

Abstract
Akt is a known client protein of heat shock protein-90 (HSP90). We have found that HSP90 is responsible for Akt accumulation in the mitochondria in unstimulated cells. Treatment of SH-SY5Y neuroblastoma cells and HEK293 cells with the HSP90 inhibitors novobiocin and geldanamycin caused substantial decreases in the level of Akt in the mitochondria without affecting the level of Akt in the cytosol. Moreover, intra-cerebroventricular injection of novobiocin into mice brains decreased Akt levels in cortical mitochondria. Knockdown of HSP90 expression with siRNA also caused a significant decrease in Akt levels in the mitochondria without affecting total Akt levels. Using a mitochondrial import assay it was found that Akt is transported into the mitochondria. Furthermore, it was found that the mitochondrial import of Akt was independent of Akt activation as both an unmodified Akt and a constitutively active mutant Akt both readily accumulated in the mitochondria in an HSP90-dependent manner. Interestingly, incubation of isolated mitochondria with constitutively active Akt caused visible alterations in mitochondrial morphology, including pronounced remodeling of the mitochondrial matrix. This effect was blocked when Akt was mostly excluded from the mitochondria with novobiocin treatment. These results indicate that the level of Akt in the mitochondria is dependent on HSP90 chaperoning activity and that Akt import can cause dynamic changes in mitochondrial configuration.
Keywords: Akt, heat shock protein-90, phosphatidylinositol 3-kinase, glycogen synthase kinase-3β, mitochondria, insulin-like growth factor-1
Introduction
The serine/threonine kinase Akt, also referred to as protein kinase B, is closely associated with growth factor signaling. Stimulation of cell surface receptors with insulin or growth factors, such as insulin-like growth factor-1 (IGF-1), results in the activation of phosphatidylinositol 3-kinase and phosphatidylinositol dependent kinases, the latter which directly phosphorylates Akt on its Thr308 site, and possibly on its Ser473 site (Alessi, 1996; Alessi, 1997; Fayard, 2005). Phosphorylation at these sites results in the full activation of Akt. Akt activation occurs at the cytoplasmic face of the plasma membrane, however subsequent to its activation Akt moves from the plasma membrane and translocates into different subcellular compartments including the cytosol, the nucleus (Andjelkovic, 1997), and mitochondria (Bijur, 2003), where it can phosphorylate compartment-specific substrates. The translocation of Akt into the mitochondria is rapid, occurring in quick succession to its activation. Although mitochondrial import of Akt is blocked by a dissipation of the mitochondrial transmembrane potential (Bijur, 2003), the molecular mechanism underlying Akt mitochondrial transport is unknown.
Akt activity is also affected by its binding to heat shock protein-90 (HSP90) (Sato, 2000). Akt is a well-known client protein of HSP90. Pharmacological inhibitors of HSP90 have provided much insight into how HSP90 functions. For example, the coumarin based compound novobiocin (NB) inhibits HSP90 by targeting the C-terminal region of HSP90 adjacent to its homodimerization domain (Marcu, 2000a). NB treatment has been associated with destabilization and proteolysis of HSP90 client proteins (Marcu 2000b; Yun, 2004). In addition, geldanamycin (GA) is a benzoquinoid ansamycin antibiotic which inhibits HSP90 by binding to its N-terminal ATPase domain (Whitesell, 1994; Panaretou, 1998). Treatment of cells with geldanamycin affects Akt by inducing its dephosphorylation (Fujita, 2002; Xu, 2003), and also induces the degradation of Akt (Kim, 2003), indicating that HSP90 is essential for stability and function of Akt. Thus, in addition to phosphorylation, Akt activity is dependent on its interaction with HSP90.
Recently it was reported that inhibition of HSP90 with NB and GA can block the import of proteins into mitochondria (Fan, 2006), indicating that HSP90 plays a major role in the translocation of proteins into the mitochondrion. The import of proteins into the mitochondria is a dynamic energy-dependent process relying on the concerted efforts of several translocase proteins resident within the mitochondria, a pore complex through which the proteins are inserted, and chaperone proteins which facilitate the import process (Pfanner, 1990; Komiya, 1997). Although many mechanistic aspects of mitochondrial import of proteins are unknown, some features have been elucidated. Two key chaperone proteins responsible for protein import into the mitochondria are heat shock proteins-90 and -70 (HSP90 and HSP70, respectively) (Young, 2003; Young, 2004). Before proteins are imported into mitochondria (preproteins) they are bound to chaperones in the cytoplasm, and notably, HSP90 was recently reported to play a major role in the mitochondrial protein import process (Fan, 2006). In the initial process of mitochondrial import, HSP90 (with its preprotein cargo) binds to one of the mitochondrial surface receptors on the translocase of the outer mitochondrial membrane (TOM) complex, the TOM70 receptor (Yano, 2004; Fan, 2006). Docking of the HSP90 to TOM70 occurs by virtue of a tetratricopeptide repeat domain on the receptor similar to the domains found on the co-chaperones of HSP90 (Young, 2003; Yano 2004). Due to the close juxtaposition of HSP90 to the TOM complex, the preprotein can make direct contact with the TOM70 receptor. Subsequently, the preprotein is imported through the outer mitochondrial membrane via the TOM40 import pore (Gabriel, 2003; Chacinska, 2004). Once a preprotein has entered the mitochondria it is further assimilated into specific mitochondrial subcompartments by numerous transport proteins, most notably the translocases of the inner membrane, commonly known as TIMs (Kerscher, 1997; Kutik, 2007). In addition, transport of proteins into the deeper compartments of the mitochondria, the inner membrane and the matrix, requires the electrochemical gradient across the inner membrane (Martin, 1991; Geissler, 2000). Thus, HSP90, in conjunction with the TOM and TIM proteins, function in the efficient translocation of proteins into the mitochondria and its subcompartments.
Given that Akt can translocate into the mitochondria, and that Akt is a well known client protein of HSP90, it was hypothesized that Akt levels in the mitochondria might be affected by HSP90 inhibitors. The present report describes how inhibition of HSP90 activity or decreased HSP90 expression causes marked decreases in the levels of Akt in the mitochondria and inhibits the translocation of Akt into the mitochondria. In addition, increased Akt signaling in the mitochondria is shown to cause remodeling of the mitochondrial cristae, indicating that Akt activity has profound affects on mitochondrial morphology.
Materials and methods
Cell Culture and Treatments
SH-SY5Y human neuroblastoma cells were grown in continuous culture RPMI media containing 10% horse serum, 5% Fetal Clone II (Hyclone, Logan, UT), 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. HEK293 human embryonic kidney cells were grown in F12/DMEM containing 10% fetal bovine serum (Invitrogen), 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. For serum withdrawal, adherent cells were rinsed twice with serum-free media supplemented with 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Prior to all treatments cultured cells were washed once with serum-free RPMI media (for SH-SY5Y cells) or F12/DMEM (for HEK293 cells) containing 2 mM L-glutamine, 100 units/mL penicillin, and 100 ug/mL streptomycin. Cells were maintained in serum-free media overnight prior to treatments with 625 μM novobiocin (NB) (Alexis Biochemicals), and 5 μM geldanamycin (GD) (AG Scientific). Cells were either subjected to a 60-minute time course with NB, or a 90 minute GA time course. Where indicated, cells were pretreated with NB or GA for 45 minutes prior to a 15 minute treatment of 50 ng/mL insulin-like growth factor-1 (IGF-1) (US Biological).
siRNA HSP90 Knockdown
Adherent HEK293 cells were transfected with 2 μg of plasmid from the pGB-HSP90 siRNA vector mix obtained from Biovision Incorporated (Mountainview, CA). Since this plasmid mix confers G418 resistance, stably transfected cells were grown in continuous culture media containing 225 μg/ml G418. Control cell lines were stably transfected with an empty pcDNA vector conferring G418 resistance. The cells were continuously cultured in serum-containing F12/DMEM media, described above.
Mitochondrial Purification
Mitochondria were isolated essentially as described in Bijur and Jope (2003) with modifications as follows. After the cells were harvested and plasma membranes disrupted by nitrogen cavitation, the cell homogenate was centrifuged once at 700 × g for 5 min to remove unbroken cells and nuclei. The crude mitochondria-containing supernatant was transferred to a new tube and centrifuged for 30 min at 17,500 × g to collect a mitochondrial pellet. The supernatant was saved as the cytosolic sample, and the pellet was washed twice with cavitation buffer followed by a 15 min centrifugation at 17,500 × g. Mitochondrial extracts were obtained by incubating mitochondria for 30 min at 4°C in lysis buffer (20 mM Tris, pH 7.5, 0.2% Nonidet P-40, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, and protease and phosphatase inhibitors). The mitochondrial extract was clarified by centrifugation at 20,800 × g for 10 min. Protein concentrations were determined by the bicinchoninic acid method (Pierce). To verify complete separation of the mitochondrial and cytosolic fraction, the two fractions were immunoblotted with β-tubulin, an abundant cytosolic protein, and pyruvate dehydrogenase, a mitochondrial protein.
Immunoblotting
Immunoblotting was performed as described previously (Bijur, 2003). Proteins separated by SDS-PAGE were transferred onto nitrocellulose membranes. The membranes were blotted with antibodies to Akt (Sigma, Cell Signaling), HSP90 (BD Transduction), HSP70, HSC70, and HSP40 (BDTransduction), phospho-Ser21/9-GSK3α/β, phospho-Ser473-Akt, phospho-Akt-substrates (Cell Signaling), pyruvate dehydrogenase, or β-tubulin (Sigma).
Mitochondrial Import Assay
Methods for the mitochondrial import assay were adapted from Fan, et al (2006) with minor modifications. Cell-free translation of wtAkt and DDAkt proteins were performed using the TNT-coupled transcription/translation rabbit reticulocyte lysate system (Promega), according to manufacturer’s directions. Briefly, T7 Polymerase (Promega) and 35S-Methionine (Amersham) were mixed with reticulocyte lysate, transfer RNAs, reaction buffer, RNAsin (Promega), and 1 μg of wtAkt or DDAkt plasmid containing the T7 promoter. The total reaction volume was adjusted to 50 μl with nuclease free water. Reactions were incubated for ninety minutes at 30°C then adjusted to 250 mM sucrose.
Mitochondria from HEK293 cells were isolated as described and aliquoted equally in import buffer (20 mM HEPES-KOH pH 7.5, 5 mM MgOAc2, 80 mM KOAc, and 250 mM Sucrose) containing 2 mM ATP, 0.4 mM ADP, 1 mM dithiothreitol, and 10 mM succinate. Mitochondria were then added to the translated proteins such that the volume of mitochondria made up 40% of the reaction volume and the exogenous protein constituted 35% of the total reaction volume. Novobiocin was added to the translation product for a final concentration of 625 μM before the mitochondria were added. The translocation reaction was then allowed to incubate for 45 min at 30°C with intermittent gentle mixing. Mitochondria were collected and half of each sample was digested for 10 minutes on ice with 250 ug/mL proteinase K to remove proteins on the outside of the mitochondria. The digested samples were then treated with 2 mM phenylmethylsulfonyl fluorine (PMSF) for 10 minutes on ice to terminate digestion. Half of each sample was left untreated. Mitochondrial lysates were obtained and mitochondrial proteins were separated by SDS-PAGE. The gels were dried and the radioactive Akt bands were visualized with a phosphorimager.
Intra-cerebroventricular (ICV) Injections
All procedures were in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines. 6–8 week old male C57BL6 mice were anesthetized using 100mg/kg ketamine and 10 mg/kg xylazine. Novobiocin (312.5 mM in 1 μL total volume) was bilaterally injected into ventricles, to achieve a final concentration of 625 μM assuming a 1mL cerebrospinal fluid volume, using a 10 μL Hamilton syringe (0.8 mm posterior and 1.6 mm left and right of Bregma). Control mice were injected with an equivalent volume of sterile 0.9 % saline solution. Following a 2 hour recovery, mice were rapidly decapitated and cortical mitochondria and cytosolic fractions were isolated and immunoblotted for Akt.
Akt Activity Assay
All reagents for the Akt activity assay, except translocation components, were obtained from Cell Signaling. Cell-free translations of wtAkt and DDAkt proteins were performed, as described above, and each reaction was adjusted to 200ul total volume with 1X Cell Lysis Buffer. 20ul immobilized Akt antibody slurry was added, and the mixture was incubated on an orbital rocker overnight at 4°C. The immobilized antibody pellets were washed two times with 1X cell lysis buffer and two times with 1X kinase buffer. Antibody pellets were incubated with 1X kinase buffer supplemented with 1ug GSK3 fusion protein and 0.2 mM ATP for 30 minutes at 30°C. Reactions were terminated with SDS sample buffer and boiled 5 minutes. Membranes were immunoblotted for phospho-GSK3α/β reactivity.
Transmission Electron Microscopy
Adherent HEK293 cells were cultured as described and either serum-starved overnight or maintained in serum media, where indicated. Functional mitochondria were isolated using nitrogen cavitation and incubated with DD-Akt protein in the presence or absence of novobiocin for 45 minutes at 30°C. Control reactions were incubated with reticulocyte lysates containing no translated protein. Following incubation, mitochondria were pelleted and fixed overnight at room temperature in 2% paraformaldehyde/2.5% glutaraldehyde in 0.1M sodium cacodylate buffer. The pellet was washed extensively with sodium cacodylate buffer, and post-fixed with 1% osmium tetroxide in sodium cacodylate buffer. Pellets were then washed for one hour with phosphate-buffered saline (PBS). The cell pellets were dehydrated with 10 minute incubations of 50% EtOH, 70% EtOH, and 95% EtOH, followed by four 15-minute incubations in 100% EtOH. Pellets were washed twice for 10 minutes with 100% propylene oxide, and placed into a 50:50 mixture of propylene oxide and embedding resin (Embed 812, Electron Microscopy Sciences, Fort Washington, PA) for 12–18 hours. The tissue was transferred to 100% embedding media for one hour, changed to fresh embedding media for an additional hour, and incubated in an oven at 60°C overnight to polymerize. Following polymerization, the resin blocks were sectioned 1–2 μm thick with a diamond knife using an ultramicrotome. The sections were stained with toluidine blue to use as a reference for thin sectioning. Thin sections were made using a diamond knife (Diatome, Electron Microscopy Sciences, Fort Washington, PA) at 70–100 nm, and the sections were placed on copper mesh grids. After drying, sections were stained with heavy metals uranyl acetate and lead citrate for contrast. Grids were allowed to dry, and then viewed on a FEI Tecnai Twin 120kv TEM (FEI, Hillsboro, OR). Digital images were taken with an AMT CCD (Advanced Microscopy Techniques) camera and the images were saved on a computer memory device. Two blinded observers who were unaware of the treatments were given several representative images of mitochondria with varying configurations as the criterion for quantitation to prevent the inclusion of fragmented mitochondria or debris in the quantitation. The grayscale settings were kept the same for all the images. The observers were told to manually and accurately circumscribe 30 or more objects on the computer screen using the ArcSoft photoimaging software, only those objects in the experiments that fit the criteria. The optical density of each object was then automatically tabulated and entered into a statistical program to determine means, standard errors, and statistical significance.
Results
Inhibition of HSP90 decreases mitochondrial Akt levels
Previously it was reported that a pool of Akt exists in the mitochondria, the level of which can flux greatly (Bijur, 2003). Two well characterized HSP90 inhibitors, NB and GA, have previously been shown to block the import of HSP90 cargo proteins into the mitochondria (Fan 2006), indicating that HSP90 plays a key role in the process of mitochondrial protein import. The goal here was to test if HSP90 inhibitors would affect the levels of Akt in the mitochondria under normal growth conditions. Akt exists mostly in the cytoplasm (Andjelkovic, 1997; Borgatti, 2000), thus it was necessary to confirm that there was no cytosolic contamination in the mitochondrial fractions. The purity of the mitochondrial fraction used to measure Akt levels was assessed by blotting for the abundant cytosolic protein β-tubulin, which could not be detected in the mitochondrial fraction (Fig 1A). The mitochondrial protein pyruvate dehydrogenase was clearly evident in the mitochondrial fraction and not in the cytosolic fraction, indicating complete separation of these two fractions. The existence of HSP90 on the mitochondria was also confirmed. Isolated mitochondria were separated into two separate aliquots and one aliquot was digested with proteinase K to strip proteins on the mitochondrial surface. Internal mitochondrial proteins are inaccessible to proteinase K activity and therefore remain intact. Figure 1B shows an abundant amount of HSP90 on the mitochondrial surface, and digestion with proteinase K removed most of this HSP90. A small amount of HSP90 was found to exist within the mitochondria. The level of ATP synthase-β which exists on the inner mitochondrial membrane was unaffected by proteinase K digestion. Thus, HSP90 resides on the mitochondrial surface.
Fig. 1.
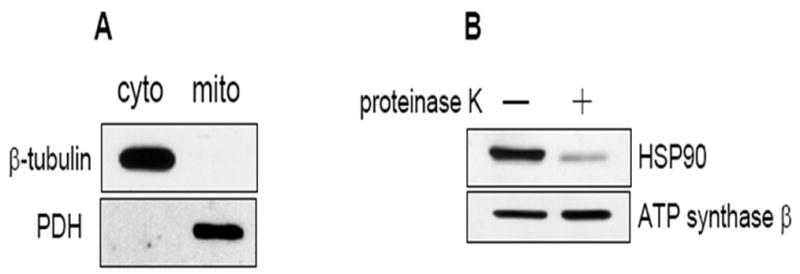
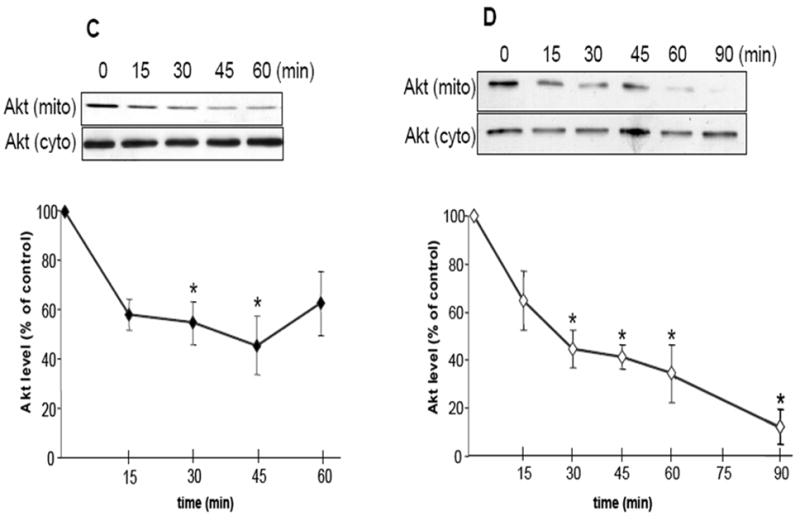
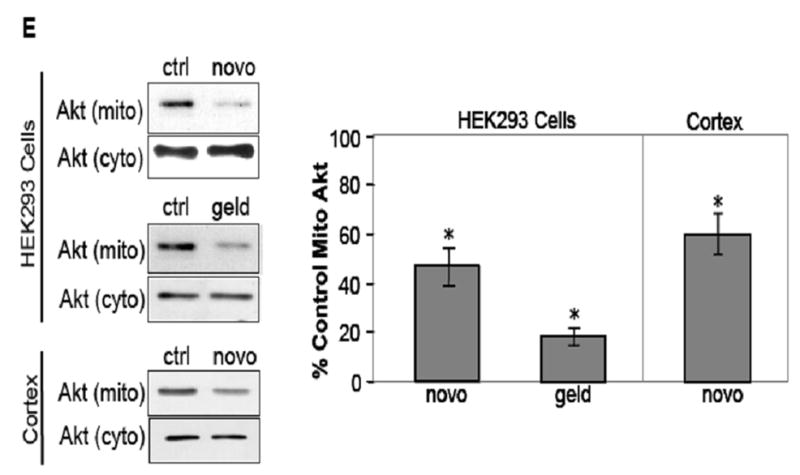
HSP90 inhibitors decrease mitochondrial Akt levels in SH-SY5Y and HEK293 cells. (A) Mitochondrial and cytosolic fractions from SH-SY5Y cells were immunoblotted for β-tubulin (a cytosolic protein) and pyruvate dehydrogenase (PDH), a mitochondrial protein, to assess mitochondrial purity. (B) Proteinase K digestion of mitochondrial fractions, followed by immunoblotting, reveals HSP90 existence on the mitochondrial surface. (C) Serum-starved SH-SY5Y cells were treated time-dependently with 625 μM novobiocin to inhibit HSP90. Cytosolic (cyto) and mitochondrial (mito) fractions were immunoblotted for Akt. The Akt bands in the mitochondria were quantitated by scanning densitometry and are shown in the corresponding graphs as percent of control, n=3, *p<0.05 compared to values from controls, ANOVA. (D) Serum-starved SH-SY5Y cells were treated with 5 μM geldanamycin to inhibit HSP90. Fractions were immunoblotted for Akt, n=3, *p<0.05 compared to values from controls, ANOVA. (E) HEK293 cells were treated for 45 min with 625 μM novobiocin or 5 μM geldanamycin to inhibit HSP90. For in-vivo studies, “cortex”, novobiocin (625 μM) was stereotaxically injected into ventricles of mice as described in “Materials and methods”. The cytosolic and mitochondrial fractions from the cells and cortical tissue were immunoblotted for Akt and protein bands were quantitated by scanning densitometry. Percent of control, n=3, *p< 0.05 compared to values from controls, Student’s t-test.
The effect of NB and GA treatments on Akt mitochondrial levels was tested in unstimulated cells. NB is a coumarin-type compound that affects the C-terminal region of HSP90 (Marcu, 2000a). NB treatment of SH-SY5Y neuroblastoma cells caused a time-dependent reduction in the level of Akt in the mitochondria (Fig 1C). After 15 min of treatment with 0.625 mM NB there was a 42% reduction of Akt in the mitochondria compared to control untreated cells. After 45 min of NB treatment, the level of Akt in the mitochondria was reduced by approximately 60% of control, and further reductions in Akt levels in the mitochondria were not seen. HSP90 inhibition results in the degradation of its client proteins including Akt, however at the concentration and the time points of NB used for this study the total Akt levels in the cytosol were not reduced (Fig 1C), suggesting that Akt degradation may not be occurring at these times and at this dose of NB treatments.
GA is a benzoquinoid ansamycin antibiotic that affects the N-terminal ATP-binding region of HSP90 (Whitesell, 1994; Panaretou 1998). Interestingly, GA does not enter the mitochondrion and accumulate in this organelle (Kang, 2007). GA (5 μM) treatment also resulted in marked reductions in Akt levels in the mitochondria (Fig 1D). After 15 min of GA treatment there was a 40% decrease in Akt levels in the mitochondria and the levels of Akt in the mitochondria were reduced further after 90 min of GA treatment, resulting in a 94% reduction of Akt in the mitochondria. The level of Akt in the cytosol was unaffected by GA treatments. Treatment of cells with higher concentrations and longer times of NB and GA treatments did result in degradation of Akt (data not shown). The effect of NB and GA treatments on Akt levels in the mitochondria was also confirmed in another cell line. Human HEK293 cells were treated for 45 min with 625μM NB or 5 μM GA, and Akt levels in the mitochondria were assessed. There were significant 50% and 80% reductions in Akt levels in the mitochondria following NB and GA treatments, respectively (Fig 1E). Thus, two chemically distinct inhibitors of HSP90 used under these specific conditions were found to markedly reduce the level of Akt in the mitochondria but not the cytosol, which was confirmed in two different cell lines.
To test if Hsp90 regulates Akt levels in brain mitochondria, 625μM NB was stereotaxically injected into both lateral ventricles of adult C57/Bl6 mice. There was a significant 40% decrease in mitochondrial Akt levels in NB injected mice compared to saline controls (Figure 1E). Levels of Akt in the cytosol were unchanged by this treatment (1E). These data indicate that, in in vivo as well as in cell culture models, inhibition of Hsp90 leads to a decrease in the level of Akt in the mitochondria.
The concern with using inhibitors of HSP90 such as NB and GA is that these agents are known to elicit the proteolysis of the HSP90 client protein such as Akt. To allay this concern, HEK293 stable cell lines were created in which HSP90 protein expression was reduced using HSP90 siRNA. HSP90 expression in the HSP90 siRNA cells was reduced by 70% of control (Fig 2A), but the expression of HSP70 and heat shock cognate-70 (HSC70) was unaffected. Interestingly, the level of HSP40 was noticeably increased. The total level of Akt was unaffected (Fig 2A), but in unstimulated HSP90 siRNA cells the Akt level in the mitochondria was significantly reduced by 50% compared to control cells (Fig 2B). Taken together, these results show that HSP90 activity markedly influences the amount of Akt accumulation in the mitochondria. However, a total blockade of Akt mitochondrial accumulation could never be achieved using any of these methods of HSP90 inhibition, suggesting that there may also be an HSP90-independent mode by which Akt accumulates in the mitochondria.
Fig. 2.
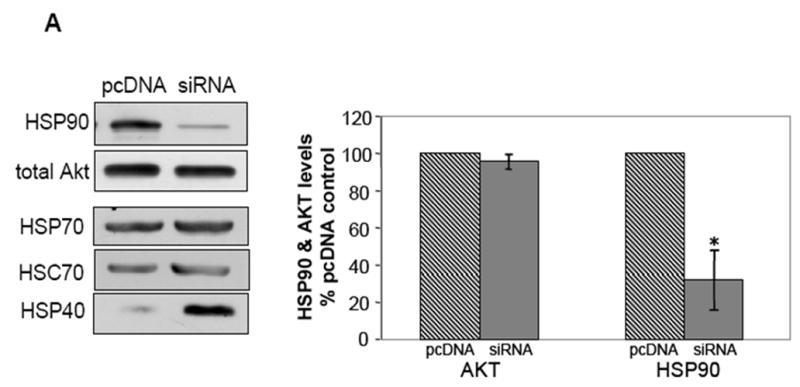
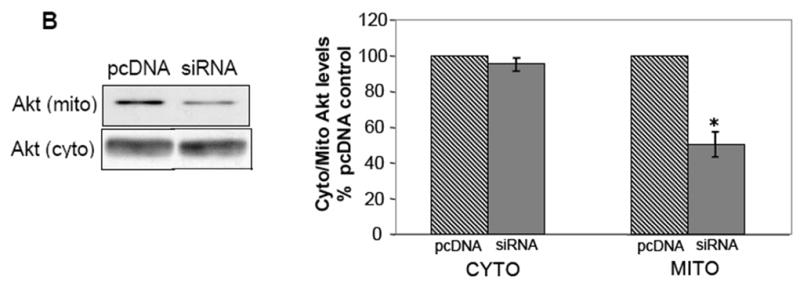
siRNA-mediated knockdown of HSP90 expression results in decreased levels of mitochondrial Akt. (A) Cells stably transfected with HSP90 siRNA were lysed and immunoblotted for expression of HSP90, Akt, HSP70, HSC70, and HSP40. *p<0.05 compared with values from control cell line, Student’s t-test. (B) Cells transfected with HSP90 siRNA or control pcDNA plasmid were fractionated into cytosolic and mitochondrial fractions and the two fractions were immunoblotted for Akt. *p<0.05 compared with values from control cell line, Student’s t-test.
HSP90 mediates import of Akt into isolated mitochondria
To directly assess Akt mitochondrial import by HSP90, an in-vitro mitochondrial import assay was employed. We also examined if the activation state of Akt affects its mitochondrial import. Two forms of Akt were tested; an unmodified wtAkt, and a constitutively-active mutant Akt in which the activity-associated phosphorylation sites at Thr308 and Ser473 sites were mutated to aspartic acid (DDAkt). These two Akt proteins were in-vitro translated using rabbit reticulocyte lysates. Initially, the relative activities of the wtAkt and DDAkt within the mitochondria were determined. Isolated intact mitochondria were incubated with the translated Akt proteins. The external mitochondrial proteins were digested with proteinase K and the mitochondria were lysed. The mitochondrial lysates were immunoblotted with a phospho-Akt substrate (PAS) antibody which detects phosphorylated substrates of Akt. Incubation of intact mitochondria with wtAkt resulted in increased PAS antibody immunoreactivity against internal mitochondrial proteins compared to the control (Fig 3A) indicating that the translated wtAkt can enter the mitochondria and has some basal kinase activity. PAS immunoreactivity of internal mitochondrial proteins was increased further with the incubation of mitochondria with DDAkt, indicating that DDAkt has greater activity than the wtAkt, and also can enter the mitochondria. The activities of the translated proteins was also measured by immunoprecipitating wtAkt and DDAkt and reacting the immobilized proteins with an Akt substrate (GSK3α/β crosstide). The Akt activity mirrored the PAS immunoreactivity (Fig 3A); the wtAkt had some basal kinase activity, while the DDAkt had increased kinase activity.
Fig. 3.
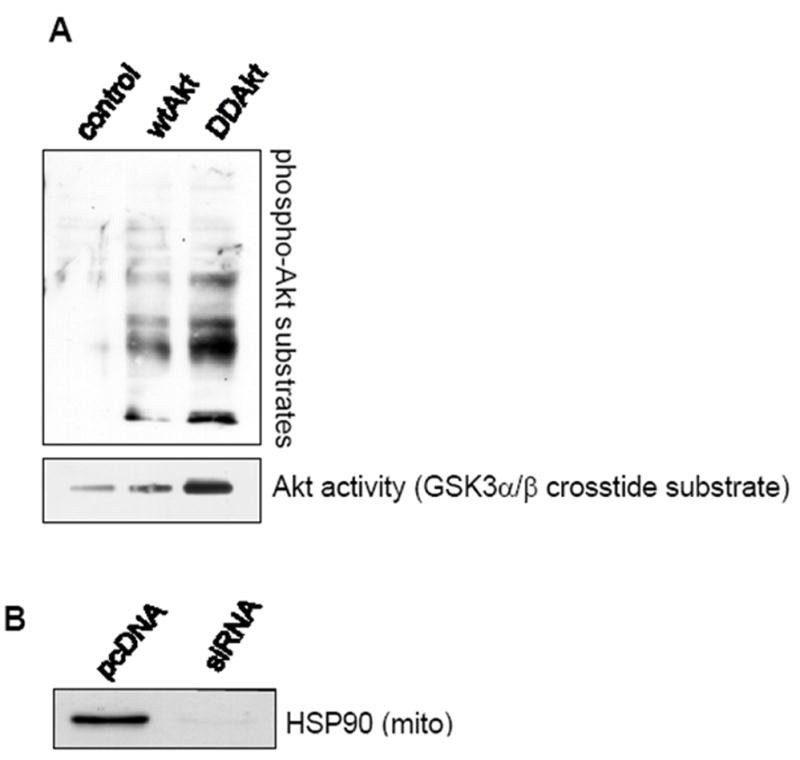
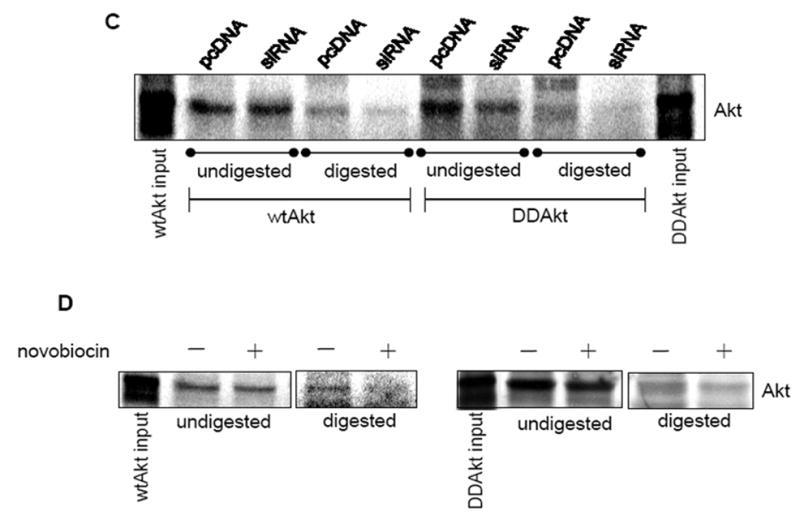
HSP90 mediates the mitochondrial translocation of Akt. (A) Top panel: Isolated mitochondria were incubated with either wild type (wt) or DDAkt, digested with proteinase K, and immunoblotted with a phospho-Akt substrate antibody. Bottom panel: In-vitro translated Akt proteins were measured for enzymatic activity as described in Material and methods. (B) Mitochondria were isolated from pcDNA control cells and HSP90 siRNA cells. Mitochondrial lysates were immunoblotted for HSP90. (C) Representative autoradiograph showing mitochondrial translocation of 35S-Methionine labeled wtAkt and DDAkt in control cells and HSP90 siRNA cells. Isolated mitochondria from each cell line were incubated with radiolabled exogenous wtAkt or DD-Akt protein. Half of each sample was digested with proteinase K, and the other half was left undigested. The Akt bands were viewed with a phosphorimager. (D) Representative autoradiograph showing mitochondrial translocation of wtAkt (left panel) and DDAkt (right panel) in the presence and absence of 625 μM novobiocin.
For the import assay, wtAkt and the constitutively active DDAkt were in-vitro translated in the presence of 35S-methionine which is incorporated into the synthesized proteins. The protein products were incubated with intact mitochondria for the import assay as described in Materials and Methods. The mitochondrial import of the 35S-methionine-labeled Akt proteins was examined in mitochondria isolated from the control pcDNA cells and the siRNA HSP90 knockdown cells. The level of HSP90 bound to the mitochondria in the HSP90 siRNA cells was first determined. Figure 3B shows the reduced level of HSP90 bound to the mitochondria in the HSP90 siRNA cells compared to the pcDNA control cells. Figure 3C shows that both the wtAkt and DDAkt were found to be attached to the surface of the mitochondria at about equivalent levels in the undigested mitochondrial samples from the pcDNA cells and the HSP90 siRNA cells. In the digested mitochondria, however, substantially less wtAkt and DDAkt were detected within the mitochondria from the HSP90 siRNA cells compared to mitochondria from the control pcDNA cells, indicating that HSP90 is mostly required for the mitochondrial import of both forms of Akt. To further test if HSP90 inhibition would block mitochondrial import of wtAkt and DDAkt, isolated mitochondria from wild-type HEK293 cells were incubated with the in-vitro translated proteins in the presence of NB. NB did not affect Akt binding to the mitochondria surface (Fig 3D), but in agreement with the previous result, NB-mediated HSP90 inhibition mostly blocked the mitochondrial import of both forms of Akt. However, in both of these cases a total blockade of Akt import into the mitochondria was not seen, further suggesting that there exists a second mechanism by which Akt is imported into the mitochondria. These results further verify that HSP90 partially facilitates Akt transport into the mitochondria.
Growth factor-stimulated mitochondrial Akt accumulation is not hampered by HSP90 inhibition
In a previous study it was shown that stimulation of HEK293 cells and SH-SY5Y cells with IGF-1, a potent activator of the PI3K signaling pathway, induced the mitochondrial translocation of Akt (Bijur, 2003). Because the mitochondrial import of constitutively active DDAkt was blocked by HSP90 inhibition, it stood to reason that mitochondrial accumulation of Akt following IGF-1 stimulation could also be blocked by HSP90 inhibition. HEK293 cells were treated with IGF-1 alone or with NB and GA prior to IGF-1 treatment. Mitochondrial lysates were immunoblotted for Akt levels. Neither NB nor GA treatments caused any diminution of Akt import into the mitochondria (Fig 4A and B). Similarly, in SH-SY5Y cells the HSP90 inhibitors had no effect on IGF-1-mediated Akt mitochondrial import (data not shown). Furthermore, in the siRNA HSP90 knockdown cells IGF-1-mediated Akt mitochondrial translocation was also not diminished compared to control cells (Fig 4C). Taken together, our data suggests that the normal flux of Akt into the mitochondria is HSP90-dependent, and appears to be distinct from growth factor-stimulated Akt mitochondrial transport, which does not appear to be dependent upon HSP90 activity.
Fig. 4.
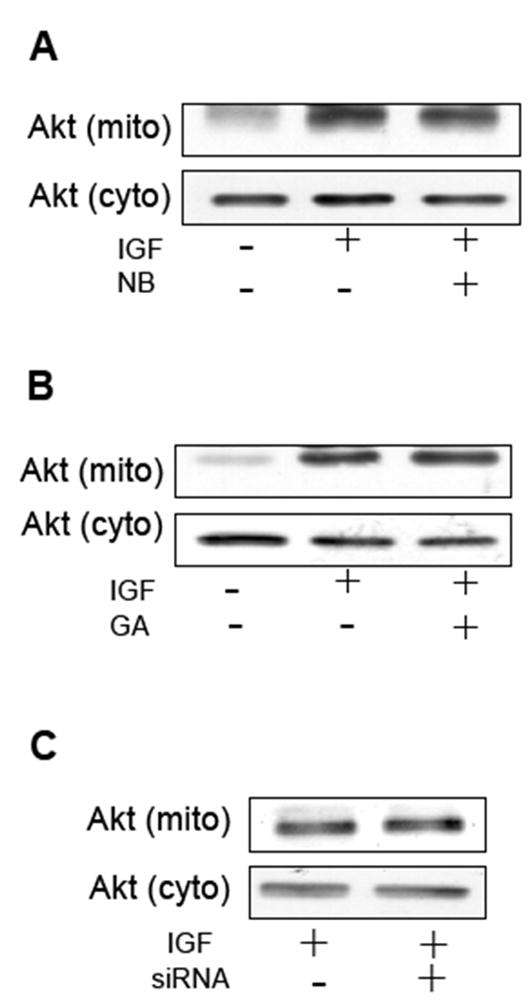
HSP90 inhibitors do not block IGF-1-induced mitochondrial Akt accumulation. (A) Serum-starved HEK293 cells were pretreated for 15 minutes with 625 μM NB prior to treatment with IGF-1, or cells were treated with IGF-1 (50 ng/mL for 15 minutes) alone. Mitochondrial and cytosolic fractions were immunoblotted for Akt. (B) HEK293 cells were treated with 50 ng/mL IGF-1 for 15 minutes, or pretreated 5μM NB for 15 min prior to treatment with IGF-1. Mitochondrial and cytosolic fractions were immunoblotted for Akt. (C) pcDNA control cells and HSP90 knockdown cells were treated for 15 minutes with 50ng/mL IGF-1. Mitochondrial and cytosolic fractions were immunoblotted for Akt.
Akt alters mitochondrial matrix configuration
The next goal was to examine how the accumulation of Akt within the mitochondria would affect mitochondrial function. Mitochondria are very dynamic and pliant organelles whose shapes change with their function; therefore, a visible feature of the mitochondrion is its morphology. For example, changes in metabolic activity, respiration state, and apoptotic signaling can markedly affect mitochondrial shape (Hackenbrock, 1966; Bossy-Wetzel, 2003; Gottlieb, 2003). In this experiment, HEK293 cells were grown in serum-free media for 24 h. Mitochondria were isolated and then incubated with the constitutively active DDAkt in the presence or absence of 625 μM novobiocin. Control reactions were incubated with rabbit reticulocyte lysates. The isolated mitochondria were viewed by transmission electron microscopy. Mitochondria from control cells in serum-free media have highly condensed cristae (indicated by an arrow), and decreased matrix density. This configuration was likely due to serum withdrawal as mitochondria from cells grown in serum typically had highly compartmentalized cristae (Fig 5A), although the size of the mitochondria was much smaller compared to mitochondria from serum-free cultured cells. Mitochondria incubated with the DDAkt had highly compartmentalized cristae and appeared to have more of an orthodox configuration. The cristae were also well defined (Fig 5B, indicated by an arrow in the middle right panel) within the mitochondria. Densitometric quantitation in a blind study revealed that DDAkt treatment caused a significant increase in the matrix density of about 115% (Fig 5C) compared to the control mitochondria. Cotreatment of the mitochondria with novobiocin and DDAkt significantly blocked the compartmentalization of the cristae (Fig 5B and C), which appears as decreased matrix density. However, some closed cristal regions could still be seen, probably indicating that some DDAkt was still entering the mitochondria and causing cristal remodeling.
Fig. 5.
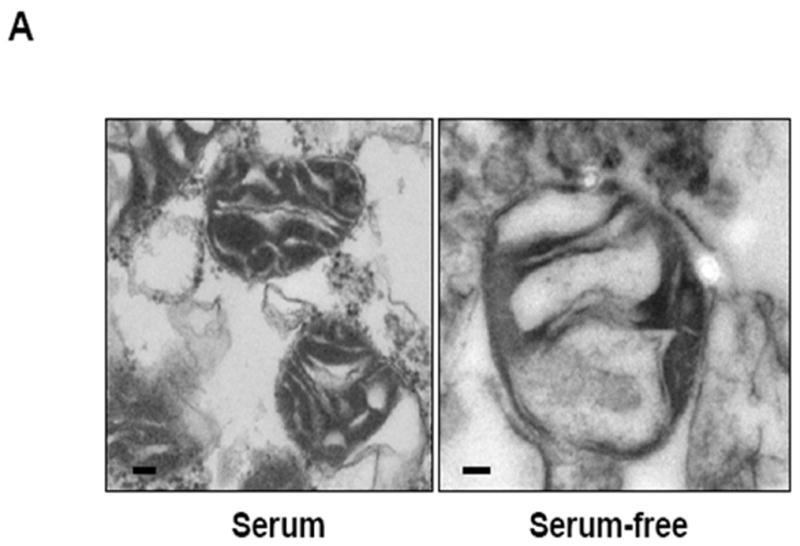
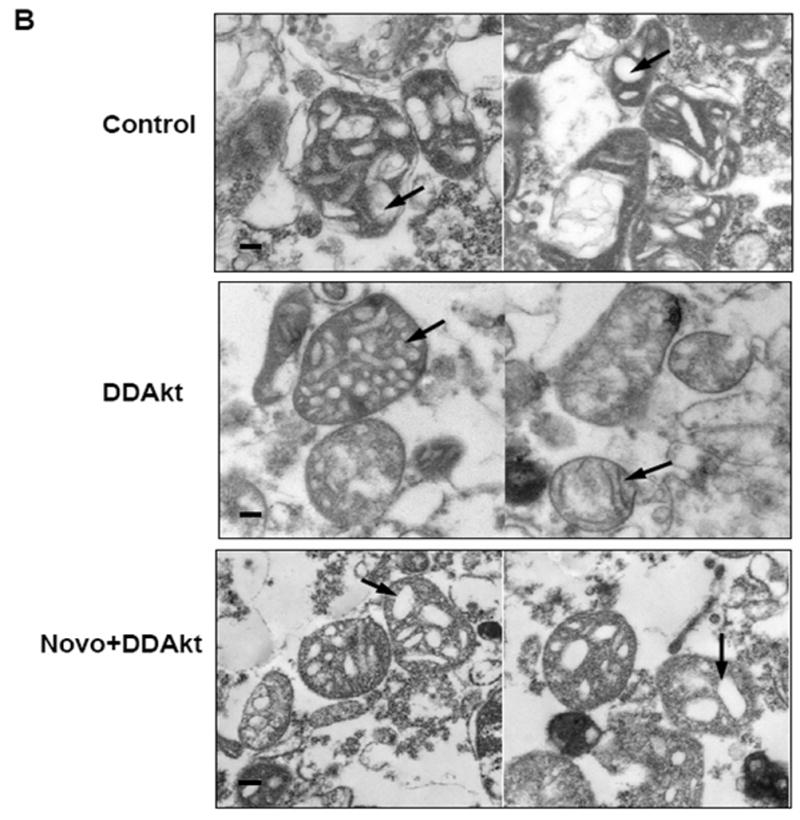
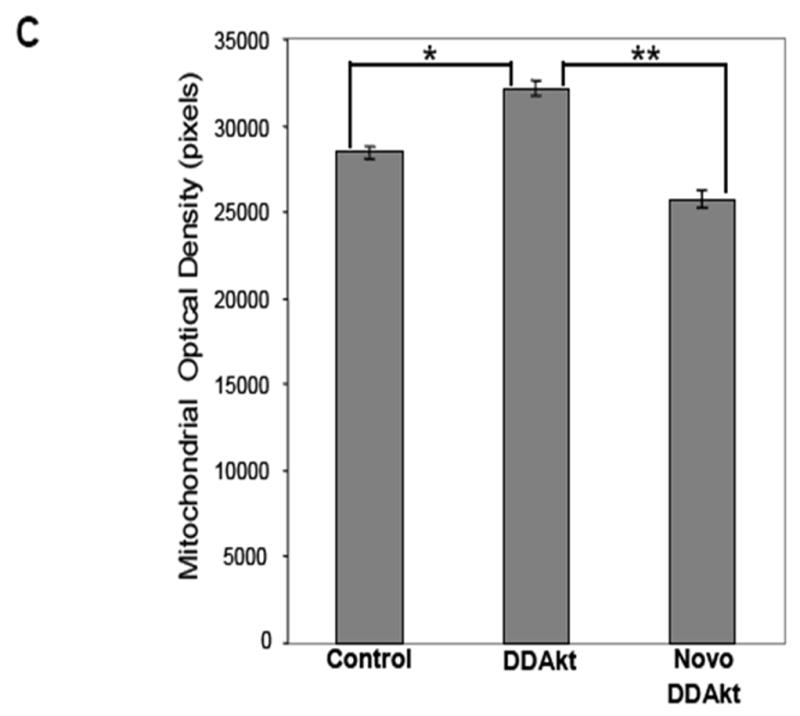
Akt affects mitochondrial morphology. (A) Mitochondrial pellets were fixed and prepared for transmission electron microscopy as described in the Materials and Methods section. Representative transmission electron micrographs of mitochondria isolated from HEK293 cells in serum-containing versus serum-containing media (size bar = 100 nm). (B) Mitochondria were isolated from serum-starved HEK293 cells and incubated with empty reticulocyte (control), DD-Akt, or 625 μM NB and DD-Akt for 45 minutes (size bar = 100 nm). Representative images are shown. (C) Optical density was quantified in a blind study from transmission electron micrographs of isolated mitochondria incubated with DD-Akt or NB + DD-Akt versus control. N=54, *p<0.05, compared to control mitochondria incubated with reticulocyte lysate. **p<0.05, compared to control mitochondria incubated with DD-Akt. ANOVA.
Discussion
Previously it was reported that Akt exists in the mitochondria (Bijur 2003). The import of some proteins into the mitochondria has been shown to be dependent on HSP90 (Young, 2003; Young, 2004; Fan, 2006). Given that Akt is a known client protein of HSP90 we theorized that HSP90 activity may influence Akt levels in the mitochondria. This turned out to be the case as the amount of Akt in the mitochondria was significantly reduced by HSP90 inhibition.
NB and GA are two well known HSP90 inhibitors which have been shown to block the import of proteins into the mitochondria (Young, 2003; Fan, 2006). Treatment of cells with both of these inhibitors resulted in the rapid reduction of Akt levels in the mitochondria. Since GA does not enter the mitochondria (Kang, 2007), it should not affect HSP90 within the mitochondria. Therefore, it can be reasoned that GA prevented endogenous cytoplasmic Akt from entering the mitochondria by only inhibiting extramitochondrial HSP90. The rapidity of the decline in mitochondrial Akt levels, occurring within 15 min of treatment with the inhibitors, raised the possibility that the reduction of Akt levels in the mitochondria may be due to Akt proteolysis. NB and GA treatments have previously been shown to cause the destabilization and degradation of Akt (Marcu, 2000b; Basso, 2002; Kim, 2003; Yun 2004). In background work for these experiments, high concentrations of NB and GA, greater than those shown in this study, did cause pronounced decreases in Akt levels in the cytosol and the mitochondria (data not shown). However, 625 μM NB and 5 μM GA for up to 60 and 90 min, respectively, did not cause reductions in the cytosolic Akt levels. The reason for the rapid decline in mitochondrial Akt level following NB and GA treatments is unclear. It is possible that Akt is normally unstable in the mitochondria and is continuously replenished by import of Akt from the cytosol. Thus, the import of Akt from the cytosol may maintain a steady-state level of Akt inside the mitochondria. The rapid decrease in Akt levels in the mitochondria following treatment with the HSP90 inhibitors may be due to an imbalance in these steady state levels, as replenishment of Akt from the cytosol is prevented. We also tested the effect of HSP90 inhibition on brain mitochondrial Akt levels by injecting NB into brains of adult mice. As seen in cell culture models, a two-hour treatment with 625 μM NB caused a significant reduction in Akt levels in mitochondria isolated from cortex of treated animals, confirming that HSP90 regulates basal mitochondrial Akt levels in-vivo.
To further test if decreased HSP90 activity affects Akt levels in the mitochondria, HSP90 expression was blocked by siRNA-mediated knockdown of HSP90. Interestingly, stable HSP90 siRNA HEK293 cell lines were able to thrive despite a significant reduction in HSP90 protein level, possibly due to a compensatory increase in another surrogate heat shock protein. For example, HSP40 was substantially increased in the HSP90 knockdown cells. Nevertheless, cytosolic Akt levels in HSP90 siRNA cell lines were unaffected, while the level of Akt in the mitochondria was found to be significantly reduced. Together these results show that decreased HSP90 activity affects the level of Akt in the mitochondria.
We could not discount the possibility that total cellular Akt levels may be decreased with HSP90 inhibition, which may be occurring imperceptibly in the cytosolic fraction, but is amplified in the mitochondrial fraction. As an alternative method to test if HSP90 affects Akt mitochondrial import, a mitochondrial import assay was employed. An unmodified wtAkt and constitutively active DDAkt were both tested for their ability to be imported into the mitochondria. Surprisingly, both forms of Akt accumulated in the mitochondria. In addition, the import of both forms was inhibited by HSP90 inhibition, and by decreased HSP90 expression. This indicates that the normal flux of Akt into the mitochondria is independent of the state of Akt activation. Indeed, Akt is constitutively bound to HSP90 regardless of its activation state (Meares, 2004), thus it is reasonable to assume that the levels of Akt in the mitochondria under unstimulated conditions are not entirely dependent on Akt activity.
Previously it was shown that IGF-1 stimulation, which induces Thr308 and Ser473 phosphorylation and activation of Akt, causes a rapid translocation of Akt into the mitochondria (Bijur, 2003). Since the DDAkt somewhat mimics Akt phosphorylation at Thr308 and Ser473, and this mutant Akt was mostly blocked from entering the mitochondria by decreased HSP90 activity, we surmised that HSP90 inhibition would also block Akt translocation into the mitochondria after IGF-1 stimulation. This was not the case, as none of the modes of HSP90 inhibition blocked IGF-1-induced import of Akt into the mitochondria. This finding clearly points to a second, HSP90-independent mode, by which Akt is imported into the mitochondria, and may partially explain why a total inhibition of mitochondrial Akt accumulation was never achievable by any of the HSP90 inhibitors. In addition to HSP90, HSP70, HSC70, and HSP40 have also been shown to mediate the import of proteins into the mitochondria (Young, 2003: Bhangoo 2007). In the HSP90 siRNA cells, the levels of HSP70 proteins were unchanged and the level of HSP40 was markedly increased. Akt has been shown to be a client protein of HSP70, and the HSP40 co-chaperones are known to interact with HSP70 (Gao, 2002; Cajo, 2006). Thus, it is conceivable that in addition to HSP90, Akt mitochondrial translocation may also be facilitated by HSP70, possibly via the HSP40 co-chaperone system described by Bhangoo et al (2007).
To determine how Akt flux affects the mitochondrion, we began by observing mitochondrial morphology. One of the most salient features of the mitochondrion is its appearance. Mitochondria are highly plastic organelles which rapidly change their configuration in response to their immediate surroundings and respiration states. In addition, substrate availability and apoptosis signaling also affect mitochondrial morphology (Gottlieb, 2003; Rasola, 2007). Mitochondria can change their size, shape, and their cristae. A normally respiring mitochondrion has a classic “orthodox” appearance (Hackenbrock, 1966; 1968), with an intact outer and inner membrane separated by an intermembrane space, and a densely-packed matrix. Mitochondria isolated from cells that have been subjected to a toxic stressor typically have highly condensed cristae with an enlarged matrix (Gottlieb, 2003). For example, serum deprivation is stressful to cultured cells and can enhance the effects of many toxic stimuli. We found that mitochondria from HEK293 cells cultured in serum-free media for 24h had enlarged mitochondria with highly condensed cristae. The addition of exogenous constitutively active Akt caused the mitochondria to acquire a more orthodox appearance, with a densely-packed cristae. The Akt-induced alteration of the mitochondria was partially blocked by the addition of NB, paralleling the finding that Akt import was also partially blocked by HSP90 inhibition.
The initial step in the execution phase of apoptosis is the release of cytochrome c from the mitochondrial intermembrane space (Haupt, 2003). It has been reported that matrix remodeling to the condensed state, as was seen in the mitochondria from cells cultured in serum-free media, exposes cytochrome c to the intermembrane space priming its release from the mitochondria during apoptosis, whereas mitochondria resistant to apoptosis maintain an orthodox conformation and this keeps cytochrome c tightly sequestered within the cristae (Gottlieb, 2003). Akt is a well known cell survival-associated protein, and overactive Akt is known to be tumorigenic and block anti-cancer treatments that activate apoptosis (Haupt, 2003). In addition, activated Akt is known to bolster neuronal survival in the face of toxic or ischemic insult (Mangi, 2003; Ohba, 2004). There are multiple mechanisms by which Akt promotes cell survival. The present study suggests that an additional mode by which Akt is protective is by maintaining mitochondria in their orthodox state, possibly facilitating the tight sequestration of cytochrome c in the cristae, and preventing its release into the cytosol to activate apoptosis.
In conclusion, this study shows that the HSP90 activity markedly influences the level of Akt in the mitochondria. Furthermore, we have shown for the first time that HSP90, via Akt, can dramatically alter mitochondrial configuration. Taken together, these results provide new insight regarding Akt signaling in the mitochondria and its effects on this organelle.
Acknowledgments
This work was supported by NIH grant NS044853. We wish to thank Dr. Richard S. Jope for the kind gift of the Akt constructs. We also wish to thank Dr. Buffie Clodfelder-Miller and Mr. Robert Mans for technical assistance in blinded studies.
Abbreviations used
- HSP90
heat shock protein-90
- NB
novobiocin
- GA
Geldanamycin
- HSP70
heat shock protein-70
- TOM
Translocase of the Outer Mitochondrial Membrane
- IGF-1
insulin-like growth factor-1
- PAS
phospho-Akt substrate
- PI3K
phosphatidylinositol 3-kinase
- GSK3β
glycogen-synthase-kinase 3β
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–51. [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol. 1997;7:261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Andjelkovi M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–24. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (HSP90) and Cdc37and is destabilized by inhibitors of HSP90 function. J Biol Chem. 2002;277:39858–66. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- Bhangoo MK, Tzankov S, Fan AC, Dejgaard K, Thomas DY, Young JC. Multiple 40-kDa heat-shock protein chaperones function in TOM70-dependent mitochondrial import. Mol Biol Cell. 2007;18:3414–28. doi: 10.1091/mbc.E07-01-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem. 2003;87:1427–35. doi: 10.1046/j.1471-4159.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgatti P, Martelli AM, Bellacosa A, Casto R, Massari L, Capitani S, Neri LM. Translocation of Akt/PKB to the nucleus of osteoblast-like MC3T3-E1 cells exposed to proliferative growth factors. FEBS Lett. 2000;477:27–32. doi: 10.1016/s0014-5793(00)01758-0. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–16. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Cajo GC, Horne BE, Kelley WL, Schwager F, Georgopoulos C, Genevaux P. The role of the DIF motif of the DnaJ (HSP40) co-chaperone in the regulation of the DnaK (HSP70) chaperone cycle. J Biol Chem. 2006;281:12436–44. doi: 10.1074/jbc.M511192200. [DOI] [PubMed] [Google Scholar]
- Chacinska A, Pfannschmidt S, Wiedemann N, Kozjak V, Sanjuan Szklarz LK, Schulze-Specking A, Truscott KN, Guiard B, Meisinger C, Pfanner N. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 2004;23:3735–46. doi: 10.1038/sj.emboj.7600389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan AC, Bhangoo MK, Young JC. HSP90 functions in the targeting and outer membrane translocation steps of TOM70-mediated mitochondrial import. J Biol Chem. 2006;281:33313–24. doi: 10.1074/jbc.M605250200. [DOI] [PubMed] [Google Scholar]
- Fayard E, Tintignac LA, Baudry A, Hemmings BA. Protein kinase B/Akt at a glance. J Cell Sci. 2005;118:5675–8. doi: 10.1242/jcs.02724. [DOI] [PubMed] [Google Scholar]
- Fujita N, Sato S, Ishida A, Tsuruo T. Involvement of HSP90 in signaling and stability of 3-phosphoinositide-dependent kinase-1. J Biol Chem. 2002;277:10346–53. doi: 10.1074/jbc.M106736200. [DOI] [PubMed] [Google Scholar]
- Gabriel K, Egan B, Lithgow T. Tom40, the import channel of the mitochondrial outer membrane, plays an active role in sorting imported proteins. EMBO J. 2003;22:2380–6. doi: 10.1093/emboj/cdg229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Newton AC. The turn motif is a phosphorylation switch that regulates the binding of HSP70 to protein kinase C. J Biol Chem. 2002;277:31585–92. doi: 10.1074/jbc.M204335200. [DOI] [PubMed] [Google Scholar]
- Geissler A, Krimmer T, Bomer U, Guiard B, Rassow J, Pfanner N. Membrane potential-driven protein import into mitochondria. The sorting sequence of cytochrome b(2) modulates the ΔΨ-dependence of translocation of the matrix-targeting sequence. Mol Biol Cell. 2000;11:3977–91. doi: 10.1091/mbc.11.11.3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10:709–17. doi: 10.1038/sj.cdd.4401231. [DOI] [PubMed] [Google Scholar]
- Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol. 1966;30:269–97. doi: 10.1083/jcb.30.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. II. Electron transport-linked ultrastructural transformations in mitochondria. J Cell Biol. 1968;37:345–69. doi: 10.1083/jcb.37.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci. 2003;116:4077–85. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific HSP90 chaperone network. Cell. 2007;131:257–70. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- Kerscher O, Holder J, Srinivasan M, Leung RS, Jensen RE. The Tim54p-Tim22p complex mediates insertion of proteins into the mitochondrial inner membrane. J Cell Biol. 1997;139:1663–75. doi: 10.1083/jcb.139.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kang J, Hu W, Evers BM, Chung DH. Geldanamycin decreases Raf-1 and Akt levels and induces apoptosis in neuroblastomas. Int J Cancer. 2003;103:352–9. doi: 10.1002/ijc.10820. [DOI] [PubMed] [Google Scholar]
- Komiya T, Rospert S, Schatz G, Mihara K. Binding of mitochondrial precursor proteins to the cytoplasmic domains of the import receptors TOM70 and TOM20 is determined by cytoplasmic chaperones. EMBO J. 1997;16:4267–75. doi: 10.1093/emboj/16.14.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutik S, Guiard B, Meyer HE, Wiedemann N, Pfanner N. Cooperation of translocase complexes in mitochondrial protein import. J Cell Biol. 2007;179:585–91. doi: 10.1083/jcb.200708199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangi AA, Noiseux N, Kong D, He H, Rezvani M, Ingwall JS, Dzau VJ. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat Med. 2003;9:1195–201. doi: 10.1038/nm912. [DOI] [PubMed] [Google Scholar]
- Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem. 2000;275:37181–6. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst. 2000;92:242–8. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- Martin J, Mahlke K, Pfanner N. Role of an energized inner membrane in mitochondrial protein import. ΔΨ drives the movement of presequences. J Biol Chem. 1991;266:18051–7. [PubMed] [Google Scholar]
- Meares GP, Zmijewska AA, Jope RS. Heat shock protein-90 dampens and directs signaling stimulated by insulin-like growth factor-1 and insulin. FEBS Lett. 2004;574:181–6. doi: 10.1016/j.febslet.2004.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba N, Kiryu-Seo S, Maeda M, Muraoka M, Ishii M, Kiyama H. Transgenic mouse overexpressing the Akt reduced the volume of infarct area after middle cerebral artery occlusion. Neurosci Lett. 2004;359:159–62. doi: 10.1016/j.neulet.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Panaretou B, Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. ATP binding and hydrolysis are essential to the function of the HSP90 molecular chaperone in vivo. EMBO J. 1998;17:4829–36. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanner N, Rassow J, Guiard B, Sollner T, Hartl FU, Neupert W. Energy requirements for unfolding and membrane translocation of precursor proteins during import into mitochondria. J Biol Chem. 1990;265:16324–9. [PubMed] [Google Scholar]
- Pfanner N, Geissler A. Versatility of the mitochondrial protein import machinery. Nat Rev Mol Cell Biol. 2001;2:339–49. doi: 10.1038/35073006. [DOI] [PubMed] [Google Scholar]
- Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2000;12:815–33. doi: 10.1007/s10495-007-0723-y. 2007. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to HSP90. Proc Natl Acad Sci U S A. 2000;97:10832–7. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91:8324–8. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yuan X, Jung YJ, Yang Y, Basso A, Rosen N, Chung EJ, Trepel J, Neckers L. The heat shock protein 90 inhibitor geldanamycin and the ErbB inhibitor ZD1839 promote rapid PP1 phosphatase-dependent inactivation of Akt in ErbB2 overexpressing breast cancer cells. Cancer Res. 2003;63:7777–84. [PubMed] [Google Scholar]
- Yano M, Terada K, Mori M. Mitochondrial import receptors TOM20 and TOM22 have chaperone-like activity. J Biol Chem. 2004;279:10808–13. doi: 10.1074/jbc.M311710200. [DOI] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones HSP90 and HSP70 deliver preproteins to the mitochondrial import receptor TOM70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–91. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- Yun BG, Huang W, Leach N, Hartson SD, Matts RL. Novobiocin induces a distinct conformation of HSP90 and alters HSP90-cochaperone-client interactions. Biochemistry. 2004;43:8217–29. doi: 10.1021/bi0497998. [DOI] [PubMed] [Google Scholar]