

Abstract
Maintenance of Th1 responses and dendritic cell (DC) functions are compromised in HIV-1 infected individuals. To better understand these immune abnormalities, we developed an HIV-1 transgenic (Tg) rat. We report that Tg DCs induce elevated levels of SOCS-1 and secrete decreased IL-12p40 and elevated levels of IL-10 following TLR-4 stimulation by LPS. This leads to further induction of SOCS-1 by IL-10 and decreased IFN-γ-mediated induction of interferon response factor (IRF)-1 and IL-12Rβ1 expression in CD4+ T cells and to decreased IL-12-induction of IFN-γ production by Th1 polarized T cells. We also show that SOCS-1 is elevated in CD4+ T cells from HIV-1 infected progressors, and is correlated with defective induction of IRF-1 following IFN-γ stimulation, compared with healthy controls and HIV-1 natural viral suppressor (NVS) patients. These results suggest a link between high levels of SOCS-1, defects in innate immunity and adaptive Th1 responses that may be reflected in the loss of Th1 immune competence observed with AIDS patients.
Keywords: HIV-1, Transgenic rat, SOCS-1, IFN-γ-IL-12 signaling, IRF-1, IL-12Rβ1
Introduction
HIV-1 infection elicits host immune responses that include dysregulation of regulatory pathways for cytokine expression that are characterized by decreased production of pro-inflammatory cytokines including IL-12 and IFN-γ and increased expression of the anti-inflammatory cytokine IL-10, the latter of which has been identified as a factor responsible for impaired innate and T helper type 1 (Th1) responses in AIDS patients (Ma & Montaner, 2000). Although the IFN-γ-IL-12 signaling network is likely pivotal to the Th1 differentiation program and immune control of HIV-1 infection (Kano, Sato et al., 2008;Murphy, Ouyang et al., 2000), the underlying mechanisms are largely uncharacterized.
Innate immunity provides the initial host protective response to infectious danger. Recognition of a wide variety of microbes depends upon toll like receptors (TLRs), which recognize conserved microbial structures. Stimulation of TLRs on DC results in secretion of pro-inflammatory cytokines and increases effector functions such as phagocytosis and the ability to present antigen to T cells. Although innate immunity represents a powerful system to combat exogenous invaders, many infections can only be cleared in combination with adaptive immunity. In this respect, IFN-γ and IL-12 play a critical role by enhancing activation of macrophages and DC and increasing antigen presentation and Th1 memory development /maintenance(Boehm, Klamp et al., 1997;Trinchieri, 2003).
During initiation of the adaptive immune response, co-stimulation of DCs with LPS or other agents induces production of IL-12, a 70kDa heterodimeric cytokine composed of 35 kDa (p35) and 40 kDa (p40) subunits. IL-12 binds to a cell surface receptor composed of β1 (IL-12Rβ1) and β2 (IL-12Rβ2) subunits (Trinchieri, 2003), leading to activation of natural killer (NK), NKT, and memory T cells to produce cytokines (particularly IFN-γ) that favor differentiation of cells that produce type-1 (Th-1) cytokines and Th1-associated classes of immunoglobins. The engagement of the IL-12R on T cells triggers activation of the Janus kinases Tyk2 and JAK2, which are associated with the IL-12Rβ1 and IL-12Rβ2 cytoplasmic domains, respectively. This results in activation of the transcription factor signal transducer and activator of transcription-4 (STAT4), which is critical for the generation of Th1 responses (Trinchieri, 2003). Through positive paracrine feedback, IFN-γ enhances IL-12 production from DCs and leads to increased T cell responsiveness to IL-12 by inducing the expression of IRF-1 which is essential for regulating the expression of IL12Rβ1(Szabo, Dighe et al., 1997;Trinchieri, 2003;Kano, Sato et al., 2008; Murphy, Terres et al., 1994). In the absence of IRF-1, naïve CD4+ T cells do not differentiate into IFN-γ producing CD4+ T cells (Kano, Sato et al., 2008). At the molecular level, IFN-γ interacts with its receptor, resulting in activation of Jak1 and Jak2 as well as phosphorylation of STAT1 which results in the induction of IFN-responsive gene transcription (Dalpke, Eckerle et al., 2003).
The IFN-γ-IL-12 signaling pathways are central in controlling both environmental and endogenous challenges (Rosenzweig & Holland, 2005). Prolonged incubation of DC with LPS and/or IFN-γ eventually results in decreased IL-12 production consequent to induction of expression of a family of eight suppressor of cytokine signaling (SOCS) proteins, each of which play a unique role in inhibiting cellular signaling. SOCS1 provides a major negative feedback for attenuating immunostimulatory functions; it negatively regulates IFN-γ, IL-2, IL-6, IL-4, TNF-alpha and IL-15 signaling through inhibition of JAKs in many lineages of immune cells (Alexander & Hilton, 2004;Kubo, Hanada et al., 2003). SOCS1 binds to the JAK activation loop through its Src-homology 2 (SH2) domain and targets JAK2 for degradation, leading to inhibition of STAT activation (Kubo, Hanada et al., 2003;Alexander & Hilton, 2004). SOCS-1 can also inhibit non-JAK-STAT signaling, such as that elicited by TNF and LPS (Chong, Thomas et al., 2002;Kinjyo, Hanada et al., 2002). The precise mechanisms by which SOCS-1 dampens LPS signaling and immune responses in DCs is not entirely clear, but may involve regulation of antigen presentation, co-stimulation and cytokine production by mature DCs via a mechanism mediated by Mal (MyD88 adapter-like protein)(Mansell, Smith et al., 2006).
It is not clear whether AIDS-related functional impairment of DCs is a result of their direct infection by HIV-1 or is indirect. Indirect mechanisms could involve exposure to virally encoded or induced proteins or chronic antigenic stimulation. Reports conflict on the extent to which DCs are infected with HIV; however, a recent report shows that a subset of myeloid DCs is infected (Granelli-Piperno, Shimeliovich et al., 2006). These infected DCs do not mature in response to a panel of stimuli, produce reduced levels of IL-12p40, increased levels of IL-10 (an inhibitor of IL-12 production) and exhibit very weak immunostimulatory functions after stimulation. However, because the extent of DC infection is generally thought to be rather limited in vivo, it is thought that indirect mechanisms play a central role in DC dysfunction in HIV disease (Ma & Montaner, 2000). During HIV infection, cytokines are hyper-expressed; many are inducers of and regulated by SOCS-1 (Alexander & Hilton, 2004;Kubo, Hanada et al., 2003).
Understanding the mechanisms by which long-lived immune responses are maintained is of intrinsic interest, but will also be important in the development of an effective HIV-1 vaccine. We have earlier reported that HIV-1 Tg rats have defects in the generation/maintenance of effector/memory phenotype T cell subsets, type 1 cytokine production and type 1 cytokine responses (Reid, Sadowska et al., 2001b;Reid, Abdelwahab et al., 2004;Yadav, Pati et al., 2006). Here we show that IL-10 induced over-expression of SOCS-1 by CD4+ T cells and DC is likely responsible for the quantitative and qualitative deficiencies in effector memory Th1 responses in Tg rats by disruption of the IL-12-IFN-γ signaling axis. We also report that IL-12p40 production following IFN-γ/LPS stimulation of BMDCs from HIV-1 Tg rats is increased by knocking down SOCS-1 expression. We show that SOCS-1 mRNA is elevated in CD4+ T cells from HIV-1 infected progressor patients and that this is correlated with decreased IRF-1 induction following IFN-γ stimulation relative to healthy controls and HIV-1 Natural Viral Suppressors (HIV infected patients who have undetectable HIV-1 viral loads without therapy). The results suggest a previously undescribed link between high levels of SOCS-1, defects in the innate immune response as determined by LPS induction of IL-12p40 in BMDCs, and dysregulation in adaptive Th1 effector memory responses.
Materials and methods
HIV-1 Tg and non-Tg animals
The construction of the HIV transgene and production of the Tg rats have been described (Reid, Sadowska et al., 2001a). Mature (12-15 month old) specific pathogen free (SPF) Tg rats and age-matched Fisher 344/NHsd non-Tg rats were used in our analyses and were housed under pathogen free conditions in microisolator cages on HEPA filtered ventilated racks. The University of Maryland Institute of Biotechnology Animal Care and Use Committee approved the experimental protocol.
Human subjects
Subjects were recruited from the Institute of Human Virology Clinical Research Unit. Written informed consent was obtained and procedures on patient material were collected under a protocol approved by the University of Maryland Committee on Human Research. All patients included in the study were adult (29- 60 years). Thirty milliliters of blood were collected for analysis. Inclusion criteria for viremic HIV+ progressors were as follows: (i) lack of prior antiretroviral therapy or lack of antiretroviral therapy for more than 60 days prior to study entry and (ii) viremia of >900 copies/ml, as well as (iii) CD4+ count of >200 cells/μl (in order to ensure that enough CD4+ T cells could be isolated for experiments). HIV-1 NVS, as described previously, 30 were included if they had both: (i) undetectable viral loads (<75 copies/ml) for at least 2 years without antiretroviral drugs) (ii) CD4+ count of >600.
Sorting CD4+ T cells
CD4+ T cells were prepared from spleens of mature (9-12 month) HIV-1 Tg rats and isogenic controls as described elsewhere (Coligan, Kruisbeek et al., 1994;Reid, Abdelwahab et al., 2004). Briefly, CD4+ T cells were isolated using negative selection StemSep™ rat cell separation kits following the manufacturer’s instructions (16052A, StemCell Technologies, Vancouver, BC). Naïve CD4+ T cells were defined by a CD45RC+/CD62L+ cell surface phenotype(Hylkema, van der et al., 2000;Ramirez & Mason, 2000). Naïve CD4+ T cells were first stained with PE conjugated mouse anti-rat CD45RC (554888, BD PharMingen), followed by anti-PE microbeads (Miltenyi Biotec, Auburn, CA) as described (Reid, Abdelwahab et al., 2004) and positively selected by AutoMacs under conditions specified by the manufacturer. The positive population was stained with FITC conjugated mouse anti-rat CD62L (PharMingen) and CD45RC+/CD62L+ purity determined by flow cytometry as described previously (Reid, Abdelwahab et al., 2004) and analyzed by FlowJo software. The sorted naïve T cell population purity was ≥85%. For adult human patient samples CD4+ T cells were separated from 40ml of blood using the negative selection RosetteSep® (15062, StemCell Technologies, Vancouver, BC) following the manufacturer’s protocol. The purity of CD4+ T cell population was 90-95% as determined by flow cytometry.
BMDC culture
Bone marrow was prepared from the femur of mature HIV-1 Tg and age-matched Fisher 344 controls as described elsewhere (Grauer, Wohlleben et al., 2002). Briefly, bone marrow cells were cultured in RPMI with 10% FBS and/or StemSpan (09650, Cell Signaling Technology, Boston, MA) in 5ng/ml rGM-CSF and rIL-4 (555111 and 555107, BD Biosciences) for 6-7 days followed by 2.5ng/ml rGM-CSF until day 8. Any contaminating B cells or macrophages were separated by positive section using AutoMacs (Miltenyi Biotec, Auburn, CA) under conditions described by the manufacturer. Generally, B cells were removed after staining with anti rat-CD45RA microbeads (Miltenyi Biotec, Auburn, CA); while contaminating macrophages were separated by staining with FITC conjugated mouse anti-rat CD68 followed by binding to anti FITC microbeads.
Cytokine analysis by ELISA
Purified BMDCs (1.0 × 106) from Tg and control Fisher rats were stimulated with 1μg/ml LPS (List Biological, Campbell, CA) and their supernatants collected. IL-12p40 and IL-10 levels were measured using ELISA cytokine detection kits (BioSource International, Camarillo, CA and R&D Systems, Minneapolis, MN, respectively) following the instructions given by the suppliers. The production of IFN-γ was measured with the Quantikine® Rat IFN-γ ELISA (R&D Systems). The detection limits of these ELISAs are 3, 10 and 10 pg/ml for IL-12, IL-10 and IFN-γ respectively. For relevant statistical processing of the data, all values below the detection limits were assigned a value extrapolated the standard curve.
Real-time PCR for IL-12Rβ1, SOCS-1, and IRF-1
Relative levels of specific mRNAs were quantified by real-time RT-PCR analyses using the IQ5 Multicolor Realtime PCR Detection System (Bio-Rad Laboratory, Hercules, CA). For IL12Rβ1 mRNA determinations, naïve CD4+ T cells were stimulated under Th1 conditions as described (Kano, Sato et al., 2008). Alternatively, purified BMDCs or CD4+ T cells were isolated from control and Tg rats and stimulated at 1.0 × 106 cell/ml for 24-48 hours with LPS at 1μg/ml or 1-3 hours with IFN-γ or rIL-10 (BD Pharmingen, San Diego, CA) at 10ng/ml or cultured 18 hours in LPS stimulated BMDC culture supernatants with or without anti IL-10 antibody (BD Pharmingen, San Diego, CA) at 50ng/ml. Total cellular RNA was prepared using an RNeasy mini kit (Qiagen, Valencia, CA). Quantitative real-time RT-PCR was carried out using a SYBR Green PCR kit (Biorad). The rat IL-12Rβ1 mRNA was amplified with: forward 5’-tcg cat aca ttg gct taa aca g-3’ and reverse 5’-gaa gtc ctc agg gtt gcg-3’; SOCS-1 mRNA was amplified with: forward 5’-agc cat cct cgt cct cgt c-3’ and reverse 5’-gcg gaa ggt gcg gaa gtg-3’; rat IRF-1 mRNA was amplified with: forward 5’-agc cac caa tgc cta tca ctc-3’ and reverse 5’-tgt tcc tgc tct ggt cct tc-3’. Human SOCS-1 mRNA was amplified with the following primers: forward 5’-ttc tgt agg atg gta ggac-3’ and reverse 5’-gag gag gag gaa gag gag-3’; human IRF-1 mRNA was amplified with: forward 5’-gac cag agc agg aac aag-3’ and reverse 5’-cca tca gag aag gta tca gg-3’. Samples were run in triplicate and the yield of IL-12Rβ1, SOCS-1 and IRF-1 PCR product was normalized to 18S RNA using an rRNA primer set. To control for DNA contamination, equal amounts of RNA were used without reverse transcriptase.
Cell Culture
Naïve CD4+ T lymphocytes were cultured in RPMI 1640 as previously described (Fujimoto, Tsutsui et al., 2002) and stimulated with plate-bound anti-CD3 plus anti-CD28 (10μg/ml and 5μg/ml respectively, BD Pharmingen, San Diego, CA) under Th1 conditions, for which anti-IL-4 antibody (10μg/ml, BD Pharmingen) and rIL-12 (1ng/ml, BD Pharmingen) were added to the culture media. For IL-2Rβ1 mRNA expression, day 3-5 Th1 polarized CD4+ T cells were harvested for RT-PCR as described above. For IL-12 induced IFN-γ, day 3-5 Th1 cells were washed, rested for 24 hours and stimulated for an additional 24 hours with rIL-12 (1ng/ml, BD Pharmingen) and supernatants collected for IFN-γ ELISA as described above. For IL-10 induced SOCS-1 mRNA analysis, non-Tg control CD4 T cells were stimulated with rIL-10 (10ng/ml BD Pharmingen) for 1 hour; additionally, CD4+ T cells from non-Tg controls were cultured for 18-hours in supernatants from LPS stimulated Tg and non-Tg control BMDC samples with/without added anti-IL-10 (50ng/ml, BD Pharmingen) antibody. Cells were harvested and analyzed by RT-PCR as described above.
Nucleofection of siRNA
BMDCs from Fisher 344/NHsd controls and aged matched HIV-1 Tg rats were cultured as described above for 8 days. For nucleofection of immature BMDCs, 2.0 × 106 cells were re-suspended in Nucleofector solution (100ul), (Human Dendritic Cell Nucleofection kit VPA-1004, Amaxa Biosystems) with 2.6ug of SOCS-1 ON-TARGET plus SMART siRNA pool (Dharmacon Inc.) and nucleofected using an Amaxa Nucleofector and program X002. As a positive and negative control cells were nucleofected with ON-TARGET plus Cyclophillin B pool (cat# D-001820-30-20) or Non-Targeting siRNA (cat# D-001810-01-20), respectively (Dharmacon Inc.) Immediately after nucleofection, cells were transferred into 500μl of pre-warmed media conditioned with 100ng/ml IL-4 and 100ng/ml GM-CSF for cells to recover for 18 hours. BMDCs were stained with 7-AAD and examined for viability by flow cytometry. BMDCs were suspended in RPMI-1640 at 1.0 × 106/ml and stimulated with 1μg/ml LPS and 10ng/ml rIFN-γ for indicated times. Cells were pelleted and supernatants collected and analyzed for IL-12p40 by ELISA. RNA was extracted from the collected cells and analyzed by RT-PCR. Nucleofection efficiency was determined by the percent Cyclophillin B mRNA inhibition. SOCS-1 mRNA inhibition was determined by comparisons of SOCS-1 expression between SOCS-1 siRNA treated groups versus non-targeting group as recommended by the manufacturer (Dharmacon Inc.). Expression of SOCS-1 mRNA by Tg and control DC was knocked down to 50% and 70%, respectively.
Statistics
Means were compared within and between two groups by the Mann-Whitney test for samples exhibiting non-normal distribution. For three groups, the Kruskal-Wallis test for groups exhibiting non-normal distribution was used and followed by the Dunn’s Multiple Comparisons post hoc test. P values were considered significant at P<0.05. Means were compared between identical groups by a paired T-test. Generally, analysis between RT-PCR reference and treatment group were considered significantly different if the lower 95% confidence limit of the treatment group was >1.
Results
HIV-1 Tg rat BMDCs express reduced IL-12 and increased IL-10 following LPS activation
The dysregulated IFN-γ production, reduced delayed type hypersensitivity and low numbers of T cells with a Th1 effector/memory phenotype seen with HIV-1 Tg rats suggested that the microenvironment in which antigen specific T cells receive activation signals was unfavorable to Th1 responses, development and/or maintenance. Since cytokine expression by DCs is critical for these processes, we tested expression by DCs of cytokines that regulate Th1 responses. Capture ELISAs for IL-12p40 and IL-10 of culture supernatants from LPS (1μg/ml)-activated purified BMDC from mature Tg and control rats showed (Figure 1A) that IL-12p40 expression by Tg rat BMDC was fourfold lower [1089 ± 807pg/ml and 1226 ± 883 after 24 hr (p=0.002) and 48 hr (p=0.002) respectively]than that of age-matched controls (4789 ± 2231pg/ml and 5825 ± 3320). IL-10 expression from Tg BMDC was 73 ± 61 pg/ml (p=0.015) 24 hr post-stimulation compared with 13 ± 11 pg/ml for age matched controls (Figure 1B). IL-12/IL-10 ratios at 24 hr post-stimulation are thus remarkably skewed (~14 for the Tg BMDC compared with ~360 for controls), suggesting that Th1/Th2 cytokine expression is highly dysregulated in a way that would lead to a lack of proper BMDC function.
Figure 1. Extracellular IL-12p40 and IL-10 expression by DC.
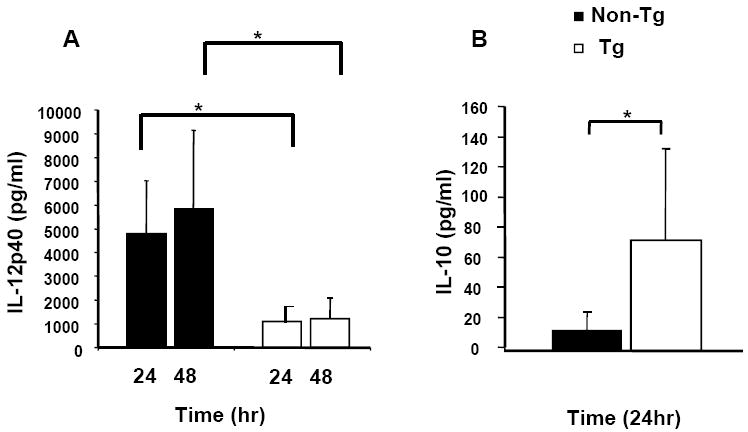
To analyze extracellular IL-12p40 and IL-10 production, culture supernatants from Tg and control rats (n=6) were assayed by capture ELISA. BMDC were stimulated with LPS and media was collected and analyzed in duplicate. (Panel A), expression of IL-12 by Tg BMDC was 1089 ± 807 pg/ml and 1226 ± 883 at 24 hr and 48 hr. stimulation compared with 4789 ± 2231 pg/ml and 5825 ± 3320 by age-matched controls at the same time points. (Panel B), IL-10 expression by Tg BMDC was 73 ± 61 pg/ml (n=6) pg/ml after 24 compared with 13 ± 11 for age matched controls. * Indicates a significant difference by Mann-Whitney test for samples exhibiting non-normal distribution.
HIV Tg rat BMDCs express elevated SOCS-1 mRNA after LPS and IFN-γ stimulation
As IL-12 production is positively regulated by IFN-γ and LPS stimulation and negatively regulated by SOCS-1(Trinchieri, 2003), we hypothesized that low expression of IL-12p40 by DC would be correlated with increased expression of SOCS-1 following stimulation with IFN-γ and/or LPS. Figures 2 A-B show that Tg BMDC cells had a 1.8 (P<0.05) and 4.5 fold, (P<0.04) increase in SOCS-1 mRNA relative to unstimulated controls following IFN-γ and LPS stimulation, respectively. Baseline SOCS-1 mRNA levels in unstimulated Tg BMDC relative to controls were also 1.5 fold higher (data not shown). These data suggest that SOCS-1 expression is dysregulated in Tg BMDCs and may down-regulate IL-12p40 expression.
Figure 2. Relative increase in SOCS-1 mRNA in BMDCs.
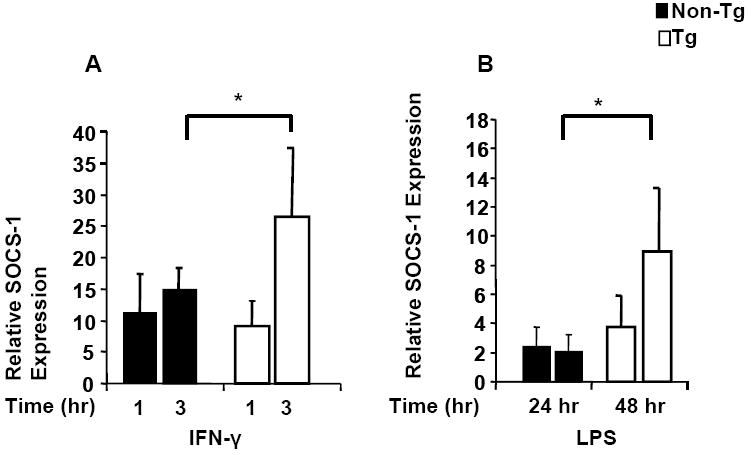
BMDC from Tg and control rats (n=4) were stimulated with 1 μg/ml of LPS for 24-48 hrs or IFN-γ at 10ng/ml for 1-3 hrs. Figure 2 (A-B) shows expression of SOCS-1 mRNA by BMDC. Levels of SOCS-1 mRNA were determined by real-time quantitative RT-PCR as described in the text. Samples were analyzed in triplicate and data normalized to the expression of 18S ribosomal RNA. SOCS-1 mRNA from BMDC of Tg was increased approximately 26 and 9.0-fold following 3 hr IFN-γ and 48 hr LPS stimulation, respectively compared with 15 and 2-fold increase in the non-Tg control. * Indicates a significant difference by Mann-Whitney test for samples exhibiting non-normal distribution.
Reduced SOCS-1 mRNA expression increases IL-12p40 production following LPS and IFN-γ stimulation of immature HIV-1 Tg rat BMDCs
As SOCS-1 negatively regulates IL-12 production following LPS stimulation and because of the positive effect of IFN-γ signaling on IL-12 production, we postulated whether siRNA knockdown of SOCS-1 expression in DC would elevate IL-12p40 expression. We nucleofected BMDCs with ON-TARGET Plus non-targeting siRNA Pool as a negative control and Cyclophilin B SMART Pool siRNA as a positive control for nucleofection/knockdown efficiency and obtained a calculated >90% transduction efficiency using the Dharmacon algorithm. Figure 3A-B shows that the ON-TARGET plus SOCS-1 siRNA SMART pool knocked down SOCS-1 mRNA expression in control and Tg DC by averages of 52 % and 47 %, respectively, at 48 hours after stimulation with LPS and IFN-γ. IL-12p40 expression was increased two-fold in both Tg and control cells, inversely proportional to the 50% knockdown of SOCS1 mRNA. Although the data show single representative control and Tg animals, Figure 3 A-B is representative of two experimental studies and have given generally similar results, suggesting that elevated SOCS-1 mRNA expression in Tg BMDC causes reduced IL-12p40 secretion in response to LPS and IFN-γ stimulation, and that this can be normalized by inhibiting SOCS-1 expression. Although IL-12p40 by Tg BMDC does not reach the level of control cells, due to their intrinsic higher levels of SOCS-1 mRNA compared to controls, more efficient SOCS1 knockdown would likely further increase IL-12p40 secretion.
Figure 3. Increase in IL-12p40 production in BMDCs following SOCS-1 siRNA nucleofection.
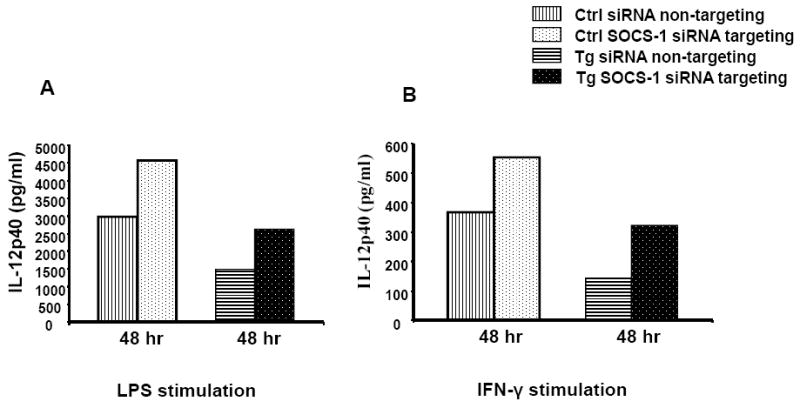
BMDC from Tg and control rats were stimulated with 1 μg/ml of LPS or IFN-γ at 10ng/ml for 48 hrs following SOCS-1 siRNA nucleofection. Figure 3 shows expression of IL-12p40 production by BMDC following siRNA treatment, determined by ELISA as described in the text. IL-12p40 production from SOCS-1 siRNA treated Tg BMDC increased approximately 2-fold following 48 hr of LPS (Figure 3A) or IFN-γ (Figure 3B) stimulation (2424 pg/ml and 322pg/ml), respectively, compared with siRNA-untargeted samples (1494 pg/ml and 143 pg/ml). siRNA-treated controls were 1.5-fold increased (4563 pg/ml and 553 pg/ml), respectively, compared with untreated samples (2974 pg/ml and 367 pg/ml). This figure is representative of two separate experimental studies.
HIV Tg rat CD4+ T cells express elevated levels of SOCS-1 and lower levels of IRF-1after IFN-γ stimulation
As IFN-γ responses are mediated through STAT-1 activation and because induction of SOCS-1 and IL-12Rβ1 by IFN-γ requires interferon regulatory factor-1 (IRF-1), a downstream target of STAT1, we asked whether high SOCS-1 mRNA expression in Tg CD4+ T cells is correlated with elevated basal expression of IRF-1. CD4+ T cells (n=5) were stimulated with 10ng/ml of IFN-γ for 0, 1hr and 3hr and analyzed by RT-PCR. Figure 4A shows that 1 hour after IFN-γ stimulation, Tg CD4+ T cells had a 4.0 fold increase (p=0.028) in SOCS-1 mRNA relative to non-Tg controls. Importantly, basal expression of SOCS-1 in unstimulated Tg CD4+ T cells was significantly elevated more than 2 fold higher than that of the controls. Figure 4B shows that IRF-1 mRNA expression in CD4+ T cells was induced 6.4 fold (p=0.028) in control cells relative to Tg cells 3 hrs after IFN-γ stimulation. However, expression of IRF-1 mRNA in unstimulated Tg cells was 1.8-fold higher than in the controls. These data suggest that the more rapid induction kinetics of SOCS-1 mRNA in Tg cells is a result of high baseline levels of IRF-1; failure to further elevate IRF-1 expression following treatment with IFN-γ is indicative of a defect in IFN-γ mediated signal transduction pathways, likely due to the elevated SOCS-1 expression in Tg CD4+ T cells.
Figure 4. Relative increase in SOCS-1 and IRF-1 mRNA in CD4+ T cells.
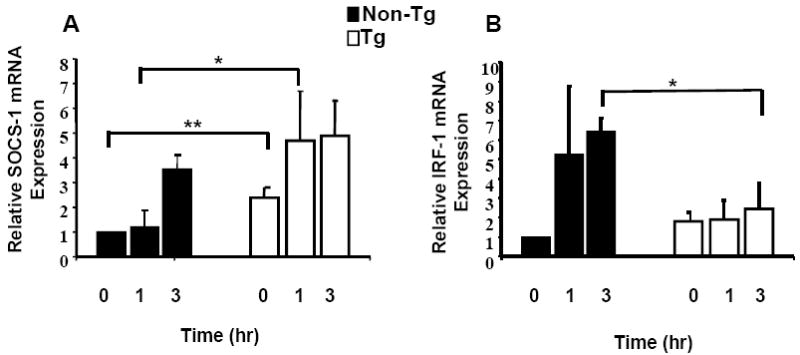
CD4+ T cells from Tg and control rats were stimulated with 10ng/ml IFN-γ for 1-3 hrs. Panel A shows expression of SOCS-1 mRNA by CD4+ T cells. Levels of SOCS-1 mRNA were determined by real-time quantitative RT-PCR as described in the text. Samples were analyzed in triplicate and data normalized to the expression of 18S ribosomal RNA. Baseline SOCS-1 mRNA from Tg CD4+ T cells was approximately 2-fold elevated compared to non-Tg control and increased 4.7-fold following 1 hr of IFN-γ treatment, compared with 1.1-fold increase in controls. Panel B shows that induction of IRF-1 in Tg CD4+ T cells was 1.9 and 2.4-fold at 1 and 3 hr respectively, versus induction in control CD4+ T cells of 5.0 and 6-fold. Baseline expression of IRF-1 in unstimulated Tg CD4+ T cells was 1.8-fold higher than controls. * Indicates a significant difference by Mann-Whitney test for samples exhibiting non-normal distribution. ** Indicates a significant difference by comparing the lower 95% confidence limit of the Tg sample (lower limit value of 1.5) to the non-Tg control (lower limit value of 1.0).
HIV-1 Tg rat Th1 polarized cells express reduced levels of IL-12Rβ1 mRNA and IFN-γ protein after stimulation
Consistent with the results in Figure 4B showing a failure to induce IRF-1 mRNA in Tg CD4+ T cells, Figure 5A shows that induction of the IRF-1 responsive gene IL-12Rβ1 was abrogated in Tg CD4+ T cells cultured for 5 (p= 0.03) days under Th1 polarizing conditions. Figure 5B demonstrates that when day 5 Th1 polarized Tg cells were washed, rested overnight and re-stimulated with rat IL-12 (1ng/ml), they showed a profound defect in IFN-γ production (p=0.035). These results suggest that SOCS-1 induction by high baseline levels of IRF-1 is defective for IL-12Rβ1-mediated IFN-γ production, IL-12-INF-γ signaling and maintenance of effector/memory Th1 lineage cells in HIV-1 Tg rats. It is tempting to speculate that a similar mechanism disrupts Th1 responses in HIV-1 infected patients.
Figure 5. Impaired IL-12Rβ 1 mRNA and secondary IFN-γ expression from Th1 polarized T cells.
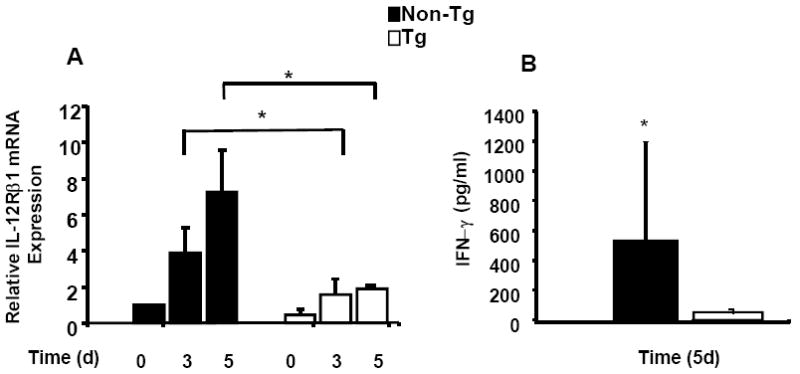
Panel A shows IL-12Rβ1 mRNA expression when purified naïve CD4 T cells were stimulated for 3 and 5 days under Th1 conditions. Samples were analyzed in triplicate and data normalized to the expression of 18S ribosomal RNA. IL-12Rβ1 mRNA from Tg CD4+ T cells (n=4) was increased 1.5 and 1.8-fold following 3 and 5 days under Th1 conditions, compared with 4.1 and 7.2 -fold increase in the non-Tg control (n=4) respectively. Panel B shows IFN-γ expression when day 5 Th1 T cells were rested overnight and then pulsed with 1ng/ml rIL-12 for 24 hours as described in the text. Samples were analyzed in duplicate for IFN-γ by ELISA. Interferon-gamma production from Tg (n=5) Th1 conditioned cells was 32 ± 38 pg/ml compared with 528 ± 660 pg/ml from non-Tg control (n=4). * Indicates a significant difference by Mann-Whitney test for samples exhibiting non-normal distribution.
IL-10 induction of SOCS-1 expression in non-Tg CD4+ T cells
Because Tg BMDC secreted elevated levels of IL-10 following LPS stimulation (Figure 1B), and because IL-10 induces SOCS-1 expression in dendritic and CD4+ T cells (Ding, Chen et al., 2003), we first measured SOCS-1 mRNA levels in control rat CD4+ T cells following 1 hr of treatment with 10ng/ml of rIL-10. SOCS-1 mRNA increased 1.5 fold (p=0.047) relative to unstimulated controls Figure 6A. Figure 6B shows that control rat CD4+ T cells cultured for 18 hours in supernatants from BMDC stimulated with LPS for 24 hr (as shown in Figure 1B) elevated SOCS-1 mRNA 2.2 and a 4.4 fold in control and Tg samples, respectively, relative to unstimulated controls. Consistent with the idea that IL-10 induces SOCS-1 expression in CD4+ T cells, treatment of these supernatants with anti-IL-10 antibody reduced SOCS-1 mRNA induction to 1.2 and 2.4-fold in control and Tg samples, respectively. Interestingly, basal expression of SOCS-1 mRNA was elevated 2.7-fold when CD4+ T cells were cultured in supernatants from unstimulated Tg BMDC relative to unstimulated control BMDC. These data suggest that the increased levels of SOCS-1 observed with Tg rats are indeed due to elevated expression of IL-10. The inability of anti-IL-10 to completely reduce SOCS-1 induction in CD4+ T cells by supernatants may be because other anti-inflammatory cytokines also play a role in SOCS-1 induction in Tg rats. Alternatively, it may be that we incompletely inhibited IL-10 activity in these samples, although we used a large excess of antibody. Elevated serum and lymph node IL-10 levels have been described in HIV infected people, and would likely have a similar effect on SOCS-1 induction to that shown here.
Figure 6. IL-10 induction of SOCS-1 mRNA in non-Tg control CD4+ T cells.
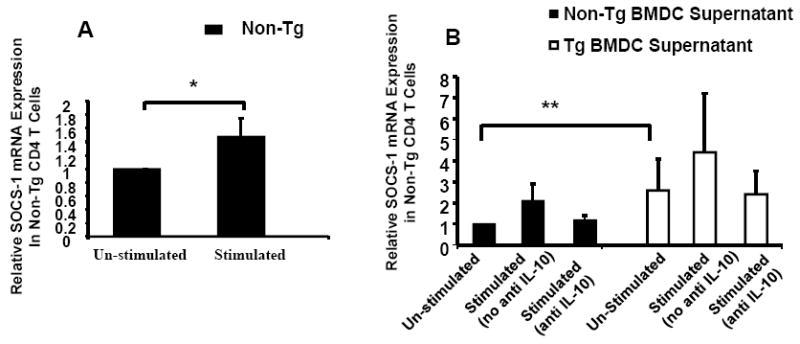
Panel A shows CD4+ T cells from non-Tg controls stimulated with rIL-10 (10ng/ml) for 1 hour. Shown is the expression of SOCS-1 mRNA in control CD4+ T cells. Levels of SOCS-1 mRNA were determined by real-time quantitative RT-PCR as described in Materials and Methods. Samples were analyzed in triplicate and data normalized to the expression of 18S ribosomal RNA. SOCS-1 mRNA from CD4+ T cells (n=4) was increased 1.5 fold relative to un-stimulated controls. Panel B shows CD4+ T cells from non-Tg controls were cultured for 18-hours in supernatants from LPS stimulated Tg and non-Tg control BMDC samples with/without added anti-IL-10 (50ng/ml) antibody. Shown is the expression of SOCS-1 mRNA in control CD4+ T cells. Levels of SOCS-1 mRNA were determined by real-time quantitative RT-PCR as described above. Samples were analyzed in triplicate and data normalized to the expression of 18S ribosomal RNA. SOCS-1 mRNA from CD4+ T cells (n=4) cultured in stimulated BMDC non-Tg control supernatant was increased 2.2 fold and decreased to 1.2 fold when anti-IL-10 antibody was added relative un-stimulated controls. SOCS-1 mRNA from CD4+ T cells (n=4) cultured in stimulated BMDC Tg supernatant was increased 4.4-fold and decreased to 2.4 fold when anti-IL-10 antibody was added. Interestingly, basal SOCS-1 mRNA expression for cells cultured in Tg un-stimulated supernatants was 2.7-fold higher than un-stimulated non-Tg controls. * Indicates a significant difference by Paired t-test. ** Indicates a significant difference by comparing the lower 95% confidence limit of CD4 T cells cultured in un-stimulated Tg BMDC Supernatant (lower limit value of 1.4) to un-stimulated Non-Tg BMDC Supernatant (lower limit value of 1.0).
HIV-1 progressor CD4+ T cells express elevated levels of SOCS-1 mRNA and lower levels of IRF-1 after IFN-γ stimulation
Because IFN-γ induction of IRF-1 was abrogated in the HIV-1 Tg rat, likely due to elevated expression of SOCS-1, we analyzed SOCS-1 and IRF-1 mRNA levels by RT-PCR following IFN-γ stimulation of human CD4+ T cells with 10ng/ml of IFN-γ for 0, 1hr and 3hr. Figure 6 A-B shows that 3 hour after IFN-γ stimulation, CD4+ T cells from individuals with progressive HIV-1 infection (n=7) showed a significantly higher SOCS-1 mRNA level and significantly lower IRF-1 mRNA relative to those from either healthy controls or NVS patients (n=8) as judged by the Kruskal Wallis test and Dunn’s multiple comparisons test: p<0.05. These data suggest that higher SOCS-1 levels in CD4+ T cells from untreated persons with progressive HIV-1 abrogate IFN-γ induction of IRF-1.
Discussion
These results demonstrate that IL-10 induces elevated levels of expression of SOCS-1 in CD4+ T cells, which appears in turn to lead to deficiencies in effector memory Th1 responses by HIV-1 Tg rats through dysregulation of the IFN-γ/IRF-1/IL-12 signaling network. In support of this idea, IL-12p40 production by BMDCs from HIV-1 Tg rats in response to IFN-γ/LPS stimulation was increased when SOCS-1 expression was inhibited. It is likely that chronic IL-10 induced over-expression of SOCS-1 leads to decreased IL-12p40 expression by BMDCs and subsequent activation-induced IFN-γ, IRF-1 and IL-12Rβ1 expression by CD4+ T cells. This would be expected to have a cumulative negative effect on IL-12/IFN-γ signal transduction, IL-12 responsiveness, and the generation and/or survival of memory CD4+ T cells with Th1 effector function. These data suggest that the situation is similar in infected humans, since SOCS-1 mRNA was elevated in CD4+ T cells from HIV-1 infected progressor patients and was correlated with defective induction of IRF-1 following IFN-γ stimulation, compared to healthy controls and HIV-1 NVS patients (HIV-infected patients with undetectable HIV-1 viral load in the absence of therapy).
These defects could have additional consequences. IL-23 is a recently identified heterodimeric cytokine consisting of the IL12p40 subunit and a novel protein (p19), and its heterodimeric receptor (IL-23R) requires expression of IL-12Rβ1 subunit for signaling. IL-17-producing T helper (Th17) cells differentiate from naïve CD4+ T cells in response to transforming growth factor-β and IL-6 but also require IL-23 for maintenance or expansion (Veldhoen, Hocking et al., 2006). IL-17 production is reduced in both HIV-1 and SIV infections (Ndhlovu, Chapman et al., 2008;Raffatellu, Santos et al., 2008). We have found that IL-17 production by Tg rat CD4+ T cells is more than three fold lower than that of control CD4+ T cells (W.R.;data not shown). Interestingly, IL-17 regulates the expression of β-defensin-2(Liang, Tan et al., 2006), a peptide with antimicrobial properties that is also an inhibitor HIV-1 infection. Sun et al. have also reported that β-defensin-2 is dramatically diminished in the oral cavity of HIV-1 infected patients, predisposing them to oral opportunistic infections(Sun, Finnegan et al., 2005). It is tempting to speculate that overexpresssion of SOCS-1 negatively regulates the development/maintenance of both Th1 and Th17 responses during HIV-1 disease.
A 50% knockdown of SOCS-1 mRNA expression was achieved in Tg BMDCs after 48 hours of stimulation with LPS and IFN-γ. IL-12p40 expression was increased about two-fold from both Tg and control cells (Figure 3 A-B), proportional to the 50% knockdown of SOCS1, and IL-10 levels in the same supernatants were reduced (data not shown). These data raise the possibility that inhibiting SOCS-1 expression would improve IL-12 responsiveness and the generation and/or survival of Th1 effector/memory T cells by increasing IFN-γ signal transduction efficiency, IRF-1 induction and IL-12Rβ1 expression. We are currently investigating this possibility.
HIV-1 infected progressor patients demonstrated elevated levels of SOCS-1 mRNA and reduced IRF-1 mRNA in response to IFN-γ signaling, suggesting that elevated SOCS-1 also inhibits IFN-γ induced responses in this group of patients relative to uninfected controls or NVS patients. We are currently investigating IL-12 responsiveness and the role of SOCS-1 in modifying these responses in humans.
Collectively, these data suggest that IL-10 induction of SOCS-1 contributes to HIV pathogenesis by affecting both innate and adaptive Th1 immune responses. Interleukin-10 is a key regulatory cytokine that suppresses immune responses and blocks IFN-γ synthesis by DC-stimulated Th1 cells, IL-5 and IL-2 production by T cells, and IL-1α, IL-1β, IL-6 IL-8, IL-12 and TNF-α production by monocytes. IL-10 induces expression of both SOCS-1 and -3 mRNA and inhibits STAT-1 activation by IFN-γ. Activated macrophages from Tg rats produce high levels of IL-10 (Chang, Beltran et al., 2007) and we show here that LPS-stimulated Tg BMDC also secrete increased IL-10, as well as decreased IL-12p40 (Figure 1). We show here that rIL-10 scientifically induces SOCS-1mRNA (Figure 6A). Although supernatants from control BMDC induce SOCS-1 expression by control CD4+ T cells, induction is not as great as with supernatants from Tg BMDC, and induction with Tg BMDC supernatants (but not that of control BMDC) cannot be completely inhibited with anti-IL-10, suggesting that other soluble factors from Tg BMDC may also contribute to SOCS-1 mRNA induction (Figure 6B). It appears that IL-10 released in vivo from both dendritic and non-dendritic sources may serve to increase constitutive levels of SOCS-1 mRNA in CD4+ T cells, similar to what is shown in Figure 6. The mechanism leading to increased Il-10 expression is obviously a key question. Although we do not yet have direct evidence for involvement of specific HIV-1 proteins, it has been shown that HIV-1 Nef may be secreted, induces SOCS-1 protein and IL-10 in human cells(Qiao, He et al., 2006;Brigino, Haraguchi et al., 1997;Haraguchi, Cianciolo et al., 1998).
In summary, defects in IL-12p40 production by HIV-1 Tg rats are likely mediated by elevated IL-10 production by BMDC, leading to induction of SOCS-1 mRNA in BMDCs and CD4+ T cells. Increased basal expression of SOCS-1 in CD4+ T cells from Tg rats is correlated with decreased IFN-γ induction of IRF-1 mRNA and IL-12Rβ1 and a decrease in IL-12-induced IFN-γ production from Th1 polarized cells. Interestingly, increased expression of SOCS-1 in HIV-1 infected progressor patients is also correlated with a decrease in IFN-γ mediated induction of IRF-1 mRNA. We also demonstrated that IL-12p40 production following IFN-γ and LPS stimulation of BMDCs from HIV-1 Tg rats is increased after inhibition of SOCS-1 expression by RNA interference. The outlined immune abnormalities suggest a causative link between IL-10 induction of SOCS-1 and defects in IL-12p40 production by BMDCs and IRF-1 and IL-12Rβ1 by CD4+ T cells following LPS and or IFN-γ signaling. This likely adversely affects IL-12 responsiveness and the development/maintenance of Th1 effector/memory responses in the Tg rats.
The decrease in IFN-γ and IL-12 responsiveness, IL-12 production and dysregulation in Th1 effector/memory responses that characterize the Tg rats recapitulate several aspects of the immune pathology associated with HIV-1 and AIDS in humans. The results suggest a previously undescribed link between high levels of SOCS-1, defects in the innate immune response as determined by LPS induction of IL-12p40 in BMDCs, and dysregulation in adaptive Th1 immune responses that could help explain the loss of Th1 immune competence in AIDS patients. Obviously, a detailed analysis using infected human cells will be important to establish the relevance of our current findings to natural infection.
Figure 7. Dysregulation of SOCS-1 and IRF-1 mRNA in CD4 T cells from HIV-1 Infected Patient Samples Following IFN-γ Stimulation.
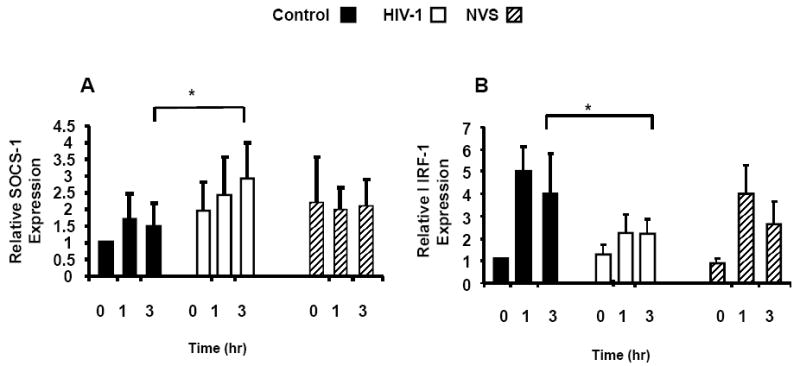
CD4+ T cells from healthy controls, HIV-1 infected patients (progressor) and HIV-1 infected NVS patents were stimulated with 10ng/ml IFN-γ for 1-3 hrs. Panel A shows expression of SOCS-1 mRNA by CD4+ T cells. Levels of SOCS-1 mRNA were determined by real-time quantitative RT-PCR as described in the text. Samples were analyzed in triplicate and data normalized to the expression of 18S ribosomal RNA. SOCS-1 mRNA induction at 1 and 3 hours following IFN-γ stimulation for healthy controls [1.7 and 1.4-fold increased] and NVS patents were [2.0 and 2.1 -fold increased]; while, HIV-1 progressive disease patients SOCS-1 levels were 3.2 and 2.2-fold increased. Panel B shows that induction of IRF-1 in HIV-1 patients CD4+ T cells with progressive disease was 3.1 and 2.2-fold at 1 and 3 hr respectively, versus induction in NVS CD4 T cells of 3.9 and 2.7 and control CD4+ T cells of 5.0 and 4.0-fold. * Indicates a significant difference by Kruskal Wallis test and Dunn’s multiple comparisons test.
Acknowledgments
We thank Drs. Charles E. Egwuagu, Gene Shearer and Marvin Reitz for their critical reading of this manuscript. We also thank members of the Core facility at the Institute of Human Virology for their technical assistance. This work was supported by grant R01 A1063171 from the NIH/NIAID.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Brigino E, Haraguchi S, Koutsonikolis A, Cianciolo GJ, Owens U, Good RA, Day NK. Interleukin 10 is induced by recombinant HIV-1 Nef protein involving the calcium/calmodulin-dependent phosphodiesterase signal transduction pathway. Proc Natl Acad Sci U S A. 1997;94:3178–3182. doi: 10.1073/pnas.94.7.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SL, Beltran JA, Swarup S. Expression of the mu opioid receptor in the human immunodeficiency virus type 1 transgenic rat model. J Virol. 2007;81:8406–8411. doi: 10.1128/JVI.00155-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong MM, Thomas HE, Kay TW. Suppressor of cytokine signaling-1 regulates the sensitivity of pancreatic beta cells to tumor necrosis factor. J Biol Chem. 2002;277:27945–27952. doi: 10.1074/jbc.M110214200. [DOI] [PubMed] [Google Scholar]
- Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W. Current Protocols in Immunology. John Wiley and Sons; New York: 1994. [Google Scholar]
- Dalpke AH, Eckerle S, Frey M, Heeg K. Triggering of Toll-like receptors modulates IFN-gamma signaling: involvement of serine 727 STAT1 phosphorylation and suppressors of cytokine signaling. Eur J Immunol. 2003;33:1776–1787. doi: 10.1002/eji.200323621. [DOI] [PubMed] [Google Scholar]
- Ding Y, Chen D, Tarcsafalvi A, Su R, Qin L, Bromberg JS. Suppressor of cytokine signaling 1 inhibits IL-10-mediated immune responses. J Immunol. 2003;170:1383–1391. doi: 10.4049/jimmunol.170.3.1383. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Tsutsui H, Yumikura-Futatsugi S, Ueda H, Xingshou O, Abe T, Kawase I, Nakanishi K, Kishimoto T, Naka T. A regulatory role for suppressor of cytokine signaling-1 in T(h) polarization in vivo. Int Immunol. 2002;14:1343–1350. doi: 10.1093/intimm/dxf094. [DOI] [PubMed] [Google Scholar]
- Granelli-Piperno A, Shimeliovich I, Pack M, Trumpfheller C, Steinman RM. HIV-1 selectively infects a subset of nonmaturing BDCA1-positive dendritic cells in human blood. J Immunol. 2006;176:991–998. doi: 10.4049/jimmunol.176.2.991. [DOI] [PubMed] [Google Scholar]
- Grauer O, Wohlleben G, Seubert S, Weishaupt A, Kampgen E, Gold R. Analysis of maturation states of rat bone marrow-derived dendritic cells using an improved culture technique. Histochem Cell Biol. 2002;117:351–362. doi: 10.1007/s00418-002-0384-4. [DOI] [PubMed] [Google Scholar]
- Haraguchi S, Cianciolo GJ, Good RA, James-Yarish M, Brigino E, Day NK. Inhibition of interleukin-2 and interferon-gamma by an HIV-1 Nef-encoded synthetic peptide. AIDS. 1998;12:820–823. [PubMed] [Google Scholar]
- Hylkema MN, van der DM, Pater JM, Kampinga J, Nieuwenhuis P, Groen H. Single expression of CD45RC and RT6 in correlation with T-helper 1 and T-helper 2 cytokine patterns in the rat. Cell Immunol. 2000;199:89–96. doi: 10.1006/cimm.1999.1607. [DOI] [PubMed] [Google Scholar]
- Kano S, Sato K, Morishita Y, Vollstedt S, Kim S, Bishop K, Honda K, Kubo M, Taniguchi T. The contribution of transcription factor IRF1 to the interferon-gamma-interleukin 12 signaling axis and TH1 versus TH-17 differentiation of CD4+ T cells. Nat Immunol. 2008;9:34–41. doi: 10.1038/ni1538. [DOI] [PubMed] [Google Scholar]
- Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, Yoshida H, Kubo M, Yoshimura A. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–591. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Montaner LJ. Proinflammatory response and IL-12 expression in HIV-1 infection. J Leukoc Biol. 2000;68:383–390. [PubMed] [Google Scholar]
- Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, Nicholson SE, Hilton DJ, O’Neill LA, Hertzog PJ. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- Murphy EE, Terres G, Macatonia SE, Hsieh CS, Mattson J, Lanier L, Wysocka M, Trinchieri G, Murphy K, O’Garra A. B7 and interleukin 12 cooperate for proliferation and interferon gamma production by mouse T helper clones that are unresponsive to B7 costimulation. J Exp Med. 1994;180:223–231. doi: 10.1084/jem.180.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KM, Ouyang W, Farrar JD, Yang J, Ranganath S, Asnagli H, Afkarian M, Murphy TL. Signaling and transcription in T helper development. Annu Rev Immunol. 2000;18:451–494. doi: 10.1146/annurev.immunol.18.1.451. [DOI] [PubMed] [Google Scholar]
- Ndhlovu LC, Chapman JM, Jha AR, Snyder-Cappione JE, Pagan M, Leal FE, Boland BS, Norris PJ, Rosenberg MG, Nixon DF. Suppression of HIV-1 plasma viral load below detection preserves IL-17 producing T cells in HIV-1 infection. AIDS. 2008;22:990–992. doi: 10.1097/QAD.0b013e3282ff884e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao X, He B, Chiu A, Knowles DM, Chadburn A, Cerutti A. Human immunodeficiency virus 1 Nef suppresses CD40-dependent immunoglobulin class switching in bystander B cells. Nat Immunol. 2006;7:302–310. doi: 10.1038/ni1302. [DOI] [PubMed] [Google Scholar]
- Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, Godinez I, Sankaran S, Paixao TA, Gordon MA, Kolls JK, Dandekar S, Baumler AJ. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. 2008;14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Mason D. Recirculatory and sessile CD4+ T lymphocytes differ on CD45RC expression. J Immunol. 2000;165:1816–1823. doi: 10.4049/jimmunol.165.4.1816. [DOI] [PubMed] [Google Scholar]
- Reid W, Abdelwahab S, Sadowska M, Huso D, Neal A, Ahearn A, Bryant J, Gallo RC, Lewis GK, Reitz M. HIV-1 transgenic rats develop T cell abnormalities. Virology. 2004;321:111–119. doi: 10.1016/j.virol.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Reid W, Sadowska M, Denaro F, Rao S, Foulke J, Jr, Hayes N, Jones O, Doodnauth D, Davis H, Sill A, O’Driscoll P, Huso D, Fouts T, Lewis G, Hill M, Kamin-Lewis R, Wei C, Ray P, Gallo RC, Reitz M, Bryant J. An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc Natl Acad Sci U S A. 2001a;98:9271–9276. doi: 10.1073/pnas.161290298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid W, Sadowska M, Denaro F, Rao S, Foulke J, Jr, Hayes N, Jones O, Doodnauth D, Davis H, Sill A, O’Driscoll P, Huso D, Fouts T, Lewis G, Hill M, Kamin-Lewis R, Wei C, Ray P, Gallo RC, Reitz M, Bryant J. An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc Natl Acad Sci U S A. 2001b;98:9271–9276. doi: 10.1073/pnas.161290298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig SD, Holland SM. Defects in the interferon-gamma and interleukin-12 pathways. Immunol Rev. 2005;203:38–47. doi: 10.1111/j.0105-2896.2005.00227.x. [DOI] [PubMed] [Google Scholar]
- Sun L, Finnegan CM, Kish-Catalone T, Blumenthal R, Garzino-Demo P, La Terra Maggiore GM, Berrone S, Kleinman C, Wu Z, Abdelwahab S, Lu W, Garzino-Demo A. Human beta-defensins suppress human immunodeficiency virus infection: potential role in mucosal protection. J Virol. 2005;79:14318–14329. doi: 10.1128/JVI.79.22.14318-14329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–824. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Yadav A, Pati S, Nyugen A, Barabitskaja O, Mondal P, Anderson M, Gallo RC, Huso DL, Reid W. HIV-1 transgenic rat CD4+ T cells develop decreased CD28 responsiveness and suboptimal Lck tyrosine dephosphorylation following activation. Virology. 2006;353:357–365. doi: 10.1016/j.virol.2006.05.026. [DOI] [PubMed] [Google Scholar]