mixture of essential (for human cells) amino acids had little effect on bacterial growth, whereas addition of a mixture of nonessential amino acids stimulated bacterial growth in a concentration-dependent manner (Fig. 10B). Further testing of nonessential amino acids demonstrated that alanine, asparagine, aspartic acid, and glycine improved bacterial growth significantly when added individually at physiological serum concentrations (Fig. 10C). Combinations of these amino acids promoted growth even more strongly, approaching the levels achieved by addition of fresh, nondepleted serum, particularly when alanine, asparagine, and glycine were used together. Thus, several nonessential amino acids in serum can promote C. rodentium growth and can account for much of the growth-promoting activity in fresh serum. Such components are likely to exude from the ulcerated mucosa into the lumen in IL-6-deficient mice, a notion consistent with the observation that these mice have impaired intestinal epithelial barrier integrity under inflammatory conditions (33).
Discussion
IL-6 is a pleiotropic cytokine whose physiological functions of which remain incompletely understood, because its contributions to inflammation and immune defense in different organs appear to depend on the specific physiological conditions (15, 16). Our studies demonstrate that IL-6 plays an important role in intestinal immune defense against the attaching/effacing lesion-inducing bacterial pathogen, C. rodentium. A protective immunological function of IL-6 has also been reported for two other enteric pathogens, Yersinia enterocolitica and Giardia lamblia (34–36), although these pathogens differ from C. rodentium in critical aspects of their interactions with the host. Y. enterocolitica are invasive bacteria that heavily colonize Peyer’s patches and mesenteric lymph nodes upon oral inoculation (37), whereas C. rodentium is only minimally invasive with limited colonization of the mesenteric lymph nodes (38). G. lamblia are noninvasive, lumen-dwelling parasites of the small intestine (39), whereas C. rodentium resides in cecum and colon in intimate contact with the epithelium (5, 40). Despite these differences, our data together with the prior reports (34, 35) indicate that IL-6 is a crucial immune mediator of immune defense against a range of different enteric pathogens. The protective role of IL-6 in gastrointestinal immunity, however, is by no means universal, because IL-6 is not involved in immune defense against the enteric viral pathogen rotavirus (41), or the gastric bacterial pathogen Helicobacter felis (19). Thus, the importance of IL-6 in host defense in the gastrointestinal tract is likely to depend on the specific nature of the host-pathogen interaction, and the resulting differential requirements for effective defense mechanisms.
We found that IL-6 deficiency had no impact on the bacterial burden in the first week after C. rodentium infection, suggesting that innate immune defenses operating early against the bacteria, such as antimicrobial peptides (42), are not IL-6-dependent. Instead, the cytokine had a critical function 2–3 wk after infection, a time period that marks the beginning of the adaptive immune response, suggesting that IL-6 might contribute to the development of specific antibacterial immunity. For example, IL-6 can regulate Ag-specific B cell responses (17), and anti-bacterial IgG has protective functions against C. rodentium (8). IL-6 is also required for the development of Th17 cells (43), for which the signature cytokine, IL-17, plays a role in clearance of C. rodentium (13). However, our data provide little support for an important function of IL-6 in regulating adaptive immunity against the bacteria, as the production of antibacterial IgG or IgM Abs was not delayed or diminished in IL-6-deficient mice. These results also suggest that IL-6 had no role in facilitating the interactions among T cells, B cells, and dendritic cells required for the development of a specific Ab response (30, 31). Furthermore, IL-6 was not necessary for expression of two key cytokines, IFN-γ and TNF-α, which regulate T cell growth and differentiation and the recruitment and activation of inflammatory cells, respectively, and which are involved in adaptive host defense against C. rodentium (11, 12). Finally, IL-6 can synergize with other stimuli to induce the production of NO (44), an antimicrobial molecule that has been shown to contribute to host defense against C. rodentium (10). However, NO production was modestly enhanced rather than attenuated in IL-6-deficient mice after infection, which indicates that NO was not an IL-6-dependent immune defense mechanism in this context.
