

Abstract
Previous studies have shown that neurokinin 1 receptor (NK1R) occurs naturally in human glioblastomas and its stimulation causes cell proliferation. In the present study we show that stimulation of NK1R in human U373 glioblastoma cells by substance P (SP) increases Akt phosphorylation by 2.5-fold, with an EC50 of 57 nM. Blockade of NK1R lowers basal phosphorylation of Akt, indicating the presence of a constitutively active form of NK1R; similar results are seen in U251 MG and DBTRG-05 glioblastoma cells. Linkage of NK1R to Akt implicates NK1R in apoptosis of glioblastoma cells. Indeed, treatment of serum-starved U373 cells with SP reduces apoptosis by 53 ± 1% (P < 0.05), and treatment with NK1R antagonist L-733,060 increases apoptosis by 64 ± 16 % (P < 0.01). Further, the blockade of NK1R in human glioblastoma cells with L-733,060 causes cleavage of Caspase-3 and proteolysis of poly (ADP-ribose) polymerase (PARP). Experiments designed to elucidate the mechanism of NK1R-mediated Akt phosphorylation revealed total involvement of non-receptor tyrosine kinase Src and PI-3-kinase, a partial involvement of epidermal growth factor receptor (EGFR), and no involvement of MEK. Taken together, the results of the present study indicate a key role for NK1R in glioblastoma apoptosis.
Keywords: NK1 receptor, Akt, apoptosis, glioblastoma
Introduction
Glioblastomas, classified as grade IV astrocytomas, are among the most aggressive and frequent primary brain tumors in adults; patients with glioblastomas have an extremely poor prognosis, with a 5-year survival rate of 1% (Davis et al. 1998; Giles and Gonzales 2001; Salcman 2001). None of the current treatments for glioblastomas (including surgery, radiation therapy, and chemotherapy) have yielded satisfactory results, and researchers are seeking to understand glioblastoma biology more fully in order to identify novel molecular targets for blocking tumor growth. One potential target is epidermal growth factor receptor (EGFR), whose gene is amplified and rearranged in a large number of glioblastomas (Shinojima et al. 2003; Karamouzis et al. 2007). Several agents (antibodies as well as small molecules) targeting EGFR in glioblastomas are currently in clinical trials (Mendelsohn and Baselga 2000; Raymond et al. 2000). Another possible target is the neurokinin 1 receptor (NK1R; previously known as substance P receptor) (Palma et al. 2000; Palma and Maggi 2000). Several lines of evidence implicate a role for NK1R in glioblastoma biology. First, in one study, NK1R was expressed in 9 out of 12 astrocytomas and 10 out of 10 glioblastomas (Hennig et al. 1995). In these tumors, the expression of NK1R correlated with the degree of malignancy, with glioblastomas expressing more receptors than astrocytomas (Hennig et al. 1995). Second, at the molecular level, NK1R stimulation in U373 MG human glioblastoma cells increases mitogenesis, cell proliferation (Luo et al. 1996), and release of interleukin-6 (IL-6) (Palma et al. 1994). Of note, IL-6 has been implicated in the progression of gliomas (Rolhion et al. 2001; Chang et al. 2005).
In the present study, we report that stimulation of NK1R in human glioblastoma cells increases the phosphorylation and activity of Akt, or Protein Kinase B (EC 2.7.11.1), a serine-threonine protein kinase which becomes activated via phosphatidyl-3-kinase (PI3K) and suppresses apoptosis (Cheng et al. 1997; Dudek et al. 1997). We also report that blockade of NK1R reduces basal activity of Akt and leads to apoptosis. This is an important finding because the presence of activated Akt in glioblastomas is linked to a poor prognosis (Choe et al. 2003; Chakravarti et al. 2004; Haas-Kogan et al. 2005). Thus, by our finding that blockade of NK1R lowers Akt phoshprylation and activity, NK1R is implicated as a source of elevated Akt phosphorylation levels seen in many glioblastoma patients (Haas-Kogan et al. 2005).
Materials and methods
Materials
Substance P (SP), human epidermal growth factor (EGF), L-733,060, L-732,138, dimethyl sulfoxide (DMSO), LY294002, wortmannin, p-coumaric acid, 5-amino-2,3-dihydro-1,4-phthalazinedione (luminol), and IGEPAL CA-630 were purchased from Sigma (St. Louis, MO). Polyvinylidene fluoride transfer membrane was obtained from Millipore Corporation (Bedford, MA). PD98059 was obtained from New England Bio labs (Beverly, MA). AG1478 and 4-Amino-5-(4-chlorphenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) were from Calbiochem (San Diego, CA). Cell Signaling Technology (Beverly, MA) supplied purified GSK-3 fusion protein as well as the following antibodies: Akt, phospho-Akt (Ser473), phospho-(Ser/Thr) Akt substrate antibody, phospho-GSK-3α/β (Ser21/9), and agarose hydrazide-conjugated Akt antibody, Caspase-3 (8G10), and poly(ADP-ribose) polymerase (PARP). Monoclonal anti-phosphotyrosine IgG (PY20) was purchased from Transduction Laboratories (Lexington, KY). Horseradish peroxidase conjugates of goat anti-rabbit and goat anti-mouse IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Tween-20 was obtained from Pierce (Rockford, IL). Cell Death Detection ELISA PLUS was obtained from Roche Diagnostics (Mannheim, Germany). All other reagents were of the highest commercial grade.
Drug solutions
Stock solutions of AG1478 (20mM), PP2 (20 mM), LY294002 (100 mM), wortmannin (10 mM), PD98059 (20 mM), L-733,060 (20 mM), and L-732,138 (20 mM) were made in DMSO. The control treatment (i.e., vehicle) for these drugs was 0.1 % DMSO. Stocks of SP (1mM) and EGF (0.1 mg/ml) were dissolved in 20 mM HEPES, pH 8.0 and dilutions were made in serum-free minimal essential medium (MEM).
Cell culture
U373 MG glioblastoma cells were obtained from ATCC (Rockville, MD). U251 MG and DBTRG-05 MG cells were kindly provided by Dr. Darell Bigner (Duke University Medical Center. Durham, NC). Minimal essential medium (MEM), Improved MEM Zinc Option (1X), non-essential amino acids, sodium pyruvate, fetal bovine serum (FBS) were obtained from GibcoBRL® Life Technologies (Grand Island, NY). U373 MG cells were cultured as previously described (Yamaguchi et al. 2005). U251 MG and DBTRG-05 MG cells were cultured in Improved MEM Zinc-option (1X) medium with 10 % FBS, and antibiotics at 37°C in an atmosphere of 5 % CO2.
Immunoblotting
Experimental treatments of U373 MG cells and immunoblotting of cell lysates were conducted as previously described (Yamaguchi et al. 2005). Antibodies against Akt, phospho-Akt (Ser473), and phospho-GSK-3α/β (ser21/9), as well as horseradish peroxidase-conjugated goat anti-rabbit IgG, were used at a 1:3000 dilution for 1 h at room temperature. Protein-bound antibodies were detected by chemiluminescence following a 2-min incubation with 100 mM Tris-HCl (pH 8.5), 0.95 % H2O2, 198 μM p-coumaric acid and 1.24 mM luminol. The results were quantified by densitometry by using a GS-800 Calibrated Densitometer with Quantity One version 4.1.1 software (BioRad Laboratories. Hercules, CA).
Akt kinase activity assay
An Akt Kinase Assay Kit (Cell Signaling Technology. Beverly, MA) was used as directed to assess Akt kinase activity in glioblastoma cells. U373 MG cells were treated as indicated in the figure legends, washed with PBS, and lysed in 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.5% IGEPAL, 2.5 mM Na4P2O7, 1 mM β-glycerophosphate, 1 mM Na3VO4, 10 μg/ml leupeptin, and 1 mM phenylmethanesulphonylfluoride (PMSF). After centrifugation of cell lysates (10,000 rpm for 10 min at 4°C), supernatants were incubated with agarose hydrazide-conjugated Akt antibody for 3 h at 4°C. Akt-agarose complexes were washed twice with lysis buffer, then twice with kinase reaction buffer (25 mM Tris, pH 7.5, 5 mM β-glycerophosphate, 2 mM dithiothreitol, 0.1 mM, Na3VO4, 10 mM MgCl2). Washed precipitates were then incubated with 200 μM ATP and 1mg of the Akt substrate, GSK-3α/β, for 30 min at 30°C. The reaction was terminated with 20 μl of 3X SDS sample buffer (187.5 mM Tris-HCl pH 6.8, 6% w/v SDS, 30% glycerol, 150 mM dithiothreitol, 0.03 % (w/v) bromphenol blue). Samples were centrifuged at 16,000 × g for 2 min and boiled for 5 min prior to 12 % SDS-PAGE. Phosphorylation of GSK-3α/β was determined by immunoblotting, as described above, using phospho-GSK-3α/β (ser21/9) antibody.
Cell death assay
Apoptosis was measured in glioblastoma cells using the Cell Death Detection ELISA PLUS kit (Roche Diagnostics. Mannheim, Germany), which detects histone-complexed DNA fragments in cell cytoplasm. U373 MG cells were seeded into a 96-well dish at a density of 1 × 104 cells/well in growth medium (i.e., containing 10% FBS), then switched to serum-free medium 18 hr later; control cells remained in 10% FBS. Drugs indicated in the figure legend were added every 12 hours over a 72 hr period. Cell lysates were prepared following the manufacturer's procedure, and incubated with an anti-histone, biotin-conjugated antibody in a microplate with streptavidin-coated walls. After washing and incubation with peroxidase substrate, histone-complexed DNA fragments in the samples were quantified by reading the plate at 405 nm on a BenchMark™Plus microplate spectrophotometer using Microplate Manager 5.2.1 software (BioRad. Hercules, CA).
Apoptosis was also assessed by immunoblotting to show cleavage of cellular Caspase-3 and PARP (Cell Signaling Technology. Beverly, MA). After serum-starving and apoptosis-inducing drug treatments described in the figure legend, growth medium and floating cells were transferred from each well into a microfuge tube, the cells were pelleted by a 5 min spin at 13,000 rpm, and the supernatant was aspirated from each tube. Lysis buffer, described above, was added to each empty well and dishes were incubated for 15 min at 4°C with shaking. The resulting lysates were transferred from the dish to their corresponding microfuge tubes, and the cell pellets were solubilized by 10-sec of sonication. After a 5 min incubation at 90°C in SDS sample buffer, the samples were subjected to 12 % SDS-PAGE.
Measurement of Substance P (SP) in Conditioned Media
The amount of SP in conditioned media from unstimulated U373 MG cells was measured using the Parameter™ Substance P Immunoassay kit (R&D Systems, Inc. Minneapolis, MN) according to the manufacturer's protocol. This assay is suitable for measuring SP in cell culture supernatant, plasma, saliva, and urine. The mean CV% are 6.2 (for intra-assay precision) 11.9 (for inter-assay precision). The minimum detectable concentration of SP by this method ranges from 16.8 − 43.8 pg/mL, with a mean of 31.5 pg/mL (∼20 pM). This level of sensitivity is more than sufficient to detect whether secreted SP is the cause of Akt phosphorylation because our data indicate that SP-stimulated Akt phosphorylation occurs at a much higher concentration (EC50 = 57 nM; Figure 1). Briefly, U373 MG cells, cultured in a 6-well dish at a density of 8 × 105 cells/well in growth medium, were incubated in 2.5 mL serum-free media per well. For assaying SP, 200 μL aliquots of the media were collected at 0, 24, and 48 hours. To ensure that the conditioned media contained no substances that might interfere with the assay, we spiked the media with known concentrations of SP. These experiments revealed that there are no interfering substances in the media.
Fig.1. Phosphorylation and activation of Akt are stimulated by SP and EGF in glioblastoma cells.
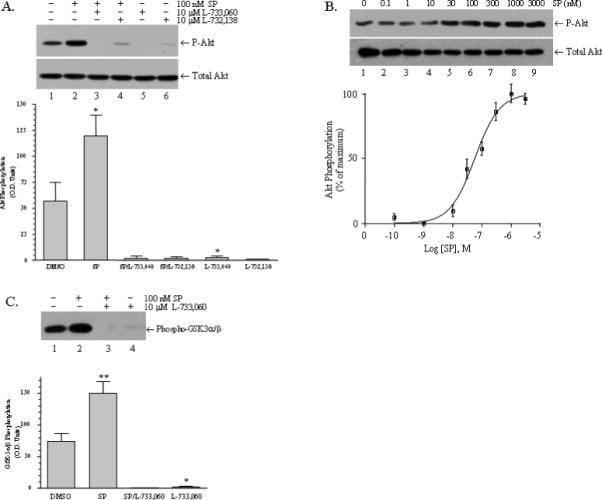
Serum-starved U373 MG cells were treated with the indicated drugs, and then phosphor-Akt (upper panel) was visualized by immunoblotting as described in “Materials and Methods.” A) Lane 1: vehicle (0.1 % DMSO); lane 2: 100 nM SP for 10 min; lane 3: 10 μM L733,060 for 30 min followed by 100 nM SP for 10 min; lane 4: 10 μM L-732,138 for 30 min followed by 100 nM SP for 10 min; lane 5: 10 μM L733,060 for 30 min; lane 6: 10 μM L-732,138 for 30 min. The lower panel shows total cellular Akt. Bar Chart: The Akt phosphorylation observed in four similar experiments was quantitated by scanning densitometry as described in “Materials and Methods.” Statistical significance was determined by paired, one-tailed t-tests; (*) represents P< 0.05. Akt phosphorylation was increased by 100 nM SP (P = 0.0448; n = 4) and the increase was blocked by L-733,060 and L732,138 (P = 0.0051, and P = 0.0043, respectively; n = 4). Basal levels of Akt phosphorylation were also reduced by both by L-733,060 (P = 0.0479) and L732,138 (P = 0.0518). B) Serum-starved U373 MG cells were stimulated for 10 min at 37°C with SP, ranging in concentration from 10−10 M to 3.0 × 10−6 M. The lower panel shows total Akt and the upper panel shows phosphor-Akt. Akt phosphorylation was quantitated by scanning densitometry and expressed as a percentage of the maximal response occurring at 3.0 × 10−6 M (n = 3). Nonlinear regression analysis was performed, fitting the data to a sigmoidal dose-response curve using GraphPad Prism version 4.0 (GraphPad Software. San Diego, CA). This analysis yielded an EC50 5.7 × 10−8 M. C) Serum-starved U373 MG cells were treated with the indicated drugs. Activated cellular Akt was immunoprecipitated, incubated with GSK-3α/β, and the resulting phosphorylation of GSK-3α/β was visualized by immunoblotting as described in “Materials and Methods.” Lane 1: vehicle (0.1 % DMSO); lane 2: 100 nM SP; lane 3: 10 μM L733,060 plus 100 nM SP; lane 4: 10 μM L733,060. Bar Chart: Phospho-GSK-3α/β from three similar experiments was quantitated by scanning densitometry as described in “Materials and Methods.” Statistical significance was determined by paired, one-tailed t-tests; (*) represent P< 0.05 and (**) represents P< 0.01. Phospho-GSK-3α/β was increased by 100 nM SP (P = 0.0087; n = 3) and the increase was blocked by L-733,060 (P = 0.0079; n = 3). Basal Akt activity was also reduced by L-733,060 (P = 0.0479; n = 3).
Statistical analysis
Results were shown by mean ± S.E.M. Statistical analysis was performed by Student's t-test.
Results
Stimulation of NK1R increases the phosphorylation of Akt
Figure 1A shows that exposure of U373 MG glioblastoma cells to SP increases the phosphorylation of Akt (lane 2), and this increased phosphorylation is blocked by NK1R antagonist, L-733,060 (lane 3). Interestingly, L-733,060 also blocks basal Akt phosphorylation (Figure 1A, lanes 1 and 3), suggesting that a fraction of NK1R is constitutively active. The lowering of basal phosphorylation of Akt is not unique to L-733,060 because it is also lowered by L-732,138 (Figure 1A, lanes 1 and 4), a chemically different NK1R antagonist. The effectiveness of both L-733,060 and L-732,138 in lowering basal phosphorylation of Akt was confirmed by using these agents without SP (Figure 1A, lanes 5 and 6, respectively). Inhibition of basal phosphorylation of Akt by NK1R antagonist L-733,060 was also seen in other glioblastoma cell lines including U251 MG and DBTRG-05 MG (data not shown).
Substance P stimulates Akt phosphorylation in a dose dependent manner in U373 MG cells
U373 MG cells were stimulated with different concentrations of substance P (SP), ranging from 0.1 nM to 3 μM and phosphorylation of Akt was examined by immunoblotting (Figure 1B). Quantitative analysis of these data indicates that SP increases the phosphorylation of Akt with an EC50 of 57 nM.
Stimulation of NK1R increases the kinase activity of Akt
We then determined whether an NK1R-mediated increase in Akt phosphorylation would also result in increased Akt kinase activity. To this end, we immunoprecipitated Akt from control and NK1R stimulated cells and measured its kinase activity against the Akt substrate, GSK3α/β. As shown in Figure 1C, Akt activity increases when cells are stimulated by SP (Figure 1C, lanes 1 and 2), and decreases below basal levels in the presence of NK1R antagonist L-733,060 (Figure 1C, lanes 1 and 3). As seen previously for the basal phosphorylation of Akt, blockade of NK1R with L-733,060 inhibits the basal kinase activity of Akt (Figure 1C, lanes 1 and 4), indicating that a fraction of NK1R is constitutively active.
Source of NK1R's constitutive activity in glioblastoma cells
NK1R's constitutive activity could arise from two potential mechanisms. First, glioblastoma cells could secrete SP or HK-1 (hemokinin-1), a recently discovered peptide agonist of NK1R (Zhang et al. 2000), which then activates NK1R in an autocrine mechanism. Second, glioblastoma cells could express a constitutively active form of NK1R. The notion that glioblastoma cells could release SP or HK-1 is supported by the finding that these cells express both the Tac1 gene, which encodes for SP, and the Tac4 gene, which encodes for HK-1 (Berger and Paige 2005). Since an assay to measure HK-1 is not available, we examined the release of SP. Using the highly sensitive radioimmunoassay for detecting SP, we did not find any SP in the conditioned media from U373 MG cells up to 48 h after placing the cells in serum-free media. As mentioned earlier, the lower limit of detecting SP by our assay was 20 pM, a concentration much lower than the EC50 of 57 nM for SP-stimulated Akt phosphorylation.
Blockade of NK1R enhances apoptosis
Since the phosphorylation state of Akt is linked to apoptosis, we examined the effect of blockade or stimulation of NK1R on apoptosis. These effects were assessed using two methods: 1) nucleosome formation, and 2) activation of Caspase-3 and proteolysis of PARP determined by western blotting. Figure 2A shows the results of an ELISA assay for nucleosome detection. Figure 2A shows that compared to untreated, serum-starved cells (control), treatment with SP reverses apoptosis by 53 ± 1% (P < 0.05), and treatment with NK1R antagonist L-733,060 increases apoptosis by 64 ± 16% (P < 0.01).
Fig.2. A. Apoptosis of serum-starved U373 MG cells is inhibited by SP and enhanced by the SPR antagonist, L-733,060.
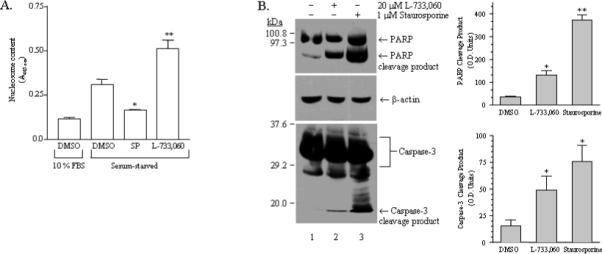
A) Subconfluent, log-phase U373 MG cells were incubated in serum-free medium for 12−15 hr then exposed to vehicle (0.1 % DMSO), 1 μM SP, or 10 μM L-733,060 for an additional 72-hr, with drugs being replaced every 12-hr. Apoptosis under these conditions was assessed by ELISA, as described in “Materials and Methods.” Cell death, indicated by nucleosome formation following drug treatments, is represented in the bar chart (n = 3). Apoptosis in U373 MG cells growing in 10% FBS (leftmost bar) was also examined for comparison with apoptosis in serum-starved cells. Statistical significance was determined using paired, one-tailed t-tests; (*) represents P< 0.05 and (**) represents P< 0.01. SP decreased apoptosis (52.6 ± 1.4 % of untreated), whereas SPR antagonist L-733,060 increases apoptosis (164.1 ± 15.5 % of untreated). B) Subconfluent, log-phase U373 MG cells were incubated for 12−15 hr in serum-free medium, then exposed to vehicle (0.1 % DMSO, lane 1), 20 μM L-733,060 for 16 hr, (lane 2), or 1 μM staurosporine for 4 hr, as a positive control (lane 3). Immunoblotting was performed as described in “Materials and Methods”; PARP (upper panel) and Caspase-3 (lower panel) antibodies were both used at a 1:1000 dilution, and incubated overnight at 4°C. Arrows indicate proteolytic cleavage fragments of PARP and Caspase-3. The transfer membranes were also probed for β-actin (middle panel), as a loading control. Bar Chart: Proteolytic cleavage of PARP or Caspase-3 was quantitated by scanning densitometry as described in “Materials and Methods.” Statistical significance was determined using paired, one-tailed t-tests; (*) represents P< 0.05 and (**) represents P< 0.01. Treatment with L-733,060 caused significant proteolytic cleavage of both PARP (P = 0.0256, n = 3) and caspase-3 (P = 0.038; n = 4).
Recent studies have demonstrated that Akt activation leads to an anti-apoptotic effect of SP in colonocytes (Koon et al. 2007) and cerebellar granule cells (Amadoro et al. 2007) but to our knowledge no study has demonstrated that blockade of NK1R increases apoptosis. Therefore, we further examined NK1R-blockade-mediated apoptosis of human glioblastoma cells by examining the activation of Caspase-3, a key executioner of apoptosis (Boatright and Salvesen 2003). Caspase-3 exists as an inactive zymogen and its activation requires proteolysis into 17 kd and 12 kd fragments which dimerize to form an active enzyme (Nicholson et al. 1995). Both the inactive zymogen and the 17 kd cleaved fragments can be detected by immunoblotting with the same antibody. The activated Caspase-3 cleaves several proteins including poly(ADP-ribose) polymerase (PARP) (Nicholson et al. 1995; Tewari et al. 1995), producing fragments that can also be detected through immunoblotting. Figure 2B shows immunoblots of control and NK1R antagonist L-733,060-treated cells with antibodies specific for Caspase-3 and PARP. As seen in this figure, blockade of NK1R leads to the formation of cleaved Caspase-3 and the proteolysis of PARP, confirming apoptosis. Figure 2B also shows Caspase-3 activation and PARP proteolysis when cells are exposed to staurosporine, a well known inducer of apoptosis (Belmokhtar et al. 2001).
Mechanism of NK1R-mediated activation of Akt
Figure 3A shows that NK1R-mediated Akt phosphorylation is almost completely blocked by the Src inhibitor PP2 (91.5% inhibition, P<0.01) and by the PI3K inhibitors wortmannin (98%, P<0.01) and LY294002 (99%, P<0.01). In contrast, NK1R-mediated Akt phosphorylation is not inhibited by the mitogen-activated protein/extracellular signal-related kinase (MEK) inhibitor, PD98059, and is only partially inhibited by the EGFR kinase inhibitor, AG1478 (40% inhibition, P< 0.01). Since stimulation of NK1R has been shown to transactivate EGFR (Castagliuolo et al. 2000), we wanted to determine whether a greater inhibition of NK1R-mediated Akt phosphorylation would occur at higher concentrations of AG1478. Figure 3B shows that while EGFR stimulated phosphorylation of Akt is almost totally inhibited by 1 μM AG1478, NK1R-mediated Akt phosphorylation is only partially inhibited even at 100 μM AG1478. These results indicate that NK1R activates Akt through both EGFR-dependent and EGFR-independent pathways.
Fig. 3. Mechanism of Akt activation by SP.
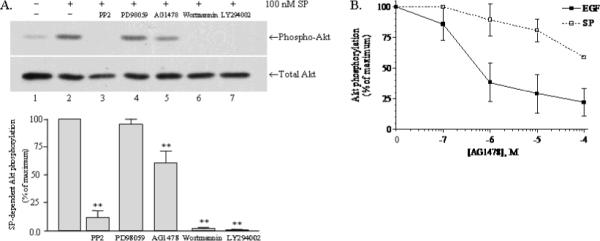
A) Stimulation of Akt phosphorylation by 100 nM SP (10 min) following a 30 min pretreatment with various drugs: lane 1, nothing; lane 2, vehicle (0.1 % DMSO); lane 3, 20 μM PP2; lane 4, 10 μM PD98059; lane 5, 10 μM AG1478; lane 6, 1 μM wortmannin; and lane 7, 50 μM LY294002. Phospho-Akt and total Akt were visualized by immunoblotting, as described in “Materials and Methods”. Bar Chart: Scanning densitometry was used to quantitate Akt phosphorylation in the experiments represented above (n = 3), and the resulting mean values are shown. Statistically significance of inhibition was determined using paired, one-tailed t-tests; (**) represents P< 0.01. B) Effect of increasing concentrations of AG1478 on SP and EGF-stimulated phosphorylation of Akt. Serum-starved U373 MG cells were stimulated for 10 min at 37°C with SP or EGF following a 1 h pretreatment with AG1478 ranging in concentration from 10−7M to 10−4 M. The resulting Akt phosphorylation was detected by immunoblotting and quantitated by scanning densitometry, then expressed as a percentage of the response obtained with SP or EGF in the absence of AG1478.
Discussion
This study shows that stimulation of NK1R with SP in human glioblastoma cells leads to the activation of Akt, resulting in the suppression of apoptosis. The antiapoptotic effects of SP through Akt activation have recently been reported in colonocytes (Koon et al. 2007) and cerebellar granule cells (Amadoro et al. 2007). Our study extends such observations of SP's antiapoptotic effects to human glioblastoma cells. Another finding of the present study is that NK1R blockade inhibits basal phosphorylation and activity of Akt. This finding indicates that the elevated phosphorylation of Akt seen in many glioblastoma patients may have its source in the constitutive activity of NK1R. Finally, we show that NK1R-mediated Akt activation occurs through Src and PI-3-kinase but only partially through EGFR kinase, and that it does not involve MAP kinase. These findings enhance our understanding of NK1R biology in human glioblastoma cells and suggest a role for NK1R antagonists in the management of glioblastomas.
Current therapies to treat glioblastomas are directed toward blocking EGFR activity (Waksal 1999; Mendelsohn and Baselga 2000; Raymond et al. 2000). However, a recent clinical trial with the EGFR kinase inhibitor erlotinib (Tarceva, Genetech, San Francisco, CA) reported no response in glioblastoma patients with tumors expressing high levels of phosphorylated Akt (Haas-Kogan et al. 2005). The finding that GBM patients with elevated levels of phosphorylated Akt did not respond to Erlotinib underscores the importance of discovering the origin of phosphorylated Akt in glioblastomas. While Hass-Kogan et al. (Haas-Kogan et al. 2005) did not speculate on the origin of phosphorylated Akt in glioblastomas, an accompanying editorial speculated that elevated levels of phosphorylated Akt may come from a lack of PTEN or from insulin-like growth factor 1 receptor (Cappuzzo 2005). We propose that elevated levels of phosphorylated Akt in glioblastomas comes from a constitutively active form of NK1R. Thus, our results suggest that a better response could be obtained by a simultaneous blockade of EGFR and NK1R.
The molecular basis of NK1R's constitutive activity is not clear. We have shown that NK1R's constitutive activity is not arising via an autocrine release of SP. The other possibility is that glioblastoma cells may express two forms of NK1R; a full-length form responsive to SP and a truncated form (NK1RΔ311) which is constitutively active. Truncated NK1RΔ311, which ends at amino acid 311 (whereas the full-length SPR has 407 amino acids), was cloned from a glioblastoma cell line (Fong et al. 1992), but is also found in normal human brain cells and peripheral tissues (Caberlotto et al. 2003). The notion that NK1RΔ311 is constitutively active is supported by a recent study which showed that NK1RΔ311 expression in non-tumorigenic breast cells leads to a transformed phenotype (Patel et al. 2005). That deletion of NK1R's carboxyl tail leads to constitutive activity is also supported by studies on truncated recombinant NK1Rs (Li et al. 1997; Richardson et al. 2003).
If the constitutive activity of NK1R detected in this study arises from truncated NK1RΔ311, the biology of this form of NK1R in glioblastomas warrants further investigation. Truncated NK1RΔ311 was first cloned from glioblastoma cells in 1992 (Fong et al. 1992). Since then, the truncated NK1RΔ311, along with the full-length NK1R, has been found in healthy CNS and peripheral tissues (Caberlotto et al. 2003), although the function of NK1RΔ311 and the factors that controlled its expression were not known. Our understanding of the biology of truncated NK1RΔ311 increased when its expression in nontumorigenic breast cells resulted in a transformed phenotype (Patel et al. 2005). More recently, the control of NK1RΔ311 expression by activation of nuclear factor kappaB (NF-κB) was described (Ramkissoon et al. 2007). This finding is particularly relevant to glioblastomas because these tumors have been known to have activated NF-κB (Nagai et al. 2002; Raychaudhuri et al. 2007). Thus, it is conceivable that when glioblastomas possess activated NF-κB, they express more NK1RΔ311, resulting in increased Akt activation. This event, in turn, could further activate NF-κB since activation of NF-κB can be triggered by Akt activation (Ozes et al. 1999; Julien et al. 2007). Whether activation of Akt activates NF-kB in glioblastomas is not known, but NK1R stimulation in glioblastomas does cause the activation of NF-κB (Lieb et al. 1997). Thus, formation of NK1RΔ311, which is constitutively active, would contribute to the formation of activated NF-κB, which would in turn increase the formation of NK1RΔ311. This positive feedback loop could be disrupted by NK1R blockade. Consequently, blocking NK1R in glioblastoma patients, either alone or in combination with an EGFR kinase inhibitor, might be an attractive strategy. The recent approval of the NK1R antagonist aprepitant makes this approach particularly promising. Aprepitant has been approved for the treatment of chemotherapy-induced emesis, but other uses, including treatment for depression, have been proposed (Kramer et al. 2004; Green et al. 2006; Keller et al. 2006). While we did not examine the ability of aprepitant to cause apotosis in glioblastoma cells, a recent study reports that aprepitant causes apoptosis of several cancer cell lines including glioblatomas (Munoz and Rosso 2009).
In summary, the present study has identified a key role for NK1R in glioblastoma apoptosis. Future studies should extend these findings to in vivo environments using both pharmacological and genetic approaches.
Acknowledgments
This study was supported by a grant from the National Institute of Health (NS 33405) and funds from the Duke University Department of Anesthesiology.
Abbreviations used
- NK1R
neurokinin 1 receptor
- PARP
poly (ADP-ribose) polymerase
- EGF
epidermal growth factor
- EGFR
EGF receptor
- SP
substance P
- PI3K
phosphatidyl-3-kinase
- PP2
4-Amino-5-(4-chlorphenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- MEM
minimal essential medium
- PMSF
phenylmethanesulphonylfluoride
- MEK
mitogen-activated protein kinase kinase
References
- Amadoro G, Pieri M, Ciotti MT, Carunchio I, Canu N, Calissano P, Zona C, Severini C. Substance P provides neuroprotection in cerebellar granule cells through Akt and MAPK/Erk activation: evidence for the involvement of the delayed rectifier potassium current. Neuropharmacology. 2007;52:1366–1377. doi: 10.1016/j.neuropharm.2007.01.020. [DOI] [PubMed] [Google Scholar]
- Belmokhtar CA, Hillion J, Segal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene. 2001;20:3354–3362. doi: 10.1038/sj.onc.1204436. [DOI] [PubMed] [Google Scholar]
- Berger A, Paige CJ. Hemokinin-1 has Substance P-like function in U-251 MG astrocytoma cells: a pharmacological and functional study. J Neuroimmunol. 2005;164:48–56. doi: 10.1016/j.jneuroim.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Caberlotto L, Hurd YL, Murdock P, Wahlin JP, Melotto S, Corsi M, Carletti R. Neurokinin 1 receptor and relative abundance of the short and long isoforms in the human brain. Eur J Neurosci. 2003;17:1736–1746. doi: 10.1046/j.1460-9568.2003.02600.x. [DOI] [PubMed] [Google Scholar]
- Cappuzzo F. Erlotinib in gliomas: should selection be based on EGFR and Akt analyses? J Natl Cancer Inst. 2005;97:868–869. doi: 10.1093/jnci/dji169. [DOI] [PubMed] [Google Scholar]
- Castagliuolo I, Valenick L, Liu J, Pothoulakis C. Epidermal growth factor receptor transactivation mediates substance P-induced mitogenic responses in U-373 MG cells. J Biol Chem. 2000;275:26545–26550. doi: 10.1074/jbc.M003990200. [DOI] [PubMed] [Google Scholar]
- Chakravarti A, Zhai G, Suzuki Y, Sarkesh S, Black PM, Muzikansky A, Loeffler JS. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J Clin Oncol. 2004;22:1926–1933. doi: 10.1200/JCO.2004.07.193. [DOI] [PubMed] [Google Scholar]
- Chang CY, Li MC, Liao SL, Huang YL, Shen CC, Pan HC. Prognostic and clinical implication of IL-6 expression in glioblastoma multiforme. J Clin Neurosci. 2005;12:930–933. doi: 10.1016/j.jocn.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Cheng JQ, Altomare DA, Klein MA, Lee WC, Kruh GD, Lissy NA, Testa JR. Transforming activity and mitosis-related expression of the AKT2 oncogene: evidence suggesting a link between cell cycle regulation and oncogenesis. Oncogene. 1997;14:2793–2801. doi: 10.1038/sj.onc.1201121. [DOI] [PubMed] [Google Scholar]
- Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, Inge L, Smith BL, Sawyers CL, Mischel PS. Analysis of the phosphatidylinositol 3'-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003;63:2742–2746. [PubMed] [Google Scholar]
- Davis FG, Freels S, Grutsch J, Barlas S, Brem S. Survival rates in patients with primary malignant brain tumors stratified by patient age and tumor histological type: an analysis based on Surveillance, Epidemiology, and End Results (SEER) data, 1973−1991. J Neurosurg. 1998;88:1–10. doi: 10.3171/jns.1998.88.1.0001. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Fong TM, Anderson SA, Yu H, Huang RR, Strader CD. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Mol Pharmacol. 1992;41:24–30. [PubMed] [Google Scholar]
- Giles GG, Gonzales MF. Epidemiology of brain tumors and factors in prognosis. In: Kaye AH, Laws ER Jr., editors. Brain Tumors. 2nd Edition Churchill Livingstone; New York: 2001. pp. 51–70. [Google Scholar]
- Green SA, Alon A, Ianus J, McNaughton KS, Tozzi CA, Reiss TF. Efficacy and safety of a neurokinin-1 receptor antagonist in postmenopausal women with overactive bladder with urge urinary incontinence. J Urol. 2006;176:2535–2540. doi: 10.1016/j.juro.2006.08.018. discussion 2540. [DOI] [PubMed] [Google Scholar]
- Haas-Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND, Baumber R, Lamborn KR, Kapadia A, Malec M, Berger MS, Stokoe D. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880–887. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- Hennig IM, Laissue JA, Horisberger U, Reubi JC. Substance-P receptors in human primary neoplasms: tumoral and vascular localization. Int J Cancer. 1995;61:786–792. doi: 10.1002/ijc.2910610608. [DOI] [PubMed] [Google Scholar]
- Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A, Larue L. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007 doi: 10.1038/sj.onc.1210546. [DOI] [PubMed] [Google Scholar]
- Karamouzis MV, Grandis JR, Argiris A. Therapies directed against epidermal growth factor receptor in aerodigestive carcinomas. Jama. 2007;298:70–82. doi: 10.1001/jama.298.1.70. [DOI] [PubMed] [Google Scholar]
- Keller M, Montgomery S, Ball W, Morrison M, Snavely D, Liu G, Hargreaves R, Hietala J, Lines C, Beebe K, Reines S. Lack of efficacy of the substance p (neurokinin1 receptor) antagonist aprepitant in the treatment of major depressive disorder. Biol Psychiatry. 2006;59:216–223. doi: 10.1016/j.biopsych.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Koon HW, Zhao D, Zhan Y, Moyer MP, Pothoulakis C. Substance P mediates antiapoptotic responses in human colonocytes by Akt activation. Proc Natl Acad Sci U S A. 2007;104:2013–2018. doi: 10.1073/pnas.0610664104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MS, Winokur A, Kelsey J, Preskorn SH, Rothschild AJ, Snavely D, Ghosh K, Ball WA, Reines SA, Munjack D, Apter JT, Cunningham L, Kling M, Bari M, Getson A, Lee Y. Demonstration of the efficacy and safety of a novel substance P (NK1) receptor antagonist in major depression. Neuropsychopharmacology. 2004;29:385–392. doi: 10.1038/sj.npp.1300260. [DOI] [PubMed] [Google Scholar]
- Li H, Leeman SE, Slack BE, Hauser G, Saltsman WS, Krause JE, Blusztajn JK, Boyd ND. A substance P (neurokinin-1) receptor mutant carboxyl-terminally truncated to resemble a naturally occurring receptor isoform displays enhanced responsiveness and resistance to desensitization. Proc Natl Acad Sci U S A. 1997;94:9475–9480. doi: 10.1073/pnas.94.17.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb K, Fiebich BL, Berger M, Bauer J, Schulze-Osthoff K. The neuropeptide substance P activates transcription factor NF-kappa B and kappa B-dependent gene expression in human astrocytoma cells. J Immunol. 1997;159:4952–4958. [PubMed] [Google Scholar]
- Luo W, Sharif TR, Sharif M. Substance P-induced mitogenesis in human astrocytoma cells correlates with activation of the mitogen-activated protein kinase signaling pathway. Cancer Res. 1996;56:4983–4991. [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–6565. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- Munoz M, Rosso M. The NK-1 receptor antagonist aprepitant as a broad spectrum antitumor drug. Invest New Drugs. 2009 doi: 10.1007/s10637-009-9218-8. [DOI] [PubMed] [Google Scholar]
- Nagai S, Washiyama K, Kurimoto M, Takaku A, Endo S, Kumanishi T. Aberrant nuclear factor-kappaB activity and its participation in the growth of human malignant astrocytoma. J Neurosurg. 2002;96:909–917. doi: 10.3171/jns.2002.96.5.0909. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- Palma C, Maggi CA. The role of tachykinins via NK1 receptors in progression of human gliomas. Life Sci. 2000;67:985–1001. doi: 10.1016/s0024-3205(00)00692-5. [DOI] [PubMed] [Google Scholar]
- Palma C, Goso C, Manzini S. Different susceptibility to neurokinin 1 receptor antagonists of substance P and septide-induced interleukin-6 release from U373 MG human astrocytoma cell line. Neurosci Lett. 1994;171:221–224. doi: 10.1016/0304-3940(94)90644-0. [DOI] [PubMed] [Google Scholar]
- Palma C, Bigioni M, Irrissuto C, Nardelli F, Maggi CA, Manzini S. Anti-tumour activity of tachykinin NK1 receptor antagonists on human glioma U373 MG xenograft. Br J Cancer. 2000;82:480–487. doi: 10.1054/bjoc.1999.0946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel HJ, Ramkissoon SH, Patel PS, Rameshwar P. Transformation of breast cells by truncated neurokinin-1 receptor is secondary to activation by preprotachykinin-A peptides. Proc Natl Acad Sci U S A. 2005;102:17436–17441. doi: 10.1073/pnas.0506351102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkissoon SH, Patel PS, Taborga M, Rameshwar P. Nuclear factor-kappaB is central to the expression of truncated neurokinin-1 receptor in breast cancer: implication for breast cancer cell quiescence within bone marrow stroma. Cancer Res. 2007;67:1653–1659. doi: 10.1158/0008-5472.CAN-06-3813. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri B, Han Y, Lu T, Vogelbaum MA. Aberrant constitutive activation of nuclear factor kappaB in glioblastoma multiforme drives invasive phenotype. J Neurooncol. 2007 doi: 10.1007/s11060-007-9390-7. [DOI] [PubMed] [Google Scholar]
- Raymond E, Faivre S, Armand JP. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs. 2000;60(Suppl 1):15–23. doi: 10.2165/00003495-200060001-00002. discussion 41−12. [DOI] [PubMed] [Google Scholar]
- Richardson MD, Balius AM, Yamaguchi K, Freilich ER, Barak LS, Kwatra MM. Human substance P receptor lacking the C-terminal domain remains competent to desensitize and internalize. J Neurochem. 2003;84:854–863. doi: 10.1046/j.1471-4159.2003.01577.x. [DOI] [PubMed] [Google Scholar]
- Rolhion C, Penault-Llorca F, Kemeny JL, Lemaire JJ, Jullien C, Labit-Bouvier C, Finat-Duclos F, Verrelle P. Interleukin-6 overexpression as a marker of malignancy in human gliomas. J Neurosurg. 2001;94:97–101. doi: 10.3171/jns.2001.94.1.0097. [DOI] [PubMed] [Google Scholar]
- Salcman M. Glioblastoma multiforme and anaplastic astrocytoma. In: Kaye AH, Laws ER Jr., editors. Brain Tumors. 2nd Edition Harcourt Publishers Limited; New York: 2001. pp. 493–523. [Google Scholar]
- Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, Oka K, Ishimaru Y, Ushio Y. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63:6962–6970. [PubMed] [Google Scholar]
- Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Waksal HW. Role of an anti-epidermal growth factor receptor in treating cancer. Cancer Metastasis Rev. 1999;18:427–436. doi: 10.1023/a:1006302101468. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Richardson MD, Bigner DD, Kwatra MM. Signal transduction through substance P receptor in human glioblastoma cells: roles for Src and PKCdelta. Cancer Chemother Pharmacol. 2005;56:585–593. doi: 10.1007/s00280-005-1030-3. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lu L, Furlonger C, Wu GE, Paige CJ. Hemokinin is a hematopoietic-specific tachykinin that regulates B lymphopoiesis. Nature Immunology. 2000;1:392–397. doi: 10.1038/80826. [see comment]. [DOI] [PubMed] [Google Scholar]