

Abstract
Our previous work suggested that collapsing the Na+ gradient and membrane potential converts the dopamine (DA) transporter (DAT) to an inward-facing conformation with a different substrate binding profile. Here, DAT expressing HEK 293 cells were permeabilized with digitonin, disrupting ion/voltage gradients and allowing passage of DAT substrates. The potency of p-tyramine and other non-catechols (d-amphetamine, β-phenethylamine, MPP+) in inhibiting cocaine analog binding to DAT in digitonin-treated cells was markedly weakened to a level similar to that observed in cell-free membranes. In contrast, the potency of DA and another catechol, norepinephrine, was not significantly changed by the same treatment, whereas epinephrine showed only a modest reduction. These findings suggest catechol substrates interact symmetrically with both sides of DAT and non-catechol substrates favor binding to outward-facing transporter. In the cocaine analog binding assay, the mutant W84L displayed enhanced intrinsic binding affinity for substrates in interacting with both outward- and inward- facing states; D313N showed WT-like symmetric binding; but D267L and E428Q showed an apparent improvement in the permeation pathway from the external face towards the substrate site. Thus, the structure of both substrate and transporter play a role in the sidedness and mode of interaction between them.
Keywords: dopamine transporter, catechol substrates, digitonin, binding sidedness, DAT conformations
Introduction
The dopamine (DA) transporter (DAT) is a member of the neurotransmitter: sodium symporter (NSS) family that includes transporters for norepinephrine (NET), serotonin (SERT), GABA (GAT), glycine, proline, or taurine (Busch and Saier, Jr. 2002). It is located on the plasma membrane of dopaminergic neurons and transports DA across the membrane. By taking up DA into neurons from the extracellular space, DAT plays a critical role in modulating dopaminergic neurotransmission (Giros et al. 1996;Rice and Cragg 2008). On the other hand, by mediating reverse transport of intracellular DA, DAT can also serve as a pathway for DA release at dopaminergic dendrites (Falkenburger et al. 2001). Direct interaction with DAT is crucial for the effects of psychostimulants with drug abuse liability such as d-amphetamine, and of neurotoxins gaining access to DA neurons through the DAT such as 1-methyl-4-phenyl-pyridinium ion (MPP+) [active metabolites of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)] (Gainetdinov et al. 2002;Amara and Sonders 1998;Bannon 2005).
d-Amphetamine, related phenethylamine psychostimulants, and MPP+ are substrates for DAT as DA itself (Chen and Justice 2000;Meiergerd and Schenk 1994;Rothman et al. 2001). Substrate interaction with monoamine transporters is thought to be defined by the alternate access model (Androutsellis-Theotokis and Rudnick 2002). Thus, the transporter protein is assumed to alternate between at least two different conformational states, which differ in the accessibility of the binding site for substrates. For inward transport, external substrates are proposed to initially bind at the outward-facing state where the binding site for substrates is exposed only to the external medium. For outward transport, internal substrates bind to the inward-facing state where the site is only exposed to the cytoplasmic fluid (Levi and Raiteri 1993;Chen and Justice, Jr. 1998). This is also supported by detailed simulation of substrate transport by the bacterial NSS homolog LeuT (Shi et al. 2008).
Gramicidin as a monovalent cation ionophore increases intracellular Na+ and decreases K+ to extracellular levels, causing membrane depolarization (Chen and Reith 2004). Our previous studies demonstrated that gramicidin treatment of cells promote accumulation of DAT in the inward-facing state (Chen and Reith, 2004; Zhen et al. 2005). With DA being relatively impermeable and DAT activity inhibited by gramicidin, internal DA levels remained very low. Gramicidin reduced DA's affinity for binding to intact cells. We speculated this was because DA had reduced access to outward-facing binding sites (measured as average affinity for multiple states interconverting rapidly within the time frame of the binding assay) (Figs. 1a and b). Furthermore, in membrane preparations with complete collapse of membrane Na+ gradient and potential, most DATs are inward-facing but DA can readily act at the side normally facing internally, and in consonance the potency of DA in inhibiting cocaine analog CFT binding was normal (Fig. 1c). Thus, DA appears to have no preference for inward- or outward-facing recognition sites. In contrast, the Ki value of non-catechol substrates such as β-phenethylamine, p-tyramine and MPP+ was increased (i.e., reduced affinity) for cells in the presence of gramicidin as well as in membrane preparations. We speculated those substrates cannot readily bind to DAT when it is facing inwardly.
Fig. 1. Cartoon of access model for DA at wild type DAT.
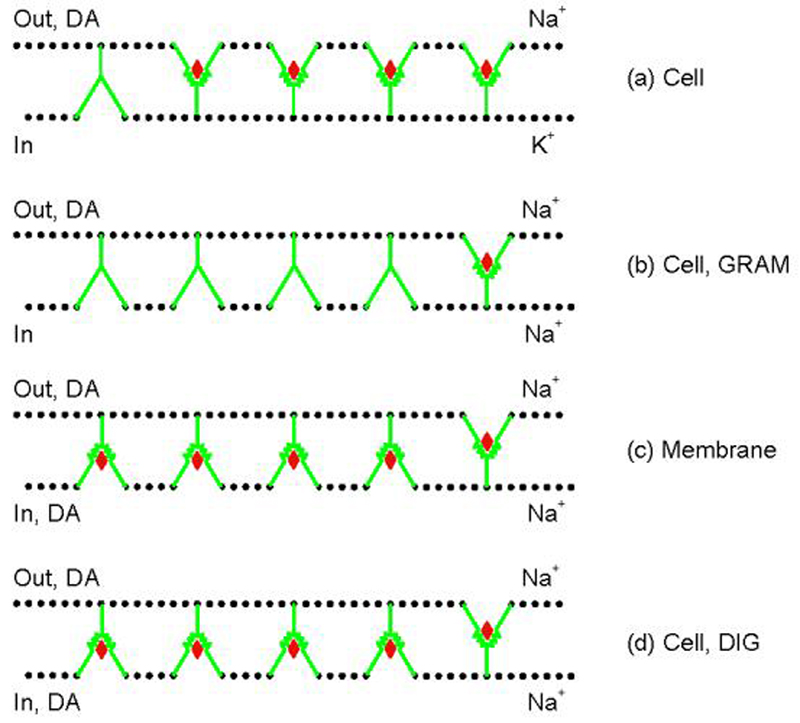
(a) In intact cells under physiological condition, most DATs reside in outward-facing state where extracellular DA binding sites are accessible. K+ prevents internally accumulated DA to bind to the few inward-facing transporters; for simplicity internal DA has been omitted from the cartoon here and in Figs. 6 and 7 (Cell Control). (b) Gramicidin (GRAM) treatment causes the collapse of membrane Na+ gradient (high [Na+]o and low [Na+]i), leading to accumulation of the transporter in inward-facing state. Under this condition, the majority of binding sites is unavailable to extracellular DA. Intracellular DA is sparse. (c) In cell-free membranes, the complete disruption of the membrane Na+ gradient causes DAT redistribution similar to that accruing in gramicidin-treated cells. Ambient DA can approach its binding site either externally or internally. (d) Digitionin (DIG) treatment causes a similar change of transporter conformation as in GRAM-treated cells or cell-free membranes, but (in contrast to the situation with GRAM) DA can access from both sides similar to the case for cell-free membranes; DIG holds the cellular milleu similar to that occurring in GRAM-treated cells. Note that all panels illustrate only the initial conformational state for ligand binding. Cartoons are drawn according to those presented for Tyt1 (Quick et al. 2006), another Na+-dependent bacterial transporter for tyrosine operating with the same gated pore mechanism for transport as the DAT.
In this work, we tested our previous speculations regarding interaction of substrates with the intracellular face of the transporter with digitonin, which in addition to creating pores for ions, also allows small molecules such as DA and other substrates to cross the membrane (Frye and Holz 1985;Wilson and Kirshner 1983;Dunn and Holz 1983). With digitonin, therefore, we can test the speculation that the binding of DA is symmetric regarding binding to the external and internal face of DAT (Fig. 1d), as opposed to the asymmetry or sidedness for the interaction of non-catechol substrates. Norepinephrine and epinephrine were tested as other catechol substrates in addition to DA, whereas d-amphetamine, β-phenethylamine, p-tyramine and MPP+ were studied as non-catechol substrates. Mutant DATs with altered DA recognition were used to study the preferential impact of external versus internal residues in DAT on externally versus internally facing substrate binding sites.