Our findings suggest an alternative mechanism of IL-6 mediated intestinal host defense, which is not dependent on the development of adaptive immunity. We pose that IL-6 operates primarily by protecting the mucosal surface against infection-induced epithelial ulceration. In its absence, mucosal ulcerations develop and can serve as a microbial niche for the infecting bacteria. Consistent with this concept, patients with inflammatory bowel disease appear to exhibit differences in the overall microbial composition and in specific bacterial species in ileum and colon compared with healthy controls (45, 46). The underlying mechanisms by which mucosal inflammation and ulcerations can alter the local intestinal microbiota and promote bacterial growth are likely to be manifold but probably include, as our data suggest, exudation of serum components. In particular, the amino acids alanine, asparagine, aspartic acid, and glycine played a major role in promoting bacterial growth under limiting nutrient conditions, which are likely to be present in the colonic lumen. Other mechanisms, such as attenuation of critical host defense functions or local surface abnormalities, could also be involved. Irrespective of the relative contributions of these mechanisms, our data suggest the concept that an intact epithelial barrier in the intestinal tract is important not only for preventing systemic access of luminal microbes (47), as evidenced by increased splenic bacterial numbers in IL-6-deficient mice with extensive colon ulceration, but also for curtailing the luminal presence of systemically available nutrients that can promote growth of subsets of luminal microbes.
IL-6 was most prominently expressed by epithelial cells and macrophages in the colon of C. rodentium-infected mice, whereas expression was not increased in other sites, including the small intestine, mesenteric lymph nodes, liver, and spleen, which are minimally colonized with bacteria in normal, immunocompetent mice. This limitation of the IL-6 response to the immediate area of infection suggests that IL-6 is induced in response to direct bacteria-host cell interaction or by bacterial factors active in close vicinity to their release. Infection of cultured colon epithelial cells with human EPEC strains, which produce attaching/effacing lesions and occupy an ecological niche similar to that of C. rodentium, activates the transcription factor, NF-κB (48), which is a key regulator of IL-6 gene transcription. A similar direct epithelial response to C. rodentium infection may also occur in the colon, although the delay in maximal IL-6 expression (wk 2) relative to maximal bacterial colonization in the colon (wk 1) would suggest that it is not a major mechanism of peak IL-6 induction. In addition, elevated IL-6 production in crypt epithelial cells, as well as mucosal macrophages, both of which are only minimally exposed to bacteria, also argues against an induction mechanism involving direct bacterial contact. Instead, the latter findings suggest that diffusible bacterial or host factors are responsible for most of the colonic IL-6 response. For example, bacterial products activate IL-6 expression in cultured macrophages through TLR2 and TLR4 (49), and TLR2 plays a role in activating IL-6 expression after C. rodentium infection (50).
IL-6 deficiency exacerbated the infection-associated epithelial ulceration and mucosal inflammation in the colon, which indicates that the cytokine limited the severity of colitis and hence had overall mucosa-protective functions under these conditions. Similarly, IL-6 attenuated acute mucosal damage upon challenge with the nonspecific irritant DSS, a finding consistent with a prior report in this model (51). In contrast, other studies have shown that IL-6 had proinflammatory functions in severe forms of DSS-induced colitis (52). However, these studies used very high doses of DSS, which might explain the difference in the role of IL-6 relative to our results. Another recent report (33), in which mice were treated with a low DSS dose, found that IL-6 deficiency impairs barrier integrity, which is consistent with our conclusion that IL-6 can be protective in acute infection-associated colitis. Several mechanisms may account for the protective functions of IL-6, including induction of critical cytoskeletal proteins (33), and, as suggested by our data, protection against apoptosis. Epithelial apoptosis is a critical early event that can lead to mucosal ulceration and inflammation, particularly in the absence of sufficient antiapoptotic counterregulation (28). Similar to the observations in colon epithelial cells, IL-6 was shown to inhibit apoptosis in lung epithelial cells (53), underlining the general importance of IL-6-dependent epithelial protection in different mucosal organs. The protection is likely to be mediated, at least in part, by the IL-6-induced up-regulation of an array of target genes encoding anti-apoptotic proteins, including Bcl-xL, Mcl-1, cIAP-2, and Bcl-3 (54), although future studies will have to reveal the relative importance of these gene products in this context.
The mucosa-protective function of IL-6 contrasts with its proinflammatory role in murine models of T cell-dependent colitis, in which IL-6-deficient mice exhibited reduced mucosal inflammation (21). In light of these findings, we conclude that in the context of infectious colitis, the contribution of IL-6 to protecting the mucosa against microbially induced damage apparently outweighed the potential proinflammatory functions of IL-6. Alternatively, the relative importance of anti- and proinflammatory activities of IL-6 may depend on the underlying pathogenetic mechanisms, which are likely to differ between infection-associated colitis and other forms of experimentally induced colitis (21). In any case, our results caution that antagonism of IL-6 as a therapeutic modality, as recently proposed for chronic intestinal inflammatory conditions (51, 55), may exacerbate colitis caused by enteric pathogens and increase susceptibility to infections with such pathogens.
Acknowledgments
We thank Irina Olshanskaya for technical assistance and Dr. Bruce Vallance for providing antisera.
Footnotes
This work was supported by National Institutes of Health Grants DK35108, AI56075, DK70867, and RR17030. S.M.D. was supported by a fellowship from the Crohn’s and Colitis Foundation, and M.E.S. was supported by a fellowship from the MFG Educative Science.
Abbreviations used in this paper: EPEC, enteropathogenic Escherichia coli; iNOS, inducible NO synthase; DSS, dextran sulfate sodium.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nougayrede JP, Fernandes PJ, Donnenberg MS. Adhesion of enteropathogenic Escherichia coli to host cells. Cell Microbiol. 2003;5:359–372. doi: 10.1046/j.1462-5822.2003.00281.x. [DOI] [PubMed] [Google Scholar]
- 3.Frankel G, Phillips AD, Novakova M, Field H, Candy DC, Schauer DB, Douce G, Dougan G. Intimin from enteropathogenic Escherichia coli restores murine virulence to a Citrobacter rodentium eaeA mutant: induction of an immunoglobulin A response to intimin and EspB. Infect Immun. 1996;64:5315–5325. doi: 10.1128/iai.64.12.5315-5325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luperchio SA, Schauer DB. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect. 2001;3:333–340. doi: 10.1016/s1286-4579(01)01387-9. [DOI] [PubMed] [Google Scholar]
- 5.Johnson E, Barthold SW. The ultrastructure of transmissible murine colonic hyperplasia. Am J Pathol. 1979;97:291–313. [PMC free article] [PubMed] [Google Scholar]
- 6.Schauer DB, Falkow S. The eae gene of Citrobacter freundii bio-type 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect Immun. 1993;61:4654–4661. doi: 10.1128/iai.61.11.4654-4661.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallance BA, Deng W, Knodler LA, Finlay BB. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect Immun. 2002;70:2070–2081. doi: 10.1128/IAI.70.4.2070-2081.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maaser C, Housley MP, Iimura M, Smith JR, Vallance BA, Finlay BB, Schreiber JR, Varki NM, Kagnoff MF, Eckmann L. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect Immun. 2004;72:3315–3324. doi: 10.1128/IAI.72.6.3315-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bry L, Brenner MB. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol. 2004;172:433–441. doi: 10.4049/jimmunol.172.1.433. [DOI] [PubMed] [Google Scholar]
- 10.Vallance BA, Deng W, De Grado M, Chan C, Jacobson K, Finlay BB. Modulation of inducible nitric oxide synthase expression by the attaching and effacing bacterial pathogen Citrobacter rodentium in infected mice. Infect Immun. 2002;70:6424–6435. doi: 10.1128/IAI.70.11.6424-6435.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simmons CP, Goncalves NS, Ghaem-Maghami M, Bajaj-Elliott M, Clare S, Neves B, Frankel G, Dougan G, MacDonald TT. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-γ. J Immunol. 2002;168:1804–1812. doi: 10.4049/jimmunol.168.4.1804. [DOI] [PubMed] [Google Scholar]
- 12.Goncalves NS, Ghaem-Maghami M, Monteleone G, Frankel G, Dougan G, Lewis DJ, Simmons CP, MacDonald TT. Critical role for tumor necrosis factor α in controlling the number of lumenal pathogenic bacteria and immunopathology in infectious colitis. Infect Immun. 2001;69:6651–6659. doi: 10.1128/IAI.69.11.6651-6659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-β induces development of the Th17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 14.Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, Kashiwamura S, Nakajima K, Koyama K, Iwamatsu A, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature. 1986;324:73–76. doi: 10.1038/324073a0. [DOI] [PubMed] [Google Scholar]
- 15.Gadient RA, Otten UH. Interleukin-6 (IL-6): a molecule with both beneficial and destructive potentials. Prog Neurobiol. 1997;52:379–390. doi: 10.1016/s0301-0082(97)00021-x. [DOI] [PubMed] [Google Scholar]
- 16.Kishimoto T. Interleukin-6: from basic science to medicine: 40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- 17.Muraguchi A, Hirano T, Tang B, Matsuda T, Horii Y, Nakajima K, Kishimoto T. The essential role of B cell stimulatory factor 2 (BSF-2/IL-6) for the terminal differentiation of B cells. J Exp Med. 1988;167:332–344. doi: 10.1084/jem.167.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Kohler G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- 19.Bromander AK, Ekman L, Kopf M, Nedrud JG, Lycke NY. IL-6-deficient mice exhibit normal mucosal IgA responses to local immunizations and Helicobacter felis infection. J Immunol. 1996;156:4290–4297. [PubMed] [Google Scholar]
- 20.Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187:461–468. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamamoto M, Yoshizaki K, Kishimoto T, Ito H. IL-6 is required for the development of Th1 cell-mediated murine colitis. J Immunol. 2000;164:4878–4882. doi: 10.4049/jimmunol.164.9.4878. [DOI] [PubMed] [Google Scholar]
- 22.Aderka D, Le JM, Vilcek J. IL-6 inhibits lipopolysaccharide-induced tumor necrosis factor production in cultured human monocytes, U937 cells, and in mice. J Immunol. 1989;143:3517–3523. [PubMed] [Google Scholar]
- 23.Ulich TR, Yin S, Guo K, Yi ES, Remick D, del Castillo J. Intratracheal injection of endotoxin and cytokines. II. Interleukin-6 and transforming growth factor β inhibit acute inflammation. Am J Pathol. 1991;138:1097–1101. [PMC free article] [PubMed] [Google Scholar]
- 24.Dalrymple SA, Lucian LA, Slattery R, McNeil T, Aud DM, Fuchino S, Lee F, Murray R. Interleukin-6-deficient mice are highly susceptible to Listeria monocytogenes infection: correlation with inefficient neutrophilia. Infect Immun. 1995;63:2262–2268. doi: 10.1128/iai.63.6.2262-2268.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ladel CH, Blum C, Dreher A, Reifenberg K, Kopf M, Kaufmann SH. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect Immun. 1997;65:4843–4849. doi: 10.1128/iai.65.11.4843-4849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romani L, Mencacci A, Cenci E, Spaccapelo R, Toniatti C, Puccetti P, Bistoni F, Poli V. Impaired neutrophil response and CD4+ T helper cell 1 development in interleukin 6-deficient mice infected with Candida albicans. J Exp Med. 1996;183:1345–1355. doi: 10.1084/jem.183.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 28.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 29.Nishihara H, Kizaka-Kondoh S, Insel PA, Eckmann L. Inhibition of apoptosis in normal and transformed intestinal epithelial cells by cAMP through induction of inhibitor of apoptosis protein (IAP)-2. Proc Natl Acad Sci USA. 2003;100:8921–8926. doi: 10.1073/pnas.1533221100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 31.Bleier JI, V, Pillarisetty G, Shah AB, DeMatteo RP. Increased and long-term generation of dendritic cells with reduced function from IL-6-deficient bone marrow. J Immunol. 2004;172:7408–7416. doi: 10.4049/jimmunol.172.12.7408. [DOI] [PubMed] [Google Scholar]
- 32.Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Srinivasan S, Theiss AL, Merlin D, Sitaraman SV. Interleukin-6 induces keratin expression in intestinal epithelial cells: potential role of keratin-8 in interleukin-6-induced barrier function alterations. J Biol Chem. 2007;282:8219–8227. doi: 10.1074/jbc.M604068200. [DOI] [PubMed] [Google Scholar]
- 34.Dube PH, Handley SA, Lewis J, Miller VL. Protective role of interleukin-6 during Yersinia enterocolitica infection is mediated through the modulation of inflammatory cytokines. Infect Immun. 2004;72:3561–3570. doi: 10.1128/IAI.72.6.3561-3570.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou P, Li E, Zhu N, Robertson J, Nash T, Singer SM. Role of interleukin-6 in the control of acute and chronic Giardia lamblia infections in mice. Infect Immun. 2003;71:1566–1568. doi: 10.1128/IAI.71.3.1566-1568.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bienz M, Dai WJ, Welle M, Gottstein B, Muller N. Interleukin-6-deficient mice are highly susceptible to Giardia lamblia infection but exhibit normal intestinal immunoglobulin A responses against the parasite. Infect Immun. 2003;71:1569–1573. doi: 10.1128/IAI.71.3.1569-1573.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koornhof HJ, Smego RA, Jr, Nicol M. Yersiniosis. II: The pathogenesis of Yersinia infections. Eur J Clin Microbiol Infect Dis. 1999;18:87–112. doi: 10.1007/s100960050237. [DOI] [PubMed] [Google Scholar]
- 38.Spahn TW, Maaser C, Eckmann L, Heidemann J, Lugering A, Newberry R, Domschke W, Herbst H, Kucharzik T. The lymphotoxin-β receptor is critical for control of murine Citrobacter rodentium-induced colitis. Gastroenterology. 2004;127:1463–1473. doi: 10.1053/j.gastro.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 39.Eckmann L. Mucosal defences against Giardia. Parasite Immunol. 2003;25:259–270. doi: 10.1046/j.1365-3024.2003.00634.x. [DOI] [PubMed] [Google Scholar]
- 40.Wiles S, Clare S, Harker J, Huett A, Young D, Dougan G, Frankel G. Organ specificity, colonization and clearance dynamics in vivo following oral challenges with the murine pathogen Citrobacter rodentium. Cell Microbiol. 2004;6:963–972. doi: 10.1111/j.1462-5822.2004.00414.x. [DOI] [PubMed] [Google Scholar]
- 41.VanCott JL, Franco MA, Greenberg HB, Sabbaj S, Tang B, Murray R, McGhee JR. Protective immunity to rotavirus shedding in the absence of interleukin-6: Th1 cells and immunoglobulin A develop normally. J Virol. 2000;74:5250–5256. doi: 10.1128/jvi.74.11.5250-5256.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iimura M, Gallo RL, Hase K, Miyamoto Y, Eckmann L, Kagnoff MF. Cathelicidin mediates innate intestinal defense against colonization with epithelial adherent bacterial pathogens. J Immunol. 2005;174:4901–4907. doi: 10.4049/jimmunol.174.8.4901. [DOI] [PubMed] [Google Scholar]
- 43.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 44.Sawada T, Falk LA, Rao P, Murphy WJ, Pluznik DH. IL-6 induction of protein-DNA complexes via a novel regulatory region of the inducible nitric oxide synthase gene promoter: role of octamer binding proteins. J Immunol. 1997;158:5267–5276. [PubMed] [Google Scholar]
- 45.Sokol H, Seksik P, Rigottier-Gois L, Lay C, Lepage P, Podglajen I, Marteau P, Dore J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:106–111. doi: 10.1097/01.MIB.0000200323.38139.c6. [DOI] [PubMed] [Google Scholar]
- 46.Masseret E, Boudeau J, Colombel JF, Neut C, Desreumaux P, Joly B, Cortot A, Darfeuille-Michaud A. Genetically related Escherichia coli strains associated with Crohn’s disease. Gut. 2001;48:320–325. doi: 10.1136/gut.48.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Porras M, Martin MT, Yang PC, Jury J, Perdue MH, Vergara P. Correlation between cyclical epithelial barrier dysfunction and bacterial translocation in the relapses of intestinal inflammation. Inflamm Bowel Dis. 2006;12:843–852. doi: 10.1097/01.mib.0000231571.88806.62. [DOI] [PubMed] [Google Scholar]
- 48.Savkovic SD, Koutsouris A, Hecht G. Activation of NF-κB in intestinal epithelial cells by enteropathogenic Escherichia coli. Am J Physiol. 1997;273:C1160–C1167. doi: 10.1152/ajpcell.1997.273.4.C1160. [DOI] [PubMed] [Google Scholar]
- 49.Schilling D, Thomas K, Nixdorff K, Vogel SN, Fenton MJ. Toll-like receptor 4 and Toll-IL-1 receptor domain-containing adapter protein (TIRAP)/myeloid differentiation protein 88 adapter-like (Mal) contribute to maximal IL-6 expression in macrophages. J Immunol. 2002;169:5874–5880. doi: 10.4049/jimmunol.169.10.5874. [DOI] [PubMed] [Google Scholar]
- 50.Gibson DL, Ma C, Rosenberger CM, Bergstrom KSB, Valdez Y, Huang JT, Khan MA, Vallance BA. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell Microbiol. 2008;10:388–403. doi: 10.1111/j.1462-5822.2007.01052.x. [DOI] [PubMed] [Google Scholar]
- 51.Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch B, Holtmann M, Becker C, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- 52.Naito Y, Takagi T, Uchiyama K, Kuroda M, Kokura S, Ichikawa H, Yanagisawa R, Inoue K, Takano H, Satoh M, et al. Reduced intestinal inflammation induced by dextran sodium sulfate in interleukin-6-deficient mice. Int J Mol Med. 2004;14:191–196. [PubMed] [Google Scholar]
- 53.Kida H, Yoshida M, Hoshino S, Inoue K, Yano Y, Yanagita M, Kumagai T, Osaki T, Tachibana I, Saeki Y, et al. Protective effect of IL-6 on alveolar epithelial cell death induced by hydrogen peroxide. Am J Physiol. 2005;288:L342–L349. doi: 10.1152/ajplung.00016.2004. [DOI] [PubMed] [Google Scholar]
- 54.Bhattacharya S, Ray RM, Johnson LR. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J. 2005;392:335–344. doi: 10.1042/BJ20050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ito H, Takazoe M, Fukuda Y, Hibi T, Kusugami K, Andoh A, Matsumoto T, Yamamura T, Azuma J, Nishimoto N, et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology. 2004;126:989–996. doi: 10.1053/j.gastro.2004.01.012. [DOI] [PubMed] [Google Scholar]