Materials and methods
Materials
The pCIN4 vector and the pCIN4-synthetic human DAT construct (pCIN4-DAT) were generous gifts from Dr. Jonathan A. Javitch (Columbia University, New York, NY, USA). [3H]CFT (84.5 Ci/mmol) were purchased from Perkin-Elmer Life Sciences (Boston, MA, USA). Unlabeled CFT was from the NIDA (Research Triangle Institute, Research Triangle Park, NC, USA). DA, p-tyramine, d-amphetamine, norepinephrine, epinephrine, and MPP+ were from Sigma Chemical Co. (St Louis, MO, USA). Digitonin and ZnCl2 were from Acros Organics Co. (Geel, Belgium).
Generation of cell lines stably expressing wild-type and mutant hDATs
The human DAT mutants were generated previously using site-directed mutagenesis (Chen et al, 2001). Briefly, site-directed mutagenesis of the hDAT was performed using the QuickChange Mutagenesis Kit (Stratagene, La Jolla, CA, USA) and confirmed by sequencing the complete coding region. Human embryonic kidney cells (HEK 293, ATCC CRL 1573) were transfected with the wild-type (WT) or mutant construct by using LipofectAMINE. Transfected cells were selected in the growth medium containing 800 μ/mL geneticin and maintained in the growth medium containing 200 μ/mL geneticin.
Treatment with detergent
Digitonin were dissolved in 100% ethanol as 2 mM stocks. The stocks were added to the incubation mixtures resulting in 0.75% ethanol and 15 μM digitonin as final concentrations. 15 min after being suspended in KRH buffer at room temperature, cells or membranes were washed before proceeding to binding assay as described for the gramicidin experiments reported by us previously (Chen and Reith, 2004).
[3H]CFT binding
Cell suspensions or cell-free membrane preparations were prepared as described previously (Chen et al. 2001;Chen et al. 2002). Modified Krebs–Ringer–HEPES (KRH) buffer was used in all experiments, which contained 10 mM HEPES (adjusted to pH 7.4 with Tris), 130 mM NaCl unless otherwise stated, 1.2 mM KH2PO4, 1.2 mM MgSO4, 1 mM CaCl2, 3 mM KCl, 10 mM glucose, 0.1 mM tropolone and 1 mM ascorbic acid. For experiments performed using “sodium-free” KRH buffer, NaCl was replaced with 130 mM N-methyl-D-glucamine chloride (NMDG-Cl), a commonly used isotonic NaCl substitute. Binding assays were conducted in 96-well plates. Cells or their membrane preparations were incubated with 2–4 nM [3H]CFT for 15 min at 21°C in a total volume of 200 μL. For saturation or competition analysis, varying concentrations of non-radioactive CFT or substrates were included in the assay mixture. Nonspecific binding was defined with 1 μM CFT. The binding reactions were terminated by filtration followed by five washes with ice-cold saline on 0.1% or 0.3% polyethyleneimine pre-soaked Wallac A filtermats (Wallac, Gaithersburg, MD, USA) with a 96-well Tomtec cell harvester (Gaithersburg, MD, USA). The radioactivity was counted in a 1405 Microbeta liquid scintillation counter (Wallac) after adding 10 ml of Betaplate Scint (Wallac) to each filtermat. Every experiment was performed in triplicate.
Data analysis
The equilibrium dissociation constant (Kd) for CFT binding, and the maximal CFT-binding capacity (Bmax) were estimated by non-linear regression fitting of data using LIGAND software (KELL program; Biosoft, Cambridge, UK). The IC50 for a compound to inhibit [3H]CFT binding was estimated by logistic fitting of data using ORIGIN software (OriginLab Co., Northampton, MA, USA). IC50 values were converted to Ki values with the Cheng-Prusoff equation.
Results
Cell permeabilization with digitonin does not affect CFT binding affinity
Digitonin is a steroid glycoside and interacts specifically with cholesterol in eukaryotic plasma membranes, creating pores that are permeable to ions and small molecules but preserving much of the membrane architecture of the intact cells (Goncalves et al. 2000). Digitonin permeablized cells allow free passage of ions and substrates. To find the best condition for permeablization in HEK293 cells, dissociated cells were incubated for 5-20 min at room temperature in KRH buffer containing 10-20 μM digitonin. The cells were tested for permeabilization using trypan blue and were considered permeabilized when less than 10% of cells excluded the dye. Treatment of cells with 15 μM digitonin for 15 min allows maximal permeability (95% of cells permeant to trypan blue) but no observable damage of CFT binding activity (data not shown). This condition was selected for further experiments, in which digitonin treatment was found not to alter CFT binding affinity for cells or membranes in WT as well as mutant DAT (Fig. 2). In addition, there was no significant difference in the Bmax value for CFT binding between permeabilized (digitonin-treated) and non-permeabilized (vehicle-treated) cell-free membranes (7.61 ± 2.90 pmol/mg and 5.35 ± 0.24 pmol/mg for WT DAT, respectively). The findings indicate digitonin does not affect the integrity of DAT or its immediate membrane environment. This makes it unlikely that digitonin exerts a direct chemical effect on the DAT that might impact its interaction with DA. The Bmax of [3H]CFT binding to cells was not changed by treatment with digitonin (data not shown).
Fig. 2. Effect of digitonin treatment on CFT and DA binding.

Intact cells (A) and cell-free membranes (B) were pretreated with 15 μM digitonin (DIG +) or vehicle (0.75% ethanol) (−) for 15 min at 21°C, and then subjected to the binding assay with 2-4 nM [3H]CFT and various concentrations of CFT or DA for 15 min at 21°C. *P < 0.05 compared with respective WT within the same treatment group (+ or − DIG) (Student's t test); #P < 0.05 compared with respective value in cell-free membranes (depicted in panel B) for same DAT construct (Student's t test). Note there is no statistically significant difference in CFT Kd between digitonin and vehicle treatment for Cell or Membrane preparations of a given DAT construct (Student's t test, paired where appropriate).
Effect of digitonin on binding affinity of catechol and non-catechol substrates
To explore the binding property of catechol and non-catechol substrates to the intracellular face of DAT, we measured the potency of various substrates in inhibiting CFT binding to digitonin treated cells and cell-free membranes. As depicted in Fig. 3, the catechol substrates (DA and norepinephrine) showed a similar Ki for cells in the absence or presence of digitonin. As predicted, there was no significant difference in Ki value between cells and cell-free membranes either with or without digitonin treatment. Thus, DA and norepinephrine do not appear to have a preference for inward or outward-facing recognition sites. Epinephrine, another catechol substrate, showed a modest 2-fold increase in Ki upon digitonin treatment of cells, with Ki values in membranes at the level of digitonin-treated cells. On the contrary, larger increases (3- to 10-fold enhanced Ki values) were observed for the non-catechol substrates (d-amphetamine, β-phenethylamine, p-tyramine and MPP+) upon digitonin treatment of cells. Moreover, the Ki of all of these non-catechol substrates was significantly enhanced (3- to 10-fold) in cell-free membranes compared to that in cells without digitonin treatment; in contrast, the same comparison for the catechols showed no difference (DA, norepinephrine) or a 2-fold increased Ki (epinephrine). Taken together, the findings suggest that non-catechol substrates have substantially weaker affinity for recognition sites when DAT is facing inwardly. Similar to the catechol substrates (DA, norepinephrine, and epinephrine), there is also no significant difference in the Ki value of these non-catechol substrates in cell membranes with or without digitonin treatment, providing further support that digitonin does not directly influence DAT-substrate interaction chemically.
Fig. 3. Potency of catechol and non-catechol substrates in inhibiting [3H]CFT binding to wild type DAT.

Intact cells (Cell) and cell-free membranes (Mem) were pretreated with 15 μM digitonin or 0.75% ethanol vehicle (control) for 15 min at room temperature, and then subjected to the binding assay with 2-4 nM [3H]CFT and various concentrations of DA, norepinephrine (NE), epinephrine (EPI), d-amphetamine (d-AMPH), β-phenethylamine (β-PE), p-tyramine (p-TYR) and MPP+ for 15 min at 21°C. #P < 0.05 compared with control Ki of respective Cell or Mem (Student's t test); *P < 0.05 compared with the Ki for Cell within the same treatment group (control or digitonin) (Student's t test). Chemical structure of each substrate is shown above the chart.
Mutant DATs and their conformational preference
Next, to test whether conformational bias of DAT influences the preference of substrates in binding to external or internal binding sites, the DAT mutants (W84L, D313N, W267L and E428Q) with altered DA recognition were used. W84 is on the extracellular side of transmembrane domain (TM) 1, whereas D313 lies close to the extracellular face of TM6. W267 and E428 are on the internal side of TM5 and TM8, respectively (Beuming et al. 2006). Previous studies show these residues might interact with intramolecular residues to mediate the hDAT oscillation between inward- and outward-facing states (Chen et al. 2001;Chen et al. 2004b;Kniazeff et al. 2008). Zn2+ increases the proportion of outward DAT and thereby enhances CFT binding (Norregaard et al. 1998;Chen et al. 2004a). We therefore used ZnCl2 to investigate the conformational preference of these mutants first. Compared with control cells, Zn2+ treatment in the absence of Na+ significantly increased the Bmax for [3H]CFT binding in WT DAT cells (Fig. 4A, see WT-Na+ bar for ratio of Bmax with Zn2+ over that without Zn2+), and this effect can be diminished by high concentration of extracellular Na+ (Fig. 4A, see WT+Na+ bar). This is consistent with the notion that the equilibrium between outward- and inward-facing states of DATs can be pushed towards the outward-facing state by a high concentration of extracellular Na+. In contrast to WT DAT, the Zn2+ effect in W84L and D313N DAT was lost in the absence of Na+ (Fig.4A, see W84L and D313N bars). This finding is consonant with the concept that these mutations promote the open state (outward facing) of the transporter (Chen et al. 2004b). On the contrary, for the W267L mutant, the Bmax ratio for [3H]CFT binding was significantly increased in conditions either with or without sodium (Fig.4A, see W267L bars). This would suggest that W267L mutation tends to hold the protein more in a closed state, such that with or without Na+, Zn2+ can enhance CFT binding. The [3H]CFT binding for the E428Q mutant was undetectable in the absence of Na+. Zn2+ had no effect on the Bmax in the presence of Na+ just as that for WT (Fig. 4A, see last bar). All data together suggest that in the presence of Na+ (condition used for all Ki determinations of substrates in the experiment described below), WT DAT and its mutants W84L, D313N and E428Q are mostly in the outward-facing state, whereas the W267L mutant is only partially in the outward conformation. It is interesting to note that in the presence of Na+, Zn2+ in general had no effect on the Kd of [3H]CFT binding, except for the observed decrease in Kd for the DAT mutants W267L and E428Q, as seen in Fig. 4B. This finding suggests Zn2+ can stabilize outward-facing conformations in these mutants with subtle differences, leading to enhanced binding affinity unlike the situation for WT.
Fig. 4. Effect of Zn2+ on transporter conformation in cells not treated with digitonin.
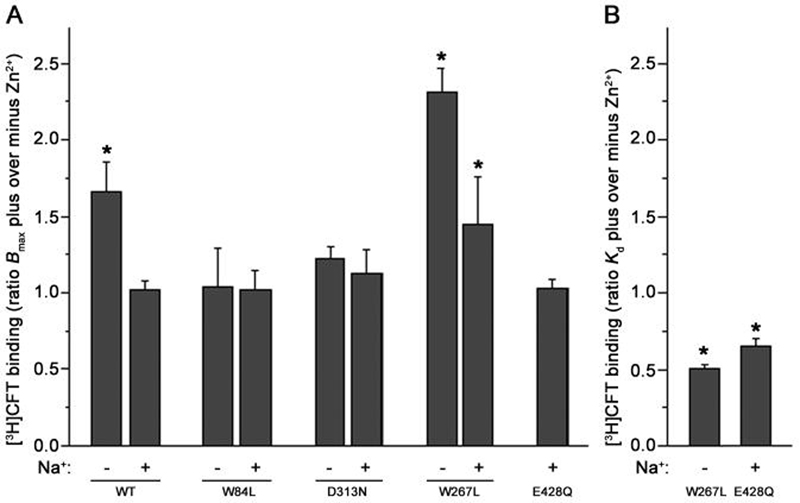
Intact cells stably expressing WT DAT or its mutants (W84L, D313N, W267L or E428Q) were subjected to the binding assay with 2-4 nM [3H]CFT and various concentrations of CFT with or without 10 μM Zn2+ for 15 min at 21°C. The ratio of the Bmax of [3H]CFT binding in the presence or absence of Zn2+ is shown in A. The ratio of the Kd in Zn2+ treated cells to that in untreated control cells is shown in B only for those changes that were statistically significant. The +Na+ and −Na+ bars indicate the presence or absence of 130 mM NaCl in the binding assay respectively, for details see Materials and Methods. Note that the E428Q mutant displayed too low [3H]CFT binding in the absence of Na+ to be analyzed. *P < 0.05 compared with ratio of unity (one-sample Student's t-test, two-tailed).
In digitonin-permeabilized cells expressing WT DAT, most transporters are inward facing even in the presence of Na+. It is, however, possible that the conformationally biased mutants used in this study differ from WT in how they are distributed between out- and inward conformational states. Fig. 5 shows that in KRH binding buffer with 130 mM Na+, digitonin-permeabilized W84L and D313N displayed a reduced increase in [3H]CFT binding with Zn2+ compared to WT. This is indicative for more outward-facing DAT in the case of these mutants, which have a bias for outward-facing conformations to begin with. In contrast, W267L and E428Q behaved similarly as WT, suggesting a similar proportion of inward-facing transporters for these mutants and WT. Thus, the tendency for W267L to favor the inward-facing state does not appear to be additive with the likewise pressure of digitonin towards the inward form.
Fig. 5. Effect of Zn2+ on transporter conformation in cells treated with digitonin.

Digitonin-permeabilized cells were incubated with 4 nM [3H]CFT and 10 μM Zn2+ in KRH buffer with 130 mM NaCl. Data are expressed as the ratio of [3H]CFT binding (at the same protein level) in the presence over that in the absence of Zn2+. *P < 0.05 compared with WT (one-way ANOVA followed by Dunnett multiple comparisons test).
Interaction of a catechol substrate with WT DAT and its mutants
Next, we analyzed the effect of external and internal residues on the binding of different substrates, and the relationship between the conformational states of these DAT mutants and the binding preference of catechol or non-catechol substrates. First, we measured the potency of DA in inhibiting binding of [3H]CFT to investigate how catechol substrates gain access to the mutant dopamine transporter. Similar to WT DAT, there was no significant difference (Ki ratio < 2) for the potency of DA in D313N and W84L between intact cells and digitonin-permeabilized cells (Table 1). Digitonin treatment also did not alter Ki values for DA in membranes, which were comparable with those in control cells (Table 1). It appears these two mutated residues, which are in the transmembrane domain but close to the extracellular loop, do not impact catechol substrate binding in a different way when the access pathway is directed from the cytosol into the transporter protein compared with when access occurs from the extracellular side. However, mutation at position 84 did affect DA binding affinity in the [3H]CFT binding assay. Compared to WT DAT, the W84L mutant displayed significantly increased binding affinity both in intact cells and cell membranes (Table 1 and Fig. 2). In the case of mutation of residues at the cytosolic side, W267L and E428Q, DA recognition was significantly enhanced by mutation only in the control cells with outward-facing DAT (Table 1 and Fig. 2). In digitonin-permeabilized cells or in membrane preparations, DA recognition was normal as in WT (Table 1). One interpretation of the data is that when these mutant DATs are inward facing, a catechol substrate accessing from the cytosolic milieu interacts with the transporter just as it does with WT DAT (see Discussion).
Table 1.
Effect of digitonin treatment on the potency of dopamine in inhibiting [3H]CFT binding to cells stably expressing wild-type or mutant DAT
| DAT preparations | Control Ki (μM) |
Digitonin Ki (μM) |
Ki ratio Digitonin/Control |
|
|---|---|---|---|---|
| WT | ||||
| Cell | 8.67 ± 1.15 (6) | 12.98 ± 2.00 (4) | 1.49 ± 0.30 | |
| Mem | 11.65 ± 1.1 (3) | 15.92 ± 2.62 (3) | 1.36 ± 0.06 | |
| W84L | ||||
| Cell | 2.67 ± 0.16 (5) * | 4.94 ± 0.27 (5) * # | 1.89 ± 0.19 | |
| Mem | 2.80 ± 0.09 (3) * | 3.12 ± 0.17 (3) * | 1.12 ± 0.09 | |
| D313N | ||||
| Cell | 16.19 ± 2.00 (6) | 19.12 ±1.58 (6) | 1.22 ± 0.07 | |
| Mem | 14.00 ± 2.15 (3) | 14.55 ± 1.89 (3) | 1.05 ± 0.04 | |
| W267L | ||||
| Cell | 1.12 ± 0.24 (5) * | 11.43 ± 2.62 (5) # | 10.58 ± 1.91 | |
| Mem | 13.15 ± 2.22 (3) | 13.74 ± 2.46 (3) | 1.05 ± 0.10 | |
| E428Q | ||||
| Cell | 2.28 ± 0.79 (6) * | 7.89 ± 1.31 (4) # | 3.46 ± 1.39 | |
| Mem | 12.00 ± 0.82 (3) | 13.88 ± 0.96 (3) | 1.18 ± 0.16 | |
Whole cells (Cell) or cell-free membranes (Mem) were pretreated with 15 μM digitonin or vehicle (0.75% ethanol) (Control) for 15 min at room temperature, and then subjected to the binding assay with 2-4 nM [3H]CFT and various concentrations of dopamine for 15 min at 21°C. Results are mean ± SEM of the number of independent experiments carried out in triplicate indicated in parentheses. Data were log-transformed where needed for similarity of SEMs of groups to be compared.
P < 0.05 compared with respective WT within Control or Digitonin group (one-way analysis of variance followed by Dunnett multiple comparisons test)
P < 0.05 compared with respective Control for Cell or for Mem (Student's t-test, paired where appropriate).
Interaction of a non-catechol substrate with WT DAT and its mutants
p-Tyramine, lacking only one of the two hydroxyl groups on the phenol ring of DA, was chosen as a non-catechol substrate for the following experiments with [3H]CFT as the radioligand for measuring substrate potency. W84L showed a binding pattern similar to that of WT: Permeabilization of cells, as well as membrane breakage, significantly increased p-tyramine Ki compared with that in control cells (Table 2). As suggested by our previous work with gramicidin (Zhen et al., 2005), the binding preference of p-tyramine appears to be directed towards the outward-facing state. This was likely also the case for W267L and E428Q since a significantly higher p-tyramine Ki was observed for cell membranes (table 2); unfortunately, the Ki for permeabilized cells stably expressing W267L or E428Q could not be determined due to their instability after digitonin treatment (Table 2). In addition, in intact cells, W267L or E428Q mutation significantly increased p-tyramine affinity (lowered Ki) compared with that for WT (Table 2). Thus mutation of W267 and E428 markedly enhanced p-tyramine affinity to the outward state, but in membrane preparations (with reduced proportion of outward-facing transporters) the affinity was similar to that in WT (Table 2). It should be noted that in all cases of blocker (CFT) and substrates (p-tyramine and DA) binding (Fig.1, Table 1, Table 2), results for digitonin treated cells were similar to that for membranes with or without digitonin treatment. Thus, digitonin-permeabilized cells mimick broken membranes in having outward and inward facing DAT exposed to the same ionic environment under conditions of membrane depolarization.
Table 2.
Effect of digitonin treatment on the potency of p-tyramine in inhibiting [3H]CFT binding to cells stably expressing wild-type or mutant DAT
| DAT preparations | Control Ki (μM) |
Digitonin Ki (μM) |
Ki ratio Digitonin/Control |
|
|---|---|---|---|---|
| WT | ||||
| Cell | 13.00 ± 2.02 (8) | 50.00 ± 3.70 (4) # | 3.84 ± 0.66 | |
| Mem | 51.81 ± 7.31 (4) § | 61.61 ± 7.42 (4) | 1.21 ± 0.04 | |
| W84L | ||||
| Cell | 7.91 ± 1.02 (4) | 23.73 ± 2.13 (4) *# | 3.05 ± 0.16 | |
| Mem | 20.06 ± 2.32 (4) * § | 24.18 ± 3.76 (4) * | 1.19 ± 0.10 | |
| W267L | ||||
| Cell | 3.69 ± 0.36 (3) * | N.T . | ||
| Mem | 57.09 ± 5.29 (3) § | 58.46 ± 2.96 (3) | 1.04 ± 0.07 | |
| E428Q | ||||
| Cell | 3.50 ± 1.06 (6) * | N.T. | ||
| Mem | 67.12 ± 10.0 (3) § | 85.75 ± 3.17 (3) | 1.36 ± 0.29 | |
Whole cells (Cell) or cell-free membranes (Mem) were pretreated with 15 °M digitonin or vehicle (0.75% ethanol) (Control) for 15 min at room temperature, and then subjected to the binding assay with 2-4 nM [3H]CFT and various concentrations of p-tyramine for 15 min at 21°C. Results are mean ± SEM of the number of independent experiments carried out in triplicate indicated in parentheses. Data were log-transformed where needed for similarity of SEMs of groups to be compared. NT, not testable due to instability with digitonin.
P < 0.05 compared with respective WT within Control or Digitonin group (one-way analysis of variance followed by Dunnett multiple comparisons test)
P < 0.05 compared with respective Control for Cell or for Mem (Student's t test, paired where appropriate)
P < 0.05 compared with respective Cell within Control group (Student's t test).
Divergency between mutation effects on DA and CFT binding
It is important to note the differential effects of mutations on DA and CFT binding. Compared with WT, both W84L and D313N cells showed higher affinity for CFT; while W84L mutation enhanced DA binding, D313N mutation did not alter the affinity for DA in a statistically significant manner (Fig. 2a). In contrast, the W267L and E428Q mutations enhanced DA binding without changing the affinity for CFT (Fig. 2a). Similar mutation-induced binding changes are also seen in membrane preparations (Fig. 2b), with the exception of the W267L and E428Q mutations, both of which affected the potency of DA in inhibiting [3H]CFT binding to cells (Fig. 2a) but not cell-free membranes (Fig. 2b). This is consistent with our previous observations that the transmembrane ion gradient in conjunction with membrane potential plays a role in DA binding to DAT (Chen and Reith 2004). Thus, changes in binding affinity of DA and CFT in cells vs. broken membranes and in different mutants are not necessarily correlated. Enhanced CFT binding does not invariably accompany enhanced DA binding and vice versa, also supporting the notion that binding sites for CFT and DA are not identical.
Discussion
Knowledge regarding the mode of interaction between ligands and DAT is important for understanding the mechanisms underlying the addictive and neurotoxic effects of cocaine and various substrate-like drugs including psychostimulants. So far, most studies have focused on how ligands bind to DAT from the extracellular side. To our knowledge, this is the first report to show different substrates have diverse ability to interact with the transporter from inside the cell with DAT facing inwardly. We present here experimental evidence supporting different models for how catechol and non-catechol substrates interact with DAT in the plasma membrane either from the outside or inside.
Our previous results with the ionophore gramicidin suggested that the catechol DA can interact with DAT from either side whereas non-catechol substrates only bind to outward-facing DAT (Zhen et al. 2005). The limitation of gramicidin as a tool is that the access pathway from the cytosol is unavailable for substrates including DA applied outside the cell (Fig. 1b). In cell-free membranes, both the ion and voltage gradient is lacking, as with gramicidin, but substrate can access from either side (Fig. 1c). However, the complete loss of the cytosolic milieu might impact the binding property of the transporter. In the present study therefore, cells were permeabilized with digitonin (for cartoons see Figs. 1d, 6, 7) to disrupt the transmembrane gradient of Na+, K+, and other ions as well as the voltage gradient, and to enable substrate to access both sides of DAT, as in membrane preparations with the important difference that the main cellular cytosolic architecture is intact.
Fig. 6. Cartoon of binding model for DA at wild-type and mutant DATs.

Conformational distributions between outward and inward states are drawn qualitatively based on the Zn2+ experiments. WT: DATs in control cells are mostly outward-facing; in digitonin (DIG)-treated cells and membranes (either with or without DIG treatment), DAT is mostly inward-facing. DA binds DAT with similar potency in both situations. W84L: Conformational preference is the same as that for WT, except for a somewhat reduced conversion to inward-facing transporters by DIG. DA is able to reach both external and internal binding sites as WT. However, the intrinsic binding affinity is enhanced at both sides (indicated by a better fit of the red diamond). D313N: Conformational preference and DA binding pattern are similar as for WT except for a somewhat reduced conversion to inward-facing transporters by DIG. D267L: In control cells, less transporter is facing outward compared with that in WT, but DA can access its binding site on DAT with greater ease (indicated by blue arrow) resulting in a higher apparent affinity compared with that at WT. In DIG-treated cells and in membranes, DAT is mostly inward-facing and the DA-DAT interaction is similar to that for WT; by analogy with the comparable case in Fig. 7, the enhanced access to outward-facing DAT at the right hand has been removed (see Fig. 7 legend). E428Q: In cells, DATs are mostly outward facing as for WT; in membranes, the reversed is true as is the case for WT. DA has free access to external and internal binding sites. However, outward-facing DAT displays higher apparent affinity for DA compared to WT because of easier access of DA to its binding site (blue arrow); by analogy with the comparable case in Fig. 7, the enhanced access to outward-facing DAT at the right hand has been removed (see Fig. 7 legend). For each cell type, the number in the left parentheses is the DA Ki in control cells; the first number in the right parentheses is the DA Ki in cells treated with DIG, the second number is the mean of DA Ki values from membranes treated with or without DIG. Changes in DA recognition or access in the mutants are drawn only when the data indicated statistically significant differences with WT.
Fig. 7. Cartoon of binding model for p-tyramine at wild-type and mutant DATs.

DAT conformational distribution in control cells, digitonin (DIG)-permeabilized cells and in membranes is the same as in Figure 5. p-Tyramine only binds to outward-facing transporters. For control cells, W84L is prone to have a higher affinity for DA compared with WT (indicated by better fit of violet diamond). W267L and E428Q have a higher apparent affinity by easier access (indicated by blue arrow). For DIG-permeabilized cells and for membranes, p-tyramine binds to WT and the mutants similarly, except in the case of W84L that displays a higher affinity (in analogy with control cells and with DA in Fig. 6 in the comparable case). Depolarization combined with disruption of the transmembrane ion gradient (panels on the right) is speculated to remove the enhanced access for W267L and E428Q. For each cell type, the number in the left parentheses is the p-tyramine Ki in control cells; the first number in the right parentheses is the p-tyramine Ki in cell treated with DIG, the second number is the mean of p-tyramine Ki values from membranes treated with or without DIG.
For WT, as studied with [3H]CFT as the radioligand, in digitonin-permeabilized cells and in broken membranes, where DAT is predominantly in the inward-facing state, DA bound with a similar Ki value as that observed in control cells where DAT is predominantly outward-facing, indicating a symmetric binding pattern (Table 1, for cartoon see Fig. 6, WT). This is different from the pattern of the non-catechol substrate p-tyramine (Table 2, Fig 7, WT). p-Tyramine only binds DAT that is outward facing, as evidenced by an increased Ki for digitonin-permeabilized cells and for membranes, where DAT is predominantly inward facing. Mutant W84L control cells showed stronger DA binding compared with WT (Table 1). This is very likely due to mutation-enhanced intrinsic binding affinity per se, rather than an improvement in the permeation pathway for substrate from the extracellular face towards the substrate binding site, which according to the crystallized bacterial homolog transporter LeuT lies midway inside the membrane in a region of unwound helix 1 and 6 (Yamashita et al. 2005). When DAT turned to be inward-facing, as in digitonin-treated cells and in membranes, DA binding was similarly improved (Fig. 6, W84L). It is easier to conceptualize a single mutation-enhanced binding affinity at the substrate site than a dual improvement in both the pathway from the extracellular as well as the pathway from the intracellular face towards the substrate site midway in the membrane. Consistent with this, p-tyamine showed enhanced binding in digitonin-permeabilized cells and in membranes with W84L, also with such a tendency in control cells (Table 2, see better fit of the violet diamond in the W84L cartoon in Fig 7, as well as better fit of the red diamond for DA in Fig. 6). For D313N, the DA binding profile was similar to that for WT: external and internal binding was symmetric (Table 1, Fig. 6). Compared with WT, the mutants W267L and E428Q in control cells, where DAT is outward facing, showed improved binding (Table 1). Our interpretation is that this is due to an improvement in the permeation pathway from the external face towards the substrate site midway inside the membrane (indicated by blue arrow in bottom left two cartoons in Fig. 6), rather than to enhanced intrinsic binding affinity. In consonance, in digitonin-permeabilized cells and in membranes, where DAT turns to be inward-facing and the permeation pathway is now directed from the inner face of the membrane towards the substrate site, DA bound DAT just as in WT (Table 1 and Fig. 6) which would not have been the case if a higher intrinsic binding affinity at the substrate site had been the underlying mechanism for enhanced affinity. A second possible interpretation is that the intrinsic binding affinity is enhanced in these two mutants, but because the internal access pathway is blocked, DA binding becomes asymmetric (preference for outward-facing state) just as the non-catechol substrate p-tyramine. However, the ratio of Ki in membranes over that in control intact cells is much higher for p-tyramine (15.1 for W267L, 22.57 for E428Q) than for DA (9.93 for W267L, 5.70 for E428Q) (calculated from data in Figs 6, 7). The p-tyramine data indicate that without binding to inward-facing DAT, the Ki for DA in membranes should increase (compared with control intact cells) to a much higher degree than what is actually seen in these two mutants, because the conversion from outward- to inward-facing DATs (by depolarization and collapse of ion gradients) is independent of the interacting ligand. A third possibility is that the mutations have a differential impact on the intrinsic binding affinities in outward- and inward-facing transporters. For reasons of simplicity we prefer the first interpretation but acknowledge that current experimental data are lacking to rule out the latter possibility.
Most DAT substrates are phenethylamine derivatives with differences in substitutions for the hydrogen atom in the backbone of phenethylamine. Work with striatal slices, where structural variants of DA were tested either as inhibitors of DA uptake or as substrates themselves, emphasized the catechol feature as mediating substrate recognition (Meiergerd and Schenk 1994), but subsequent work with cells expressing human DAT has been equivocal (Appell et al. 2004;Wang et al. 2003). In addition, the classical view of the catechol hydroxyls interacting with serine residues in TM7 of DAT (Kitayama et al. 1992) has been questioned (Wang et al. 2003;Xhaard et al. 2008). Our previous results from [3H]CFT competition studies suggest that DA and the substrates with a modified catechol moiety differ in their binding interactions with DAT as revealed under conditions where the intracellular part of DAT is facing higher [Na+]in, (Zhen et al. 2005;Chen and Reith 2004). Here in [3H]CFT binding assays on digitonin-permeabilized cells, where substrates can go inside the cells freely, catechol substrates (DA and norepinephrine) show binding activity similar as in intact cells (symmetric binding). On the contrary, the non-catechol substrates p-tyramine, d-amphetamine, ß-phenethylamine, and MPP+ have a much lower affinity for DAT with digitonin, consonant with their lack of interaction at the inward face of the transporter (asymmetric binding) under the conditions used (Fig. 3). Thus, even the lack of just one of the two phenolic hydroxyl groups has a dramatic effect on binding of substrates to the inward-facing transporter, and this will impact their ability to undergo reversed transport. This would provide an additional explanation as to how amphetamine, while it is itself accumulated intracellularly, causes efflux of DA (catechol) rather than of amphetamine (non-catechol) through DAT, with DA efflux known to be severely impacted by the absence of DAT in preparations from DAT-KO mice (Jones et al. 1998). Be that as it may, at high enough intracellular concentrations, even non-catechol substrates will be able to leave the cell through DAT in the reversed mode. It can also be thought that for asymmetrically acting substrates lipohilicity will be a crucial additional property enabling passage through the lipid phase of the plasma membrane diminishing a role for DAT as the passage conduit. Another example of the relevance of the catechol group in determining ligand interaction with proteins important in monoaminergic neurotransmission relates to the β2-adrenergic receptor (Swaminath et al. 2005) where the aromatic ring of the noncatechol salbutamol (m-methylhydroxyl instead of hydroxyl) occupies a binding space that does not overlap with that for catecholamines. Our results do not suggest much impact on binding affinity for outward facing DAT in intact cells upon removing one hydroxyl (p-tyramine) or two hydroxyls (β-phenethylamine) from the catechol dopamine (Fig. 3, see cell control bars), but the catechol feature appears essential for the interaction with inward-facing DAT as deduced from the digitonin experiments (Fig. 3, see cell digitonin and membrane bars). This could be thought to be related to the proposed coordination of catechol hydroxyls by Na+ (Xhaard et al., 2008) if one postulates that this chelation process plays different roles depending on DAT conformation. There was a modest decrease in the potency of epinephrine for inward-facing DAT in digitonin-permeabilized cells or in membranes (Fig. 3), which we interpret as resulting from the extra methyl group on the aliphatic chain portion of the molecule. Although the catechol feature is crucial for symmetric binding, additional structural features appear to impact substrate recognition such that the same domain when switching from the outward- to the inward-facing state, looses some of its binding strength.
All the residues mutated in this work are highly conserved in the superfamily of Na+/Cl−-dependent transporters (Chen et al. 2001). The mutations are located at the external (W84L and D313N) or internal (W267L and E428Q) side of the transporter, they tend to constitutively hold transporter in the outward (W84L and D313N) or inward (W267L) facing state, and the residue mutated is either a tryptophan (W84L and W267L), which is prominent in providing cation-π interactions, or an acidic amino acid (E428Q and D313N), which provides a negative charge. None of these mutations cause affinities for DA in membranes or digitonin-treated cells lower than those observed in WT as measured via [3H]CFT binding (Table 1), and none of the speculated mechanisms (Figs. 6, 7) require an impediment or enhancement for DA in the ease of accessing the substrate binding site from the internal side through the internal permeation pathway in order to describe the data (Table 1). It will be important to assess what residues in DAT line this permeation pathway, as well as the external pathway spanning the external side and the substrate site in the center of the plasma membrane. According to a recent study on the serotonin transporter (Forrest et al. 2008), the external and internal permeation pathway is made up of TMs 1b, 3, 6a, 10 and 1a, 5, 6b, 8, respectively, which contain the residues studied here (84 and 313 in TM 1b and 6a, respectively, and 267 and 428, in TM 5 and 8, respectively). The same study also indicates which residues line the internal pathway based on modeling and subsituted cysteine accessibility to methanethiosulfonate: W282 in SERT, equivalent to W267 in DAT, does not face the internal permeation pathway, but E444, corresponding to E428 in DAT, does. Such information is not available for residues lining the external pathway. Residues 84, 313, 267, or 428 in DAT are clearly not part of the substrate binding site (Yamashita et al. 2005). W84 is next to R85, which likely forms the extracellular gate by ion pairing with D476; this ion pairing helps holding the transporter in the inward-facing form, and therefore mutation of the neighboring W84 can be thought to disrupt the 85-476 ionic bond leading to destabilization of the inward-facing conformation as observed in our experiments. E428 is modeled to interact with Y335, which is part of an intracellular interaction network with bonds between residues assisting in holding the DAT in an outward-facing conformation (Kniazeff et al., 2008). Nevertheless, mutation of E428 to Q did not promote an inward form in our Zn2+ experiments (Fig. 4), but it is possible an effect could have been uncovered if lower Na+ concentrations had been tested. Three out of the four mutations studied here displayed enhanced apparent affinity for DA binding from the external side (Fig. 6), and given their locations in relation to the primary substrate site as described above, these effects appear to be indirect.
Treatment of cells with digitonin is known to increase the membrane permeability of cells to ions and proteins (Frye and Holz 1985;Wilson and Kirshner 1983;Dunn and Holz 1983). Thus, proteins as large as phenylethanolamine-N-methyltransferase (38 kDa) and lactic dehydrogenase (134 kDa) pass through membranes with digitonin, and therefore substrates of less than 200 Da as used in the present study can easily permeate. CFT will also pass through easily, but it always does (regardless of the presence of digitonin) due to its lipophilicity as does its parent compound, cocaine. Digitonin permeabilization might cause perturbations other than the intended membrane leakiness for ions and phenethylamine-related compounds and MPP+, potentially adding complexity to the interpretation of the result. Acute regulation of the DAT includes a receptor- or protein kinase-mediated internalization of the DAT (Melikian and Buckley 1999;Boudanova et al. 2008;Mortensen et al. 2008;Sorkina et al. 2006;Zapata et al. 2007;Bolan et al. 2007). In experiment involving digitonin, one could consider that changes in intracellular ion concentration and membrane potential cause reduced availability of the transporter at the cell surface impacting DAT measurements. If that is the case, one should observe a low Bmax in cells after their treatment with digitonin, which is contrary to the present results. In addition, our previous work with gramicidin-treated cells shows that after similar ionic and membrane potential changes, DAT mostly remains on cell surface and its CFT binding characteristics (Kd and Bmax) undergo only minor changes (Chen and Reith 2004). It should be noted that in broken membranes, digitionin had no effect on the binding of various substrates in different cell types (Tables 1 and 2), indicating a lack of direct effect on substrate-DAT interactions by the low concentration (15 μM) and short time (15 min) of digitonin exposure used in this study. Digitonin-permeabilized cells have also been used successfully to analyze the intracellular topology of the serotonin transporter (SERT) (Androutsellis-Theotokis and Rudnick 2002). Most importantly, the ability of DAT and SERT to bind the cocaine analogs CFT and β-CIT is preserved in digitonin-solubilized striatal membranes or digitonin-permeabilized cells (Androutsellis-Theotokis and Rudnick 2002;Gracz and Madras 1995). There are also evidences that digitonin renders cell permeable but still preserves membraneous protein function and physiological activity. Thus, plasma membranes of adrenal medullary chromaffin cells can be made leaky by digitonin allowing free entry of extracellular Ca2+ and ATP, but importantly these cells are still able to undergo Ca2+-induced exocytosis (Frye and Holz 1985).
As p-tyramine appears to bind only to outward-facing DAT, and CFT has been speculated to prefer the outward-facing conformation as other cocaine-like compounds (Chen and Reith 2004;Loland et al. 2008;Chen et al. 2004b;Reith et al. 1992), it is instructive to compare the change in their binding affinities for DAT-expressing intact cells upon shifting the equilibrium from outward- towards inward-facing transporters. In [3H]CFT binding assays, digitonin reduced the affinity of p-tyramine for WT and W84L 3- to 4-fold (Table 2), and for W267L 15-fold if the membrane affinity is taken to represent the digitonin-intact cell value as is generally true for substrates (Tables 1 and 2, Fig. 3). In contrast, the affinity of CFT was virtually unchanged by digitonin in WT, W84L, and W267L (Fig. 2). This suggests that CFT binding is much less susceptible to the conformational state of DAT than substrate binding. This is consonant with the modest effect of gramicidin on [3H]CFT binding (1.3-1.5-fold reduction) observed in our previous studies (Chen and Reith 2004;Zhen et al. 2005) under comparable conditions of membrane depolarization and ion gradient collapse. It could be considered that closure of the DA permeation pathway on the extracellular side with DAT facing inwardly, does not close off access to cocaine-like compounds, even though the cocaine site has been modeled to overlap with the substrate site by Beuming et al. (2008). In this context, the effects of DAT mutations themselves are of course complex in that those can affect both the conformational equilibrium of DAT and the intrinsic binding affinity at the substrate or cocaine binding site (or access through entry pathway to binding pocket). Thus, the W84L and W267L mutants both show enhanced affinity for p-tyramine compared with WT (Table 2), but only W84L displays higher affinity for CFT (Fig. 2). With W84L tending towards an outward-facing and W267 towards an inward-facing conformation, there clearly are mutation-dependent effects beyond conformational impacts. The DAT mutant Y335A studied by Loland et al. (2008) tends to be inward facing, and the potency of CFT in inhibiting [3H]DA uptake into this mutant is reduced as much as 100-fold compared with WT, again emphasizing mutational effects on recognition in addition to conformation.
The present Zn2+ effects deserve a comment in comparison with available literature from other labs and with our own previous results. In the present work, Zn2+ generally affected [3H]CFT binding through changes in Bmax (Fig. 4). This is consonant with observations by Norregaard et al. (1998) and can be rationalized by considering that Zn2+ fixates a portion of the transporters in a conformation with high affinity for [3H]CFT, thereby reducing the contribution of low-affinity [3H]CFT binding sites that can escape detection by rapid dissociation during the filtration process for capturing binding. However, there is also the oscillation between various conformations that normally gives a time-averaged measure over the window of the binding assay; and Zn2+ could be thought to increase this average affinity consonant with our previous observations (Chen et al. 2004a;Wu et al. 1997). A recent study (Pifl et al. 2008) shows that Zn2+ regulation of DAT is additionally susceptible to the influence of membrane potential and ion (K+, Cl−) concentration. Such complexities may contribute to observations for [3H]CFT binding of either Bmax and/or Kd changes with Zn2+. In this context, there were also some minor differences between the present comparisons between WT and D313N or E428Q cells in binding affinity for DA or CFT, respectively, and those reported by us previously (Chen et al. 2001;Chen and Reith 2004). Generally, the protocols followed in the present experiments were the same as in the previous studies, but small methodological differences cannot be ruled out. Overall, however, conclusions are not impacted by these differences.
The elegant study by Shi et al. (2008) postulates a secondary substrate site, a vestibular site situated between the primary substrate site in the center of the plasma membrane (in the region of the unwound portions of helices 1 and 6) and the cytoplasmic end of transmembrane domains 1, 3 and 10 capped by the tip of extracellular loop 4. This vestibular site is also the binding site for tricyclic antidepressants in the bacterial homolog transporter, the LeuT (Singh et al. 2007;Zhou et al. 2007). If the secondary site also exists in human DAT, the question is how it would affect the interpretation of the present results. This site would bind DA with or without Na+ from the external side in cells in analogy with LeuT (Shi et al. 2008) or in membranes from the side of the DAT normally facing external medium. In the context of the present results, one needs to know the relationship between the secondary (and primary) substrate site to the binding pocket for CFT, as [3H]CFT is the radioligand used here for assessing substrate affinities. If the computational model of Beuming et al. (2008) and a recent photoaffinity labeling study favoring the same model (Parnas et al. 2008) are correct, DA and [3H]CFT interact at the primary binding site in the center of the membrane. It could be thought that DA binding to the secondary, more extracellularly located site reduces the affinity of CFT for the primary site as it also weakens the binding of DA at that site (Shi et al. 2008). What argues against this idea is the lack of complexities observed in inhibition of [3H]CFT binding by DA, which rather appears to obey general kinetics of competitive inhibition in both rat brain tissue and human DAT expressing cells (Reith et al. 1992;Amejdki-Chab et al. 1992;Li and Reith 1999), as well as the lack of effect of DA on the dissociation rate of [3H]mazindol binding (Zimanyi et al. 1989) which is known to overlap with CFT binding (Reith and Selmeci 1992;Tidjane et al. 2001). So far, the data support a simple interaction of CFT and DA at the primary substrate site, with the caveat that the above kinetic evidence does not prove that CFT physically binds to the primary DA site itself (just that binding of DA and CFT is mutually exclusive). Based on these considerations, we suggest that the secondary substrate site, if it exists in human DAT, plays no role in the present experiments.
Asymmetry for DAT properties, as reported here for non-catechol interactions, is not a novel phenomenon for DAT. Thus, outward and inward transport of DA are regulated differentially as revealed by human DAT constructs with alanine substituted for S528 (Chen and Justice 2000) or with substitutions of five N-terminal serines (Khoshbouei et al. 2004). It is possible that in addition to differences in recognition and translocation constants, different external and internal environments in contact with the DAT play a role. The present experiments with digitonin are an attempt to equalize two parts of these environments: the ionic composition and the membrane potential. Our previous study showed the importance of simultaneous influx of Na+ and membrane depolarization in turning the DAT inward (Chen and Reith, 2004). Such an increase in intracellular Na+ under depolarizing conditions impedes binding and uptake of extracellular DA while facilitating outward transport of intracellular DA. In brain, such events could be thought to be triggered by depolarizing Na+ influx through voltage-gated Na+ channels resulting from pre- or post-synaptic excitation. In future studies it will be of interest to define the role of Na+ influx via Na+ channels in DAT activity. Prolonged opening of Na+ channels has been implicated in inherited diseases of neuronal hyperexcitability, glutamate excitotoxicity, and brain ischemia (Catterall 2000;Hammarstrom and Gage 2002;Sugawara et al. 2001).
Acknowledgements
The study was supported by National Institute of Drug Abuse Grants DA13261 and DA019676.
Abbreviations used
- CFT
2b-carbomethoxy-3b-(4-fluorophenyl) tropane
- DA
dopamine
- DAT
dopamine transporter
- HEK
human embryonic kidney
- KRH
Krebs/Ringer/HEPES buffer
- MPP+
1-methyl-4-phenylpyridinium
- NMDG-Cl
N-methyl-D glucamine chloride
- PBS
phosphate-buffered saline
- TM
transmembrane
- WT
wild-type
Reference List
- Amara SG, Sonders MS. Neurotransmitter transporters as molecular targets for addictive drugs. Drug Alcohol Depend. 1998;51:87–96. doi: 10.1016/s0376-8716(98)00068-4. [DOI] [PubMed] [Google Scholar]
- Amejdki-Chab N, Costentin J, Bonnet JJ. Kinetic analysis of the chloride dependence of the neuronal uptake of dopamine and effect of anions on the ability of substrates to compete with the binding of the dopamine uptake inhibitor GBR 12783. J. Neurochem. 1992;58:793–800. doi: 10.1111/j.1471-4159.1992.tb09327.x. [DOI] [PubMed] [Google Scholar]
- Androutsellis-Theotokis A, Rudnick G. Accessibility and conformational coupling in serotonin transporter predicted internal domains. J. Neurosci. 2002;22:8370–8378. doi: 10.1523/JNEUROSCI.22-19-08370.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appell M, Berfield JL, Wang LC, Dunn WJ, III, Chen N, Reith ME. Structure-activity relationships for substrate recognition by the human dopamine transporter. Biochem. Pharmacol. 2004;67:293–302. doi: 10.1016/j.bcp.2003.09.013. [DOI] [PubMed] [Google Scholar]
- Bannon MJ. The dopamine transporter: role in neurotoxicity and human disease. Toxicol. Appl. Pharmacol. 2005;204:355–360. doi: 10.1016/j.taap.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat. Neurosci. 2008;11:780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuming T, Shi L, Javitch JA, Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 2006;70:1630–1642. doi: 10.1124/mol.106.026120. [DOI] [PubMed] [Google Scholar]
- Bolan EA, Kivell B, Jaligam V, Oz M, Jayanthi LD, Han Y, Sen N, Urizar E, Gomes I, Devi LA, Ramamoorthy S, Javitch JA, Zapata A, Shippenberg TS. D2 receptors regulate dopamine transporter function via an extracellular signal-regulated kinases 1 and 2-dependent and phosphoinositide 3 kinase-independent mechanism. Mol. Pharmacol. 2007;71:1222–1232. doi: 10.1124/mol.106.027763. [DOI] [PubMed] [Google Scholar]
- Boudanova E, Navaroli DM, Stevens Z, Melikian HE. Dopamine transporter endocytic determinants: carboxy terminal residues critical for basal and PKC-stimulated internalization. Mol. Cell Neurosci. 2008;39:211–217. doi: 10.1016/j.mcn.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch W, Saier MH., Jr. The transporter classification (TC) system, 2002. Crit Rev. Biochem. Mol. Biol. 2002;37:287–337. doi: 10.1080/10409230290771528. [DOI] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Chen N, Justice JB., Jr. Cocaine acts as an apparent competitive inhibitor at the outward-facing conformation of the human norepinephrine transporter: kinetic analysis of inward and outward transport. J. Neurosci. 1998;18:10257–10268. doi: 10.1523/JNEUROSCI.18-24-10257.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Justice JB. Differential effect of structural modification of human dopamine transporter on the inward and outward transport of dopamine. Brain Res. Mol. Brain Res. 2000;75:208–215. doi: 10.1016/s0169-328x(99)00288-0. [DOI] [PubMed] [Google Scholar]
- Chen N, Reith ME. Interaction between dopamine and its transporter: role of intracellular sodium ions and membrane potential. J. Neurochem. 2004;89:750–765. doi: 10.1111/j.1471-4159.2004.02409.x. [DOI] [PubMed] [Google Scholar]
- Chen N, Rickey J, Berfield JL, Reith ME. Aspartate 345 of the dopamine transporter is critical for conformational changes in substrate translocation and cocaine binding. J. Biol. Chem. 2004a;279:5508–5519. doi: 10.1074/jbc.M306294200. [DOI] [PubMed] [Google Scholar]
- Chen N, Sun L, Reith ME. Cationic interactions at the human dopamine transporter reveal binding conformations for dopamine distinguishable from those for the cocaine analog 2 alpha-carbomethoxy-3 alpha-(4-fluorophenyl)tropane. J. Neurochem. 2002;81:1383–1393. doi: 10.1046/j.1471-4159.2002.00941.x. [DOI] [PubMed] [Google Scholar]
- Chen N, Vaughan RA, Reith ME. The role of conserved tryptophan and acidic residues in the human dopamine transporter as characterized by site-directed mutagenesis. J. Neurochem. 2001;77:1116–1127. doi: 10.1046/j.1471-4159.2001.00312.x. [DOI] [PubMed] [Google Scholar]
- Chen N, Zhen J, Reith ME. Mutation of Trp84 and Asp313 of the dopamine transporter reveals similar mode of binding interaction for GBR12909 and benztropine as opposed to cocaine. J. Neurochem. 2004b;89:853–864. doi: 10.1111/j.1471-4159.2004.02386.x. [DOI] [PubMed] [Google Scholar]
- Dunn LA, Holz RW. Catecholamine secretion from digitonin-treated adrenal medullary chromaffin cells. J. Biol. Chem. 1983;258:4989–4993. [PubMed] [Google Scholar]
- Falkenburger BH, Barstow KL, Mintz IM. Dendrodendritic inhibition through reversal of dopamine transport. Science. 2001;293:2465–2470. doi: 10.1126/science.1060645. [DOI] [PubMed] [Google Scholar]
- Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proc. Natl. Acad. Sci. U. S. A. 2008;105:10338–10343. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye RA, Holz RW. Arachidonic acid release and catecholamine secretion from digitonin-treated chromaffin cells: effects of micromolar calcium, phorbol ester, and protein alkylating agents. J. Neurochem. 1985;44:265–273. doi: 10.1111/j.1471-4159.1985.tb07140.x. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Sotnikova TD, Caron MG. Monoamine transporter pharmacology and mutant mice. Trends Pharmacol. Sci. 2002;23:367–373. doi: 10.1016/s0165-6147(02)02044-8. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Goncalves CA, Gottfried C, Dunkley PR. The use of permeabilized cells to assay protein phosphorylation and catecholamine release. Neurochem. Res. 2000;25:885–894. doi: 10.1023/a:1007533927813. [DOI] [PubMed] [Google Scholar]
- Gracz LM, Madras BK. [3H]WIN 35,428 ([3H]CFT) binds to multiple charge-states of the solubilized dopamine transporter in primate striatum. J. Pharmacol. Exp. Ther. 1995;273:1224–1234. [PubMed] [Google Scholar]
- Hammarstrom AK, Gage PW. Hypoxia and persistent sodium current. Eur. Biophys. J. 2002;31:323–330. doi: 10.1007/s00249-002-0218-2. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J. Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A, Javitch JA. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS. Biol. 2004;2:E78. doi: 10.1371/journal.pbio.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitayama S, Shimada S, Xu H, Markham L, Donovan DM, Uhl GR. Dopamine transporter site-directed mutations differentially alter substrate transport and cocaine binding. Proc. Natl. Acad. Sci. U. S. A. 1992;89:7782–7785. doi: 10.1073/pnas.89.16.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeff J, Shi L, Loland CJ, Javitch JA, Weinstein H, Gether U. An intracellular interaction network regulates conformational transitions in the dopamine transporter. J. Biol. Chem. 2008;283:17691–17701. doi: 10.1074/jbc.M800475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi G, Raiteri M. Carrier-mediated release of neurotransmitters. Trends Neurosci. 1993;16:415–419. doi: 10.1016/0166-2236(93)90010-j. [DOI] [PubMed] [Google Scholar]
- Li LB, Reith ME. Modeling of the interaction of Na+ and K+ with the binding of dopamine and [3H]WIN 35,428 to the human dopamine transporter. J. Neurochem. 1999;72:1095–1109. doi: 10.1046/j.1471-4159.1999.0721095.x. [DOI] [PubMed] [Google Scholar]
- Loland CJ, Desai RI, Zou MF, Cao J, Grundt P, Gerstbrein K, Sitte HH, Newman AH, Katz JL, Gether U. Relationship between conformational changes in the dopamine transporter and cocaine-like subjective effects of uptake inhibitors. Mol. Pharmacol. 2008;73:813–823. doi: 10.1124/mol.107.039800. [DOI] [PubMed] [Google Scholar]
- Meiergerd SM, Schenk JO. Striatal transporter for dopamine: catechol structure-activity studies and susceptibility to chemical modification. J. Neurochem. 1994;62:998–1008. doi: 10.1046/j.1471-4159.1994.62030998.x. [DOI] [PubMed] [Google Scholar]
- Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. J. Neurosci. 1999;19:7699–7710. doi: 10.1523/JNEUROSCI.19-18-07699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen OV, Larsen MB, Prasad BM, Amara SG. Genetic complementation screen identifies a mitogen-activated protein kinase phosphatase, MKP3, as a regulator of dopamine transporter trafficking. Mol. Biol. Cell. 2008;19:2818–2829. doi: 10.1091/mbc.E07-09-0980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norregaard L, Frederiksen D, Nielsen EO, Gether U. Delineation of an endogenous zinc-binding site in the human dopamine transporter. EMBO J. 1998;17:4266–4273. doi: 10.1093/emboj/17.15.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas ML, Gaffaney JD, Zou MF, Lever JR, Newman AH, Vaughan RA. Labeling of dopamine transporter transmembrane domain 1 with the tropane ligand N-[4-(4-azido-3-[125I]iodophenyl)butyl]-2beta-carbomethoxy-3beta-(4-chlorophenyl) tropane implicates proximity of cocaine and substrate active sites. Mol. Pharmacol. 2008;73:1141–1150. doi: 10.1124/mol.107.043679. [DOI] [PubMed] [Google Scholar]
- Pifl C, Wolf A, Rebernik P, Reither H, Berger ML. Zinc regulates the dopamine transporter in a membrane potential and chloride dependent manner. Neuropharmacology. 2008 doi: 10.1016/j.neuropharm.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Quick M, Yano H, Goldberg NR, Duan L, Beuming T, Shi L, Weinstein H, Javitch JA. State-dependent conformations of the translocation pathway in the tyrosine transporter Tyt1, a novel neurotransmitter:sodium symporter from Fusobacterium nucleatum. J. Biol. Chem. 2006;281:26444–26454. doi: 10.1074/jbc.M602438200. [DOI] [PubMed] [Google Scholar]
- Reith ME, de CB, Rice KC, Jacobson AE. Evidence for mutually exclusive binding of cocaine, BTCP, GBR 12935, and dopamine to the dopamine transporter. Eur. J. Pharmacol. 1992;227:417–425. doi: 10.1016/0922-4106(92)90160-w. [DOI] [PubMed] [Google Scholar]
- Reith ME, Selmeci G. Cocaine binding sites in mouse striatum, dopamine autoreceptors, and cocaine-induced locomotion. Pharmacol. Biochem. Behav. 1992;41:227–230. doi: 10.1016/0091-3057(92)90087-v. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. Dopamine spillover after quantal release: rethinking dopamine transmission in the nigrostriatal pathway. Brain Res. Rev. 2008;58:303–313. doi: 10.1016/j.brainresrev.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol. Cell. 2008;30:667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- Sorkina T, Miranda M, Dionne KR, Hoover BR, Zahniser NR, Sorkin A. RNA interference screen reveals an essential role of Nedd4-2 in dopamine transporter ubiquitination and endocytosis. J. Neurosci. 2006;26:8195–8205. doi: 10.1523/JNEUROSCI.1301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc. Natl. Acad. Sci. U. S. A. 2001;98:6384–6389. doi: 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminath G, Deupi X, Lee TW, Zhu W, Thian FS, Kobilka TS, Kobilka B. Probing the beta2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J. Biol. Chem. 2005;280:22165–22171. doi: 10.1074/jbc.M502352200. [DOI] [PubMed] [Google Scholar]
- Tidjane CA, Do-Rego JC, Costentin J, Bonnet JJ. Differential sensitivity to NaCl for inhibitors and substrates that recognize mutually exclusive binding sites on the neuronal transporter of dopamine in rat striatal membranes. Neurosci. Res. 2001;39:319–325. doi: 10.1016/s0168-0102(00)00230-3. [DOI] [PubMed] [Google Scholar]
- Wang W, Sonders MS, Ukairo OT, Scott H, Kloetzel MK, Surratt CK. Dissociation of high-affinity cocaine analog binding and dopamine uptake inhibition at the dopamine transporter. Mol. Pharmacol. 2003;64:430–439. doi: 10.1124/mol.64.2.430. [DOI] [PubMed] [Google Scholar]
- Wilson SP, Kirshner N. Calcium-evoked secretion from digitonin-permeabilized adrenal medullary chromaffin cells. J. Biol. Chem. 1983;258:4994–5000. [PubMed] [Google Scholar]
- Wu Q, Coffey LL, Reith ME. Cations affect [3H]mazindol and [3H]WIN 35,428 binding to the human dopamine transporter in a similar fashion. J. Neurochem. 1997;69:1106–1118. doi: 10.1046/j.1471-4159.1997.69031106.x. [DOI] [PubMed] [Google Scholar]
- Xhaard H, Backstrom V, Denessiouk K, Johnson MS. Coordination of Na(+) by monoamine ligands in dopamine, norepinephrine, and serotonin transporters. J. Chem. Inf. Model. 2008;48:1423–1437. doi: 10.1021/ci700255d. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Zapata A, Kivell B, Han Y, Javitch JA, Bolan EA, Kuraguntla D, Jaligam V, Oz M, Jayanthi LD, Samuvel DJ, Ramamoorthy S, Shippenberg TS. Regulation of dopamine transporter function and cell surface expression by D3 dopamine receptors. J. Biol. Chem. 2007;282:35842–35854. doi: 10.1074/jbc.M611758200. [DOI] [PubMed] [Google Scholar]
- Zhen J, Chen N, Reith ME. Differences in interactions with the dopamine transporter as revealed by diminishment of Na(+) gradient and membrane potential: dopamine versus other substrates. Neuropharmacology. 2005;49:769–779. doi: 10.1016/j.neuropharm.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317:1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimanyi I, Lajtha A, Reith ME. Comparison of characteristics of dopamine uptake and mazindol binding in mouse striatum. Naunyn Schmiedebergs Arch. Pharmacol. 1989;340:626–632. doi: 10.1007/BF00717737. [DOI] [PubMed] [Google Scholar